
Recent Approaches in Transition Metal-Catalyzed Chalcogenative Heteroannulation of Alkenes and Alkynes
 , ,
, , 
{kind=link}
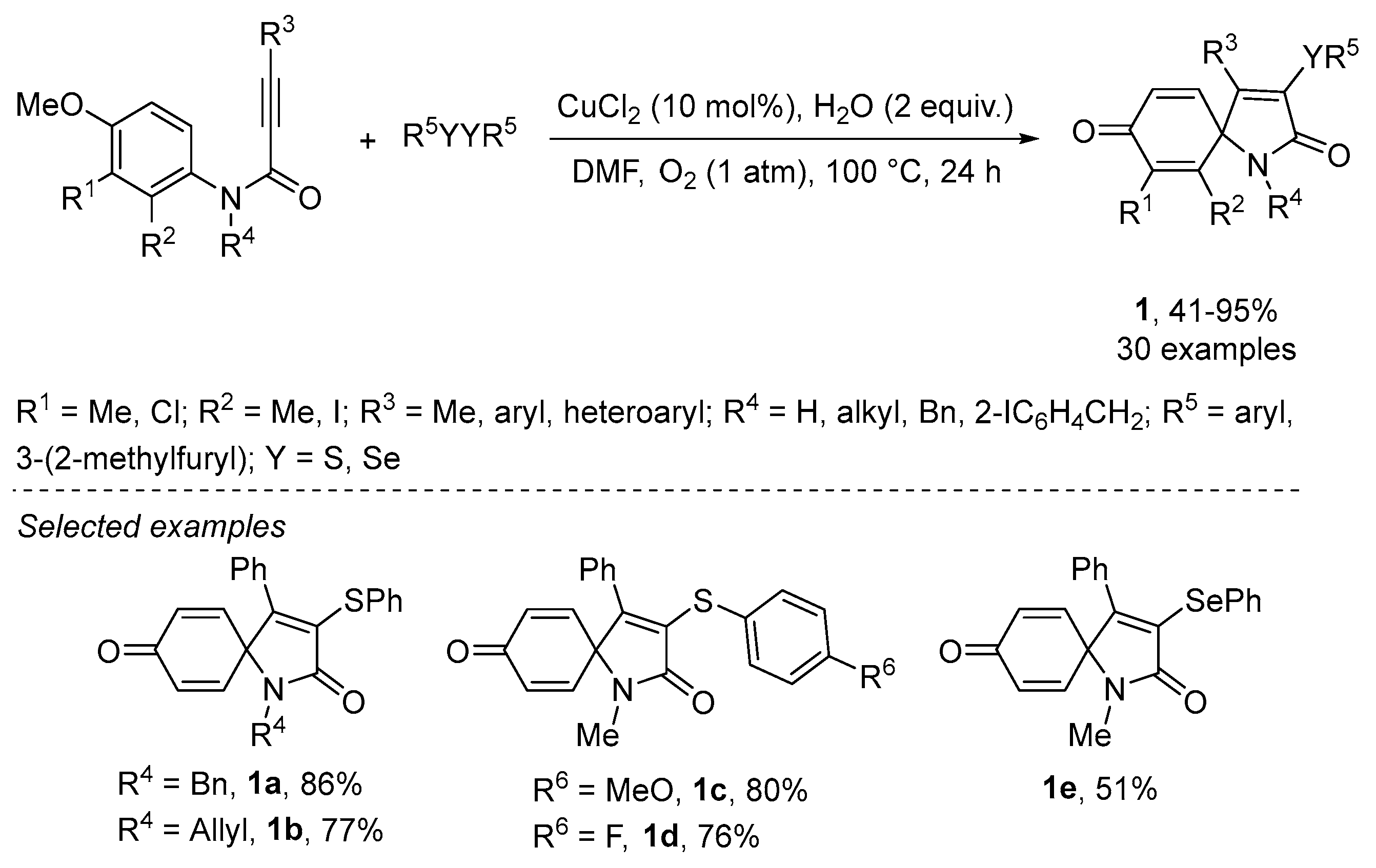{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
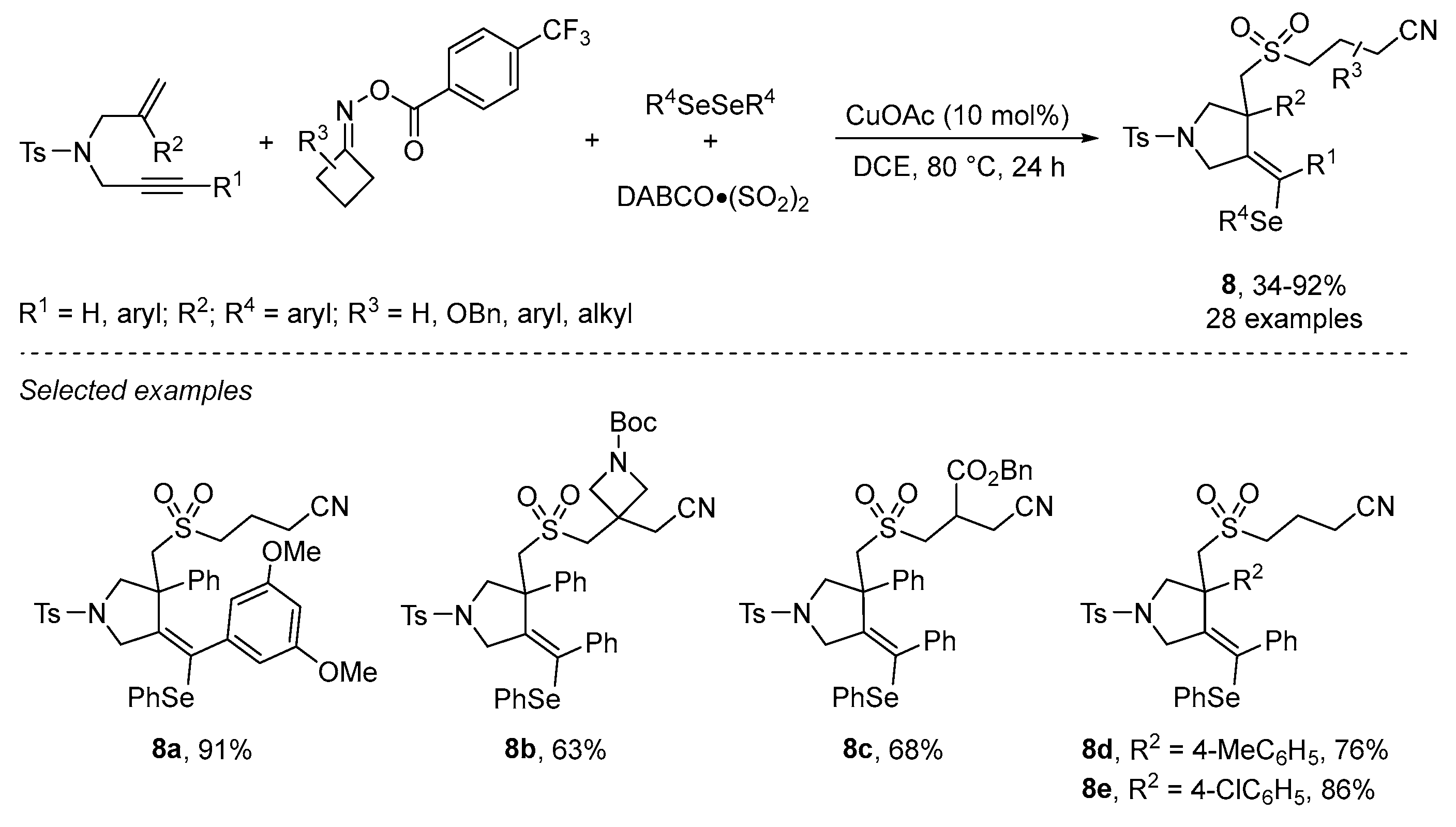{kind=link}
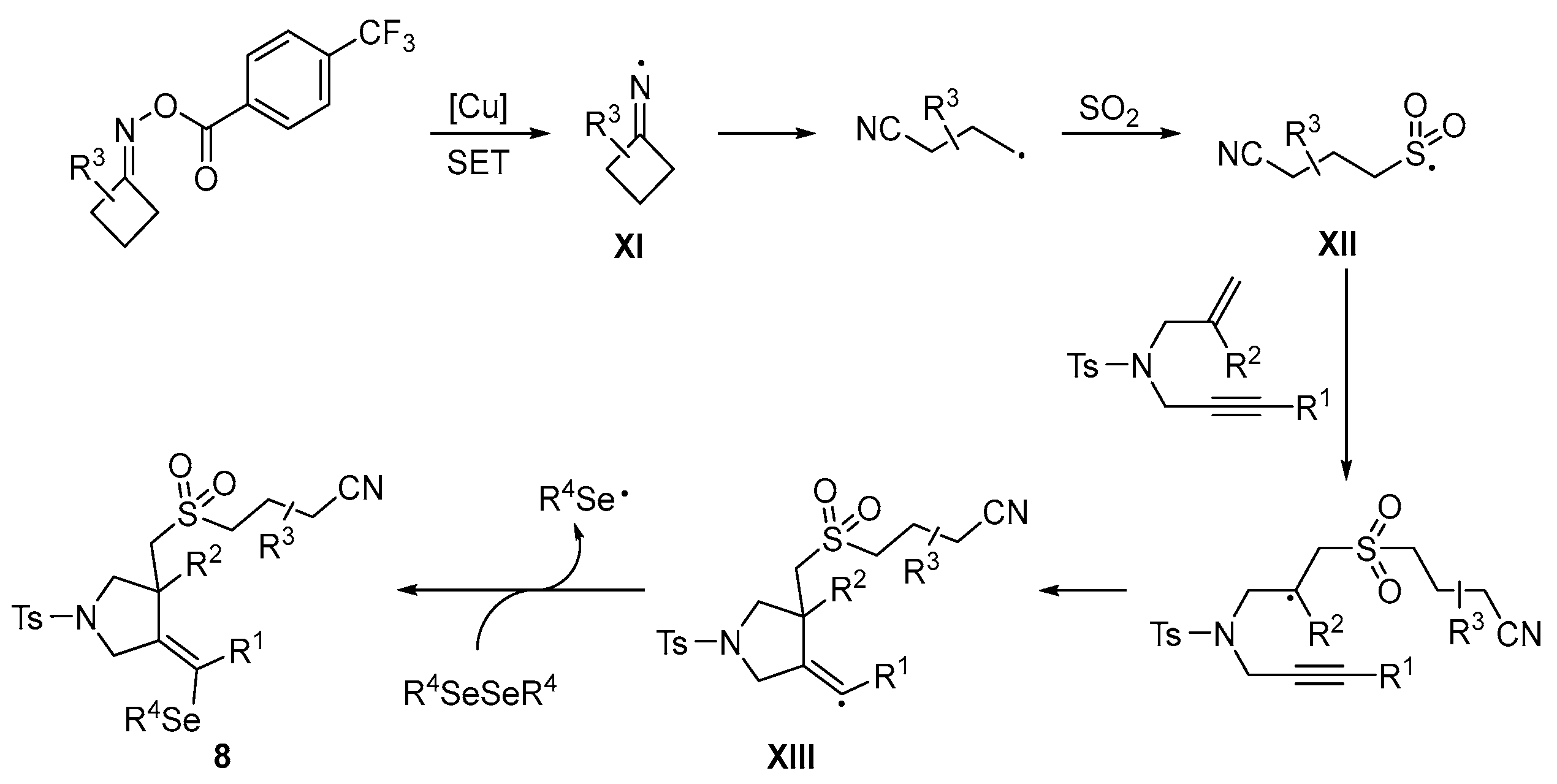{kind=link}
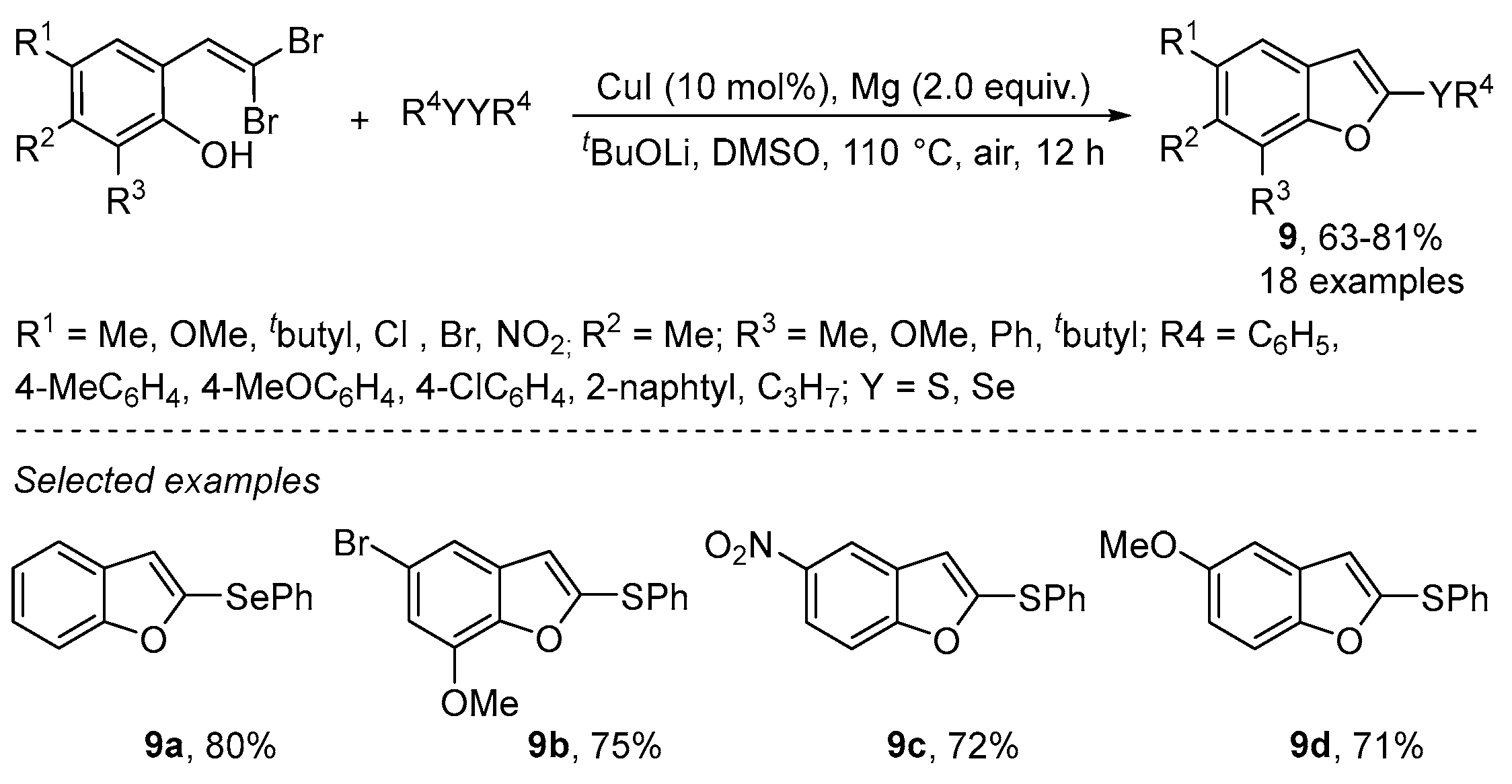{kind=link}
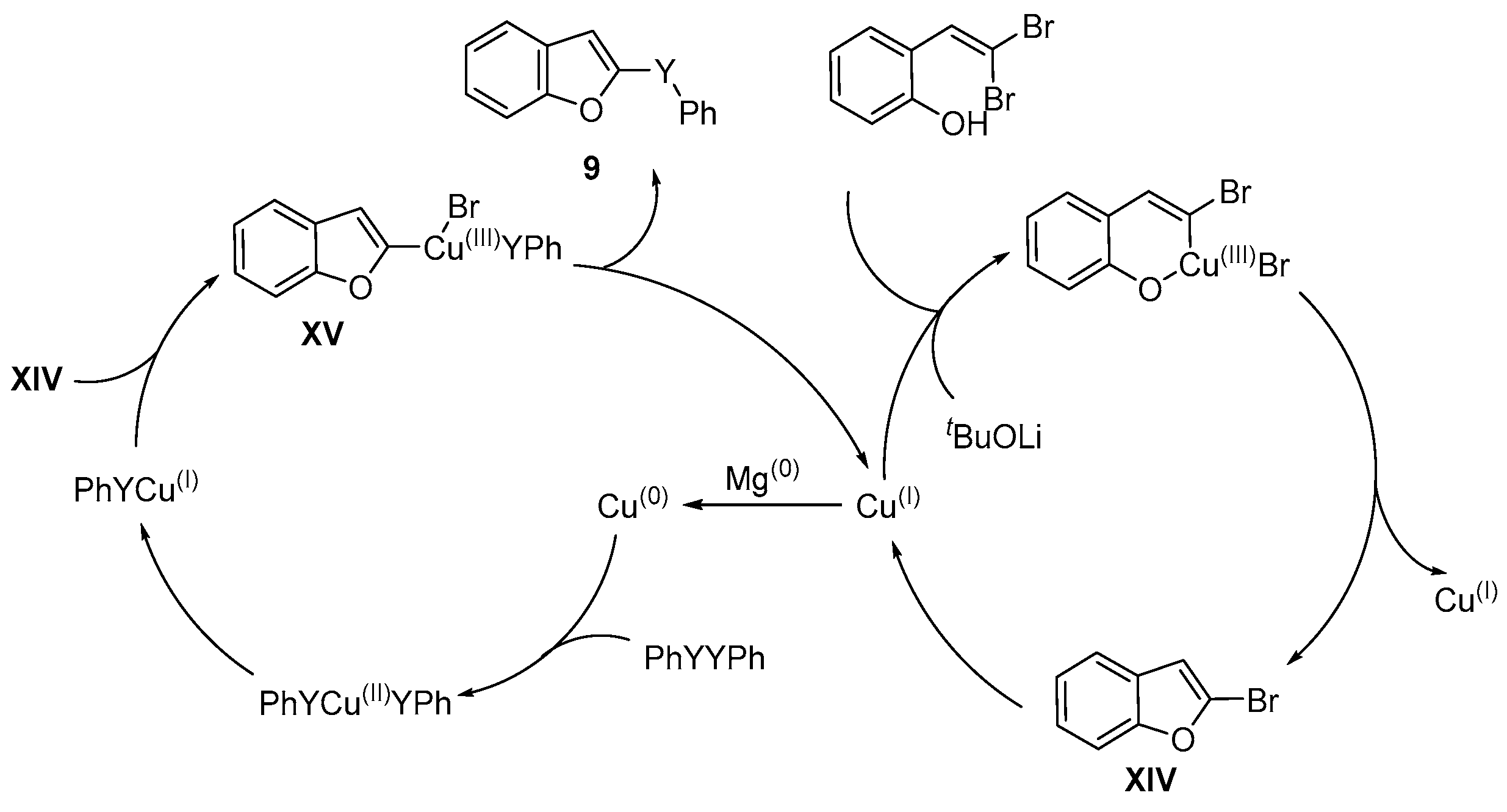{kind=link}
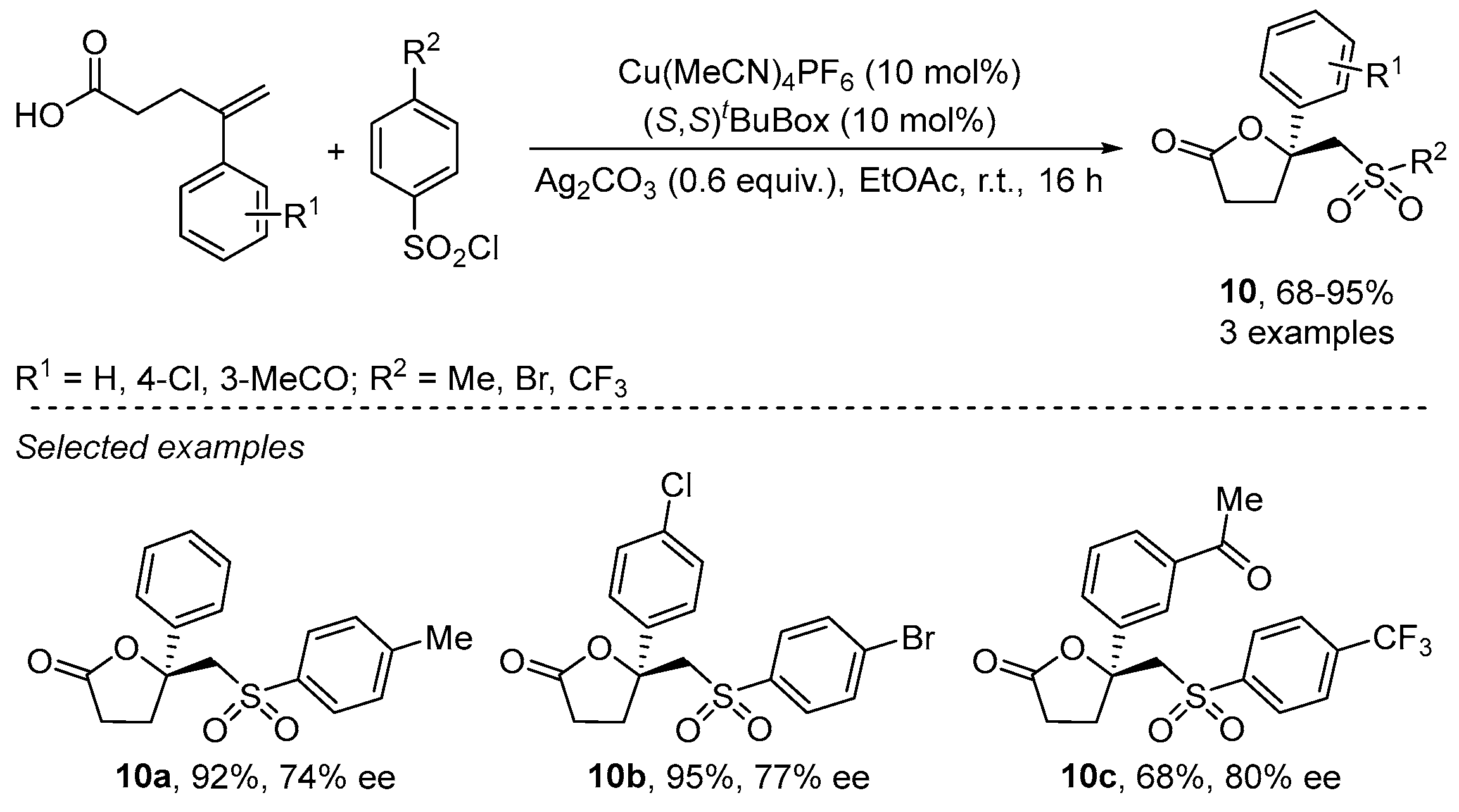{kind=link}
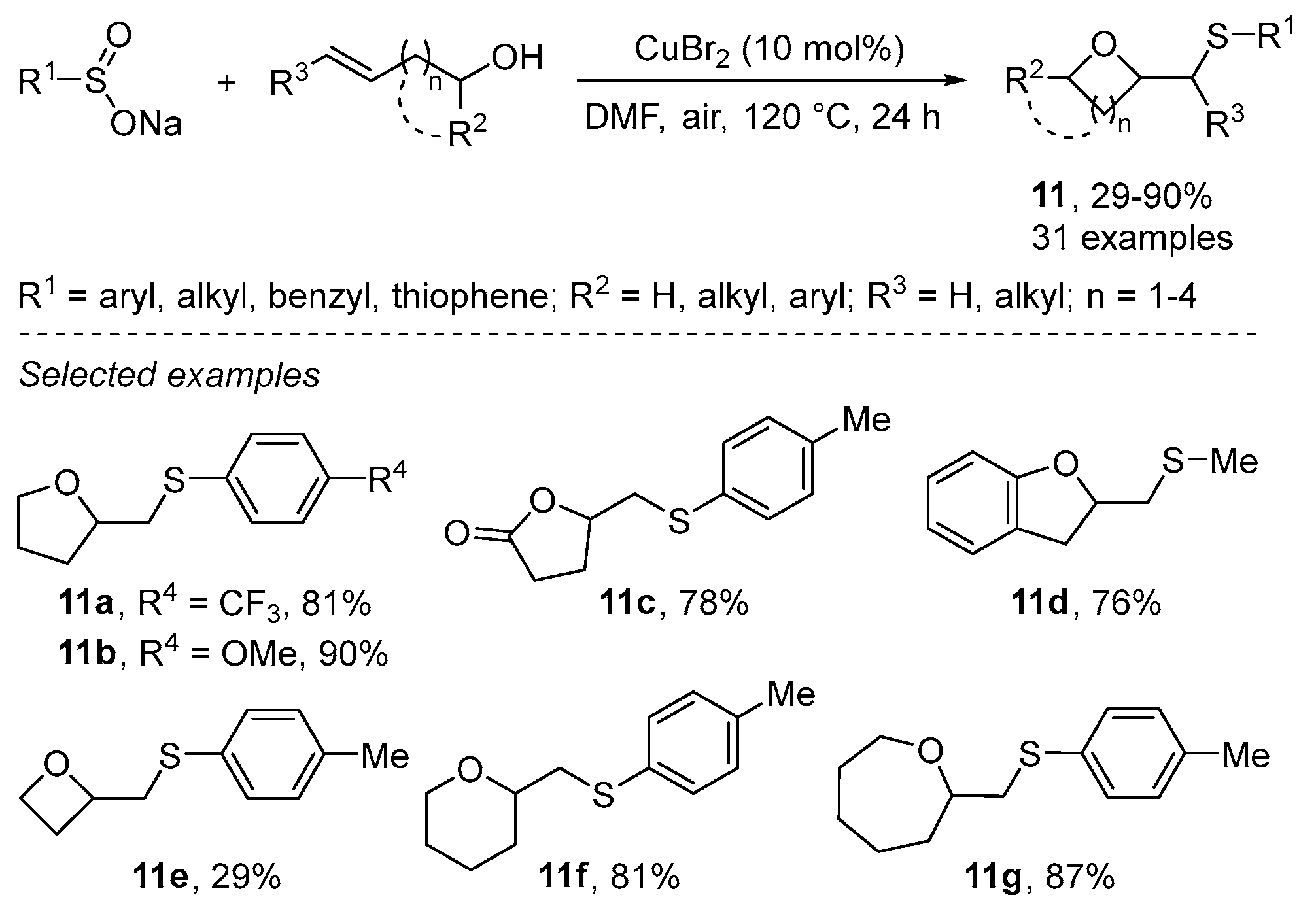{kind=link}
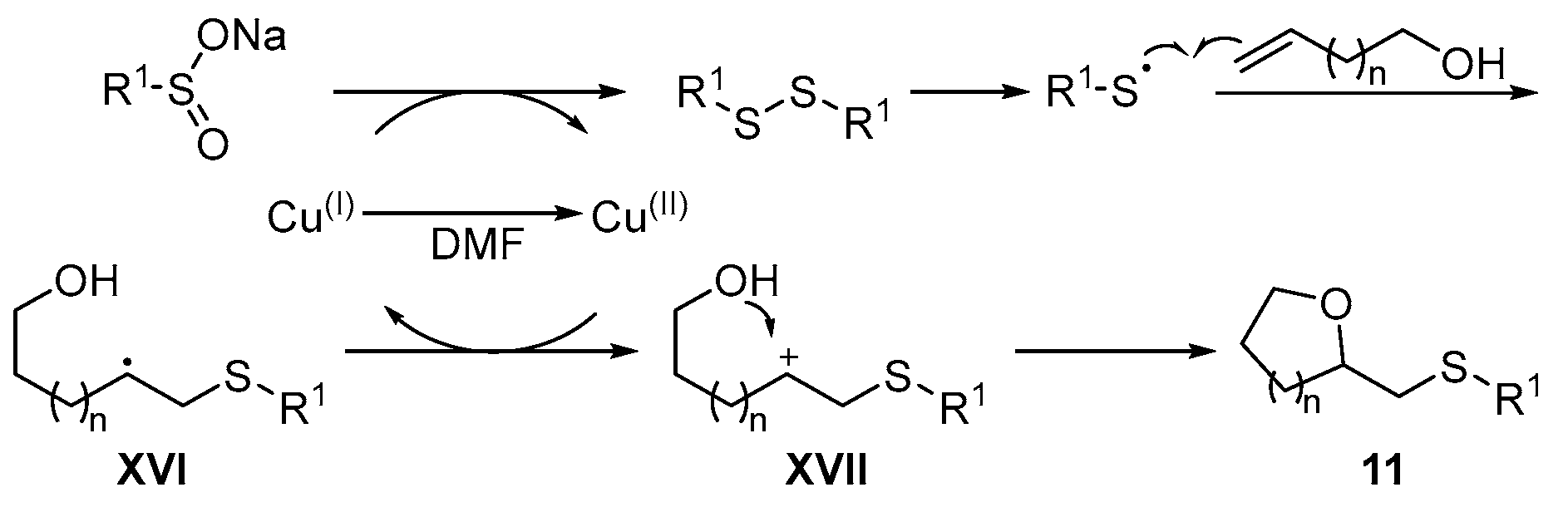{kind=link}
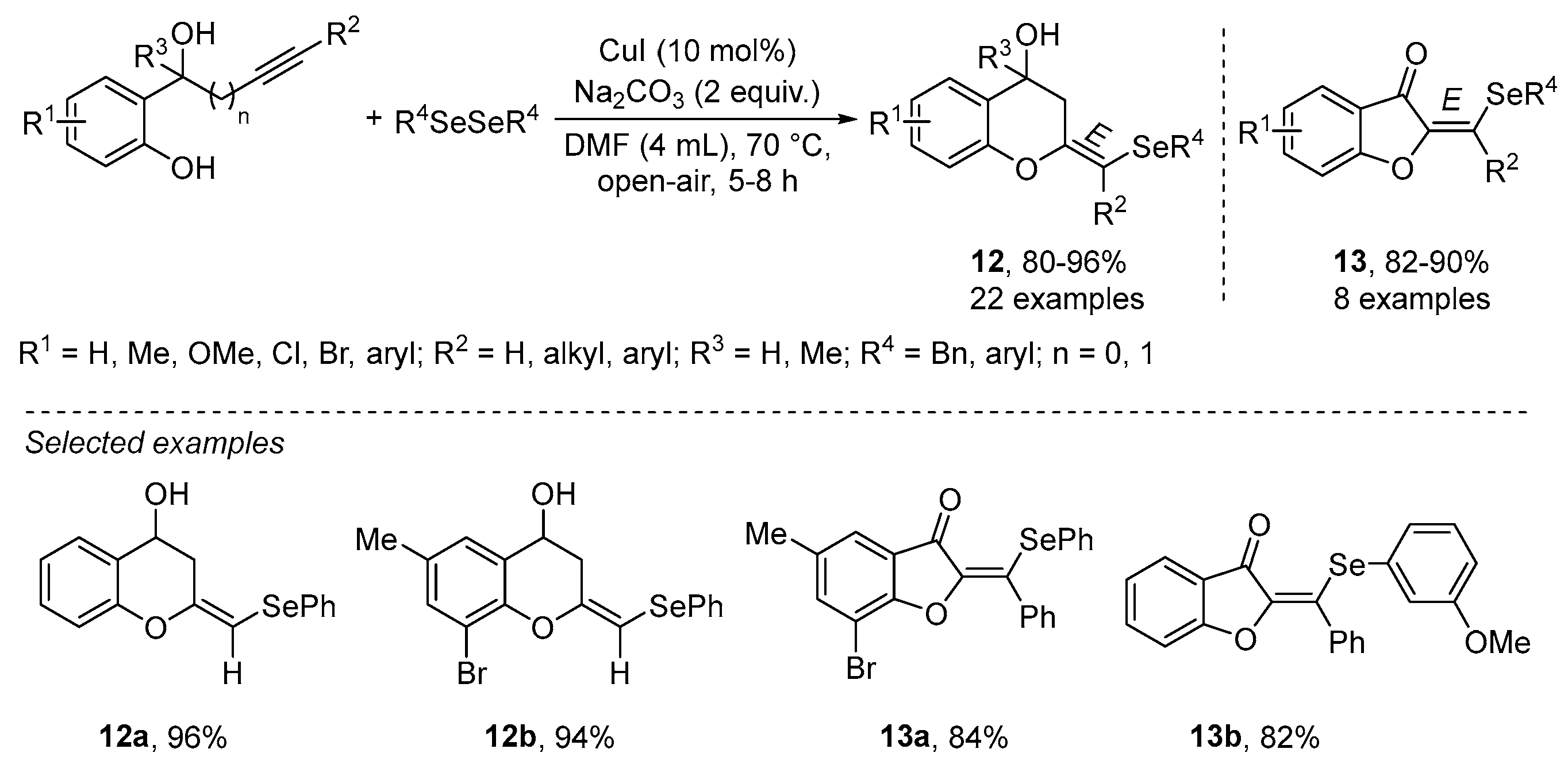{kind=link}
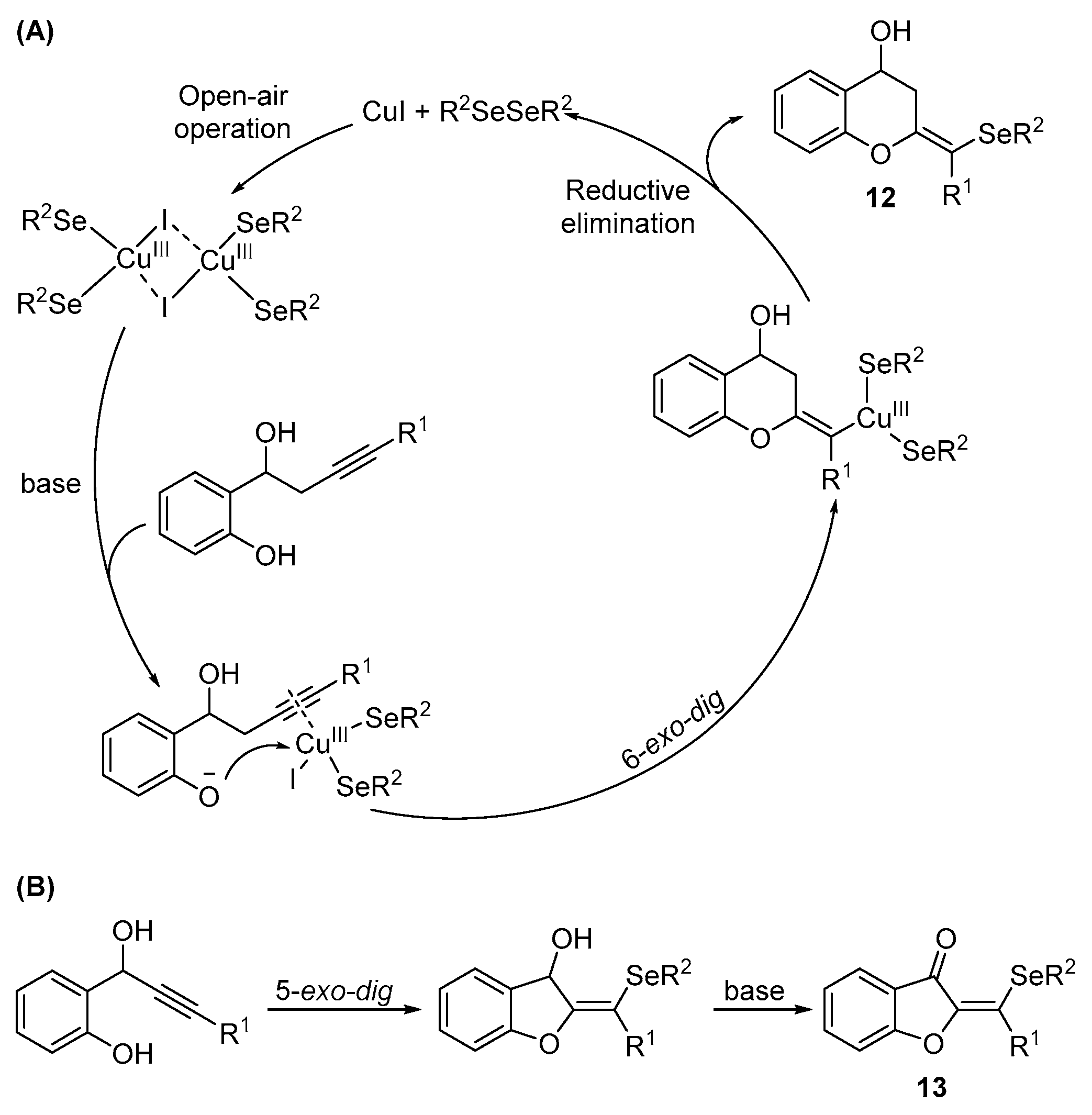{kind=link}
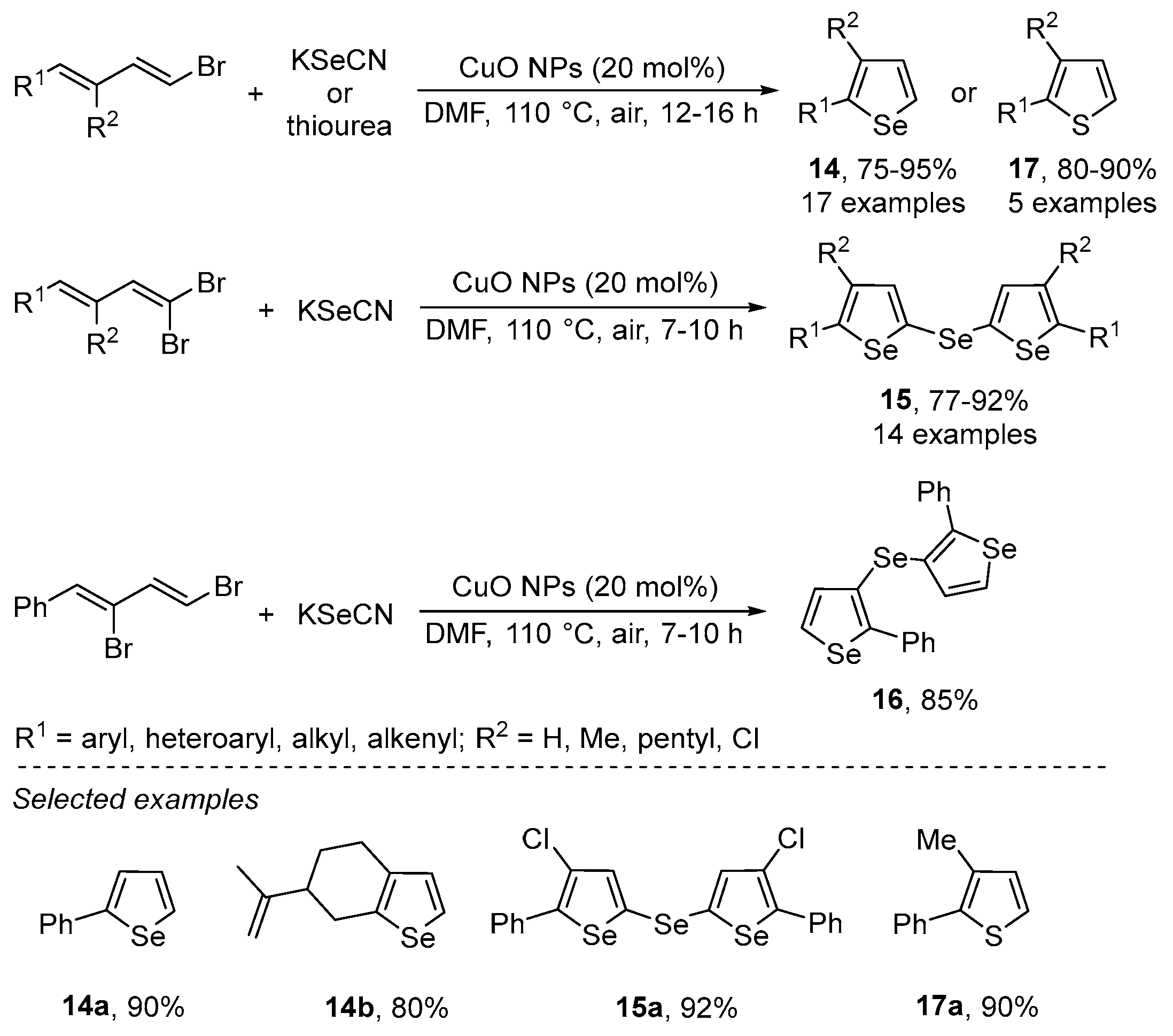{kind=link}
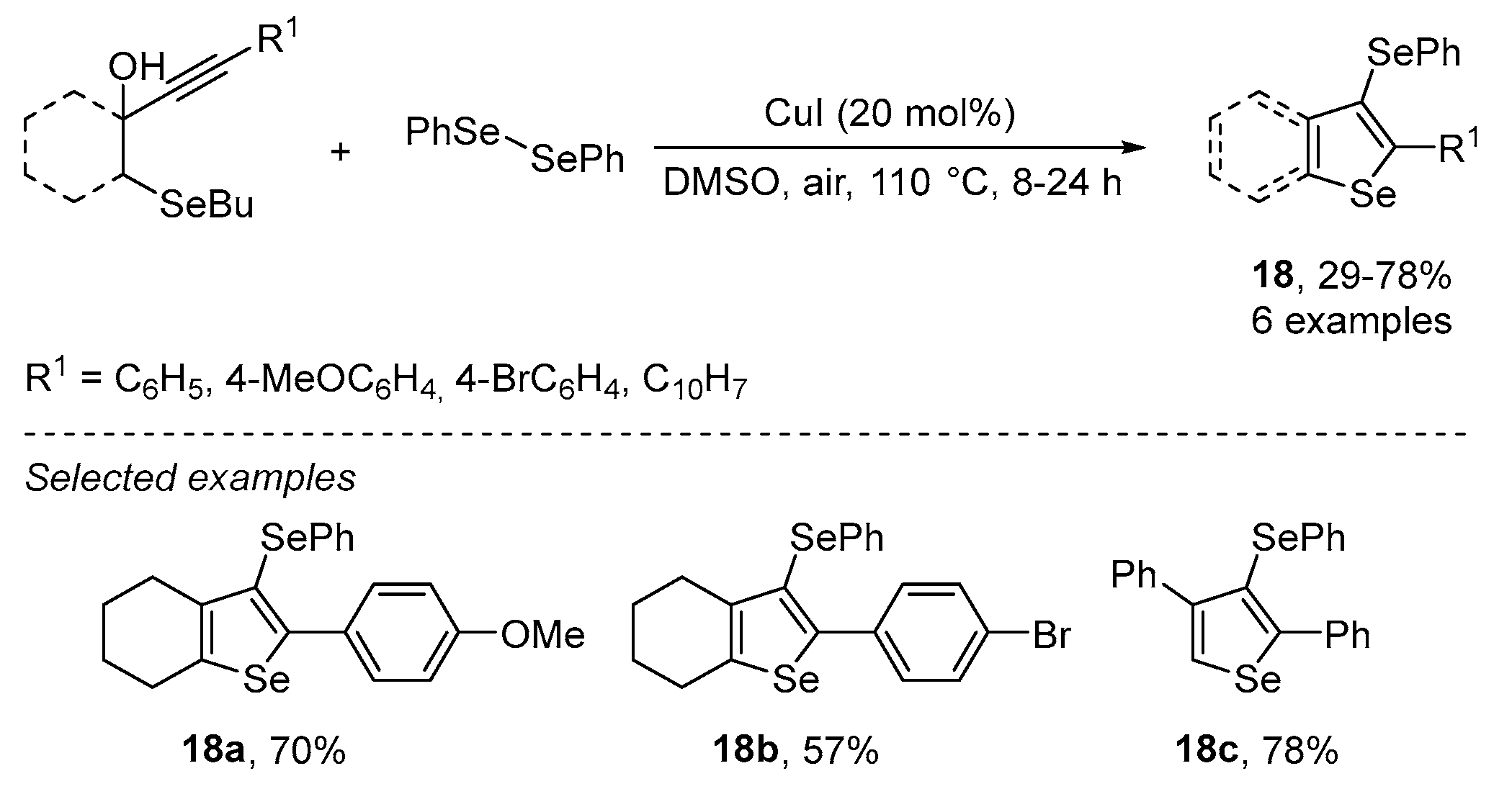{kind=link}
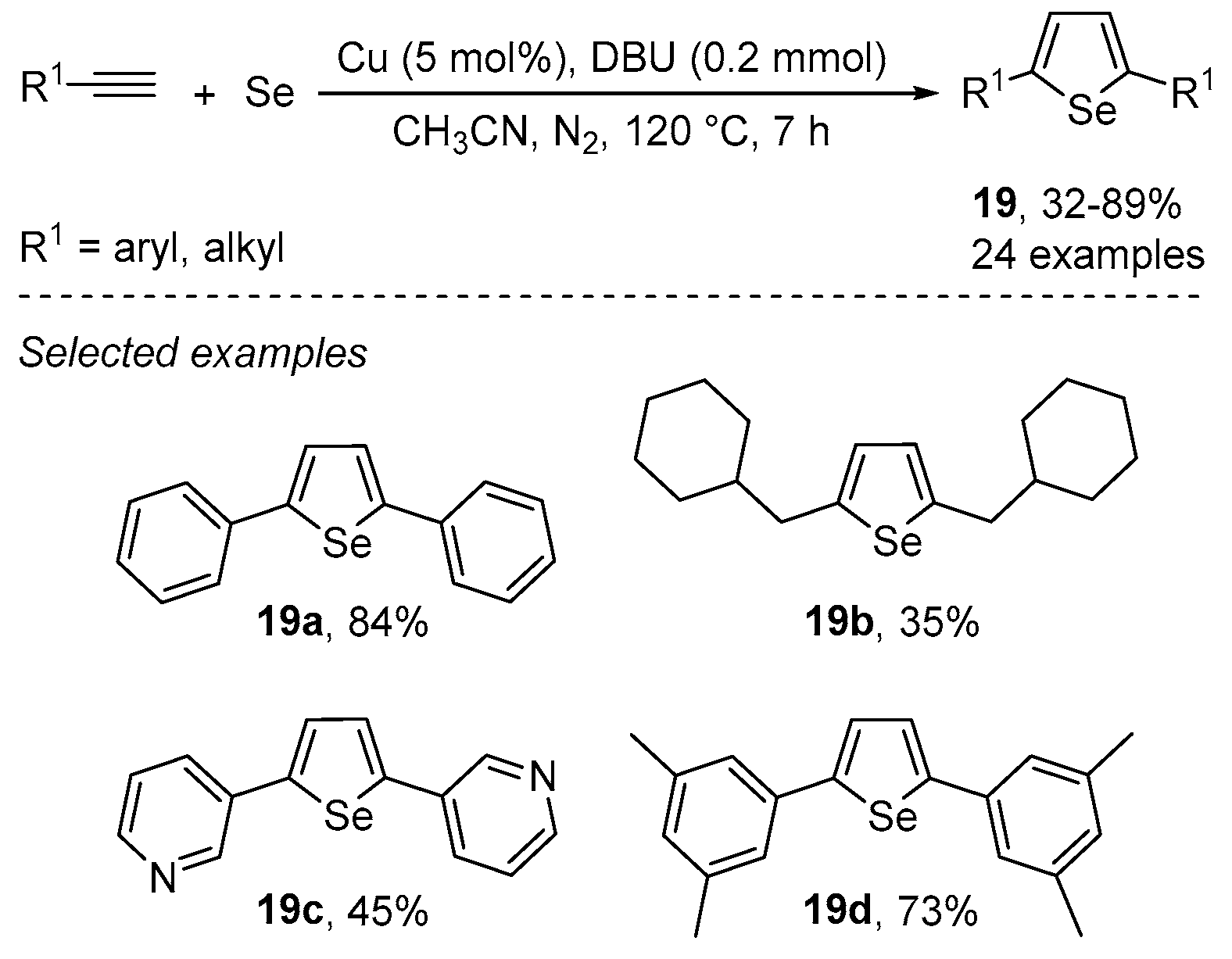{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
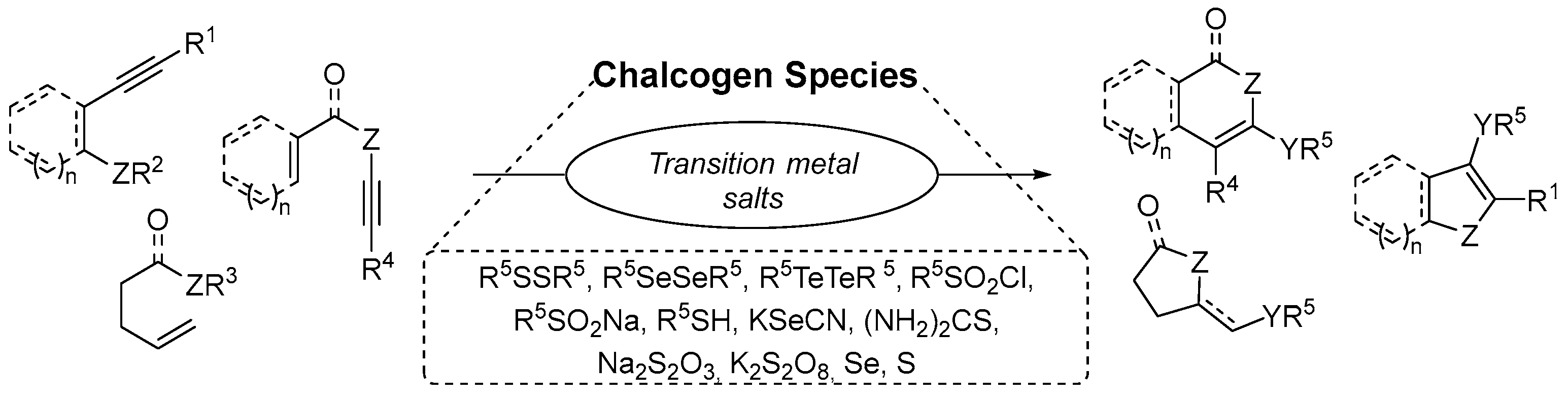
:1. Introduction
2. Transition Metal-Catalyzing Chalcogenative Heteroannulation
2.1. Using Copper as Catalyst
2.1.1. N-Based Heterocycles
2.1.2. O-Based Heterocycles
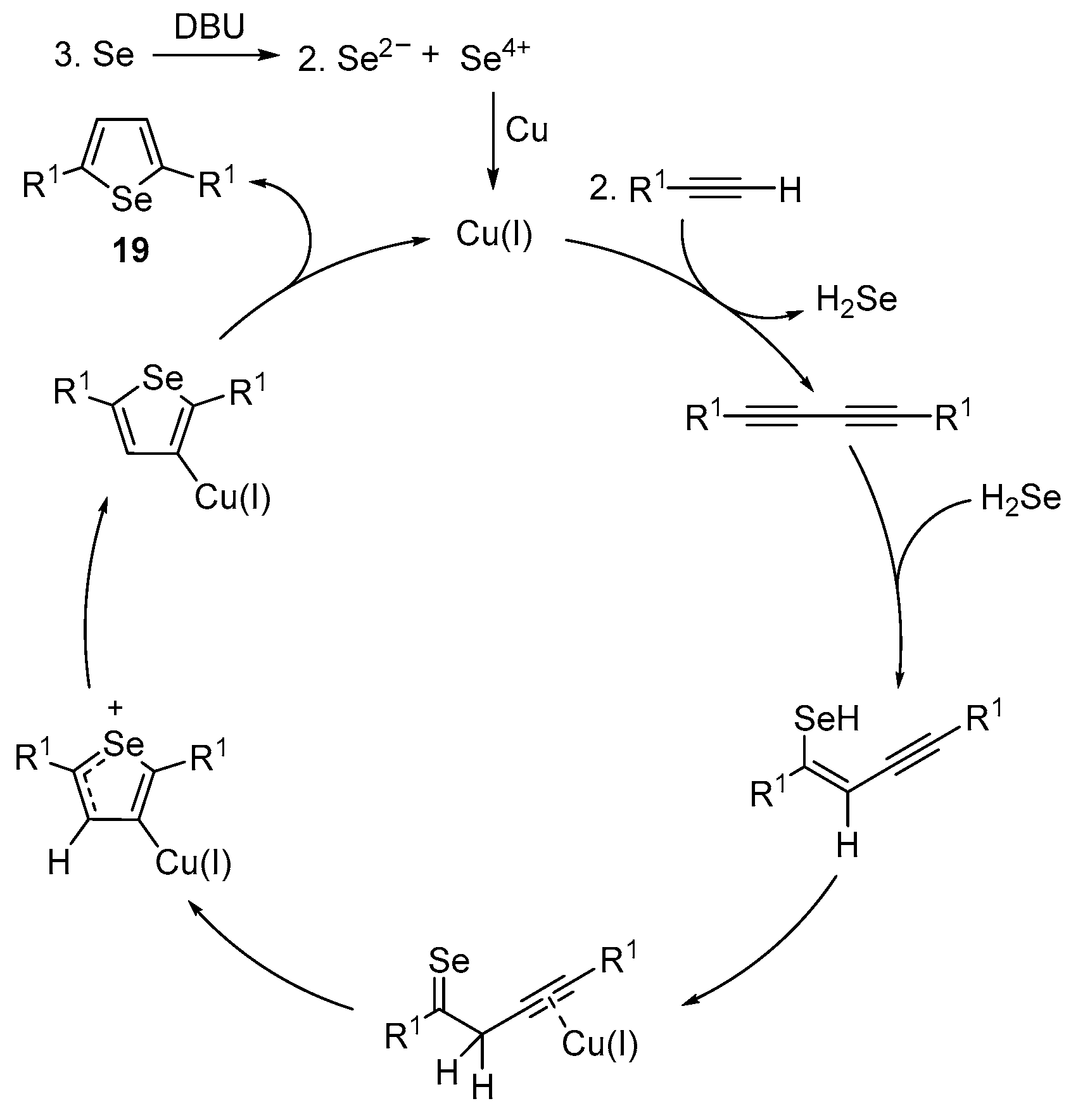
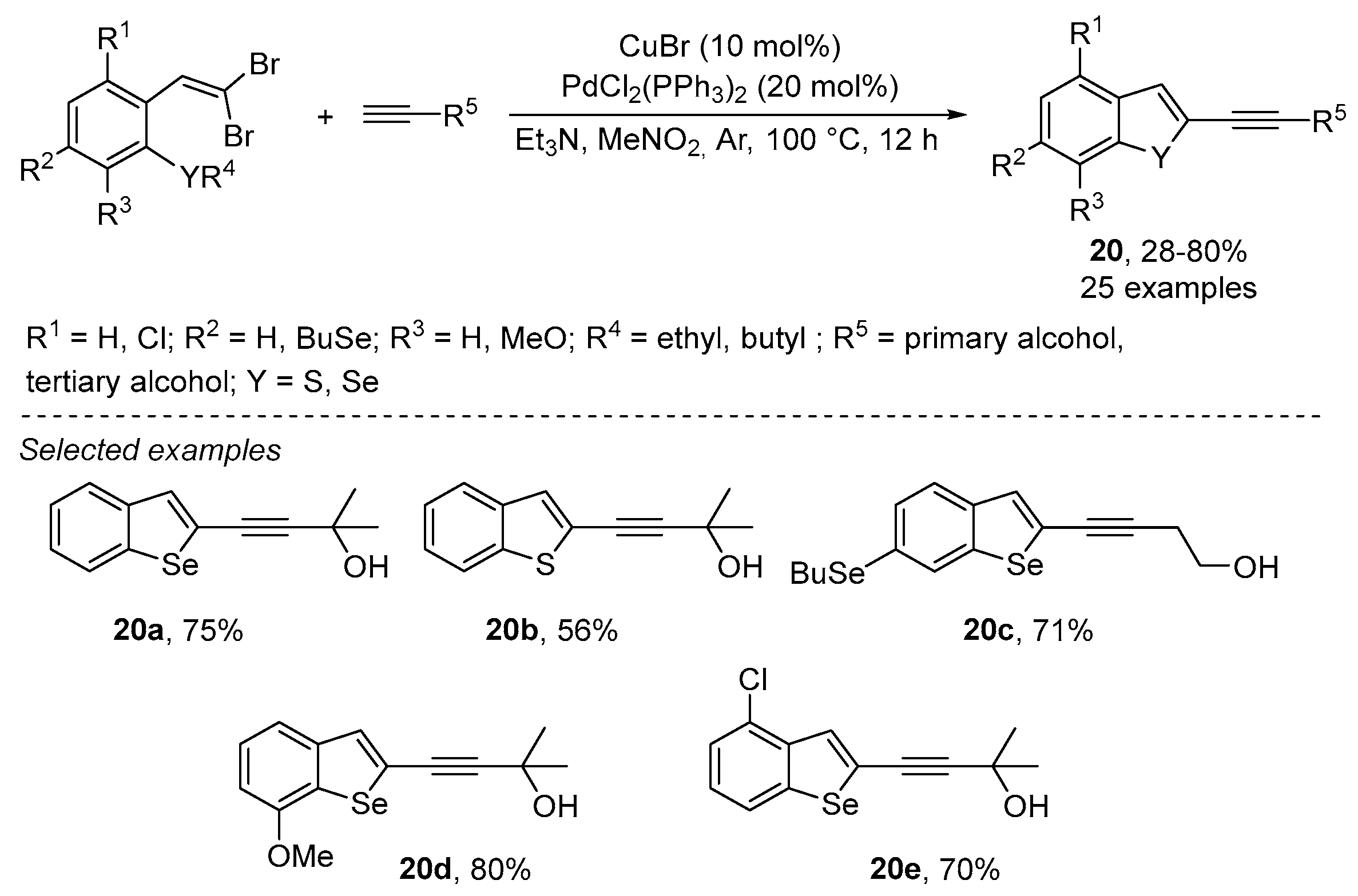
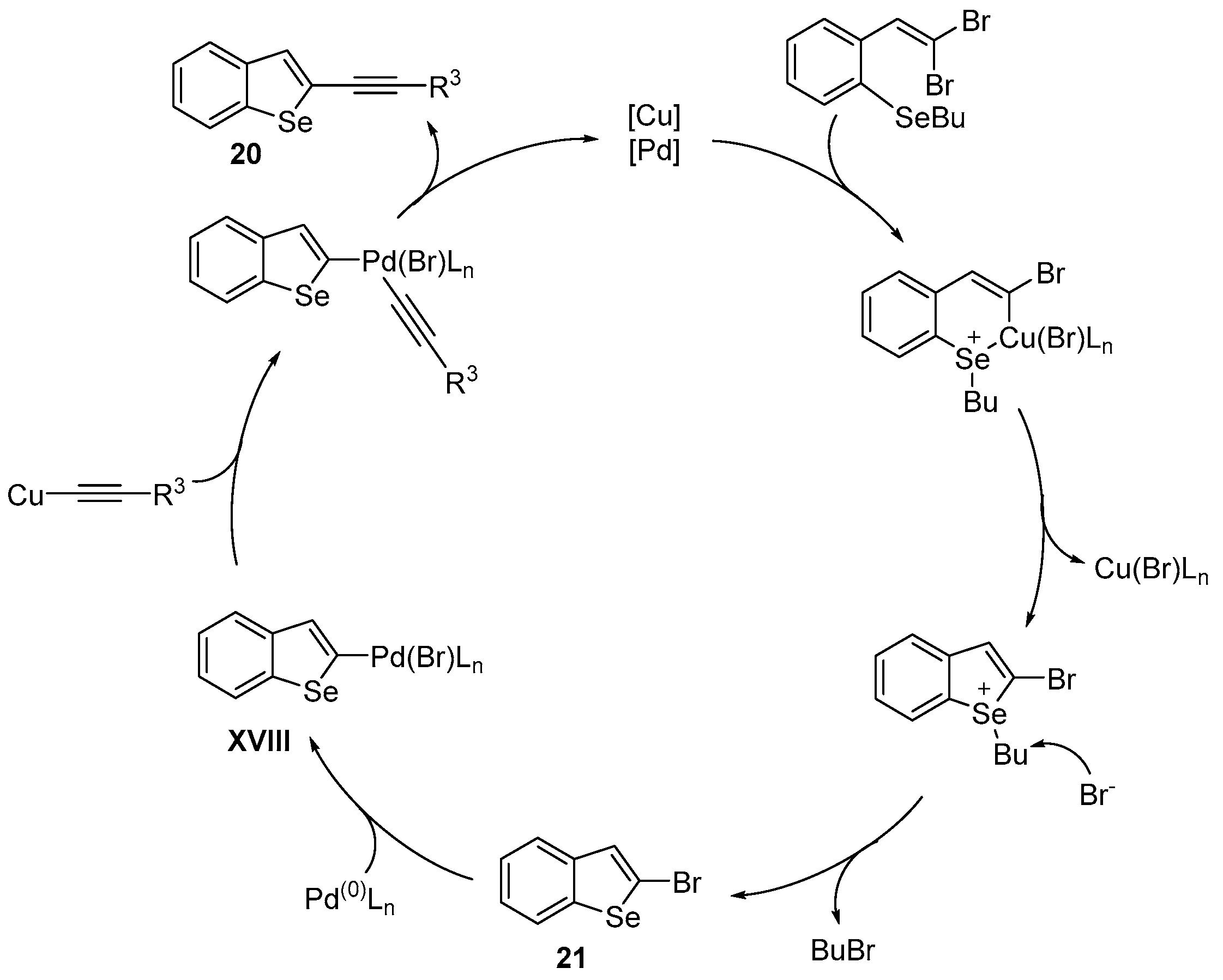
2.1.3. Chalcogen-Based Heterocycles
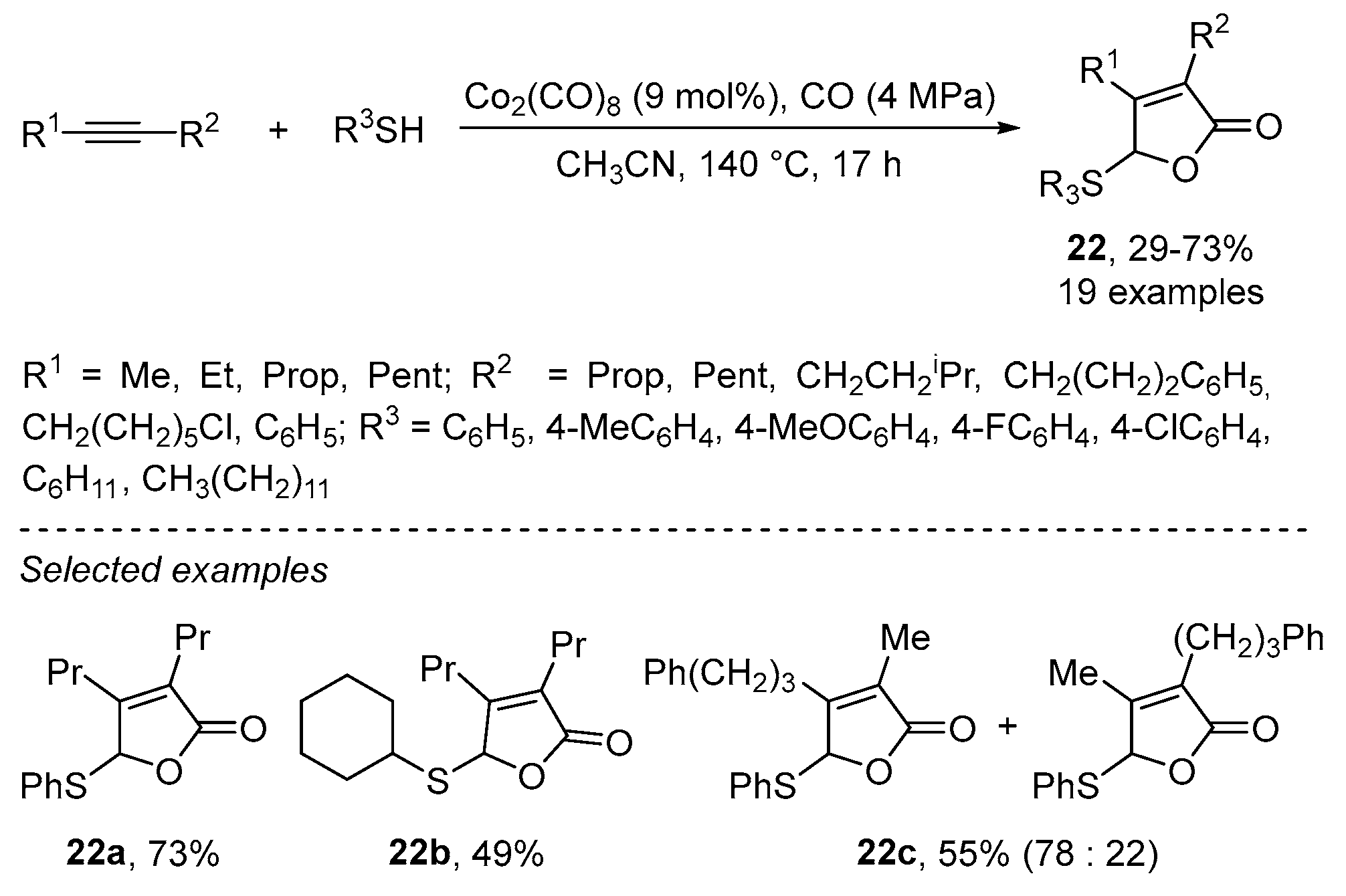
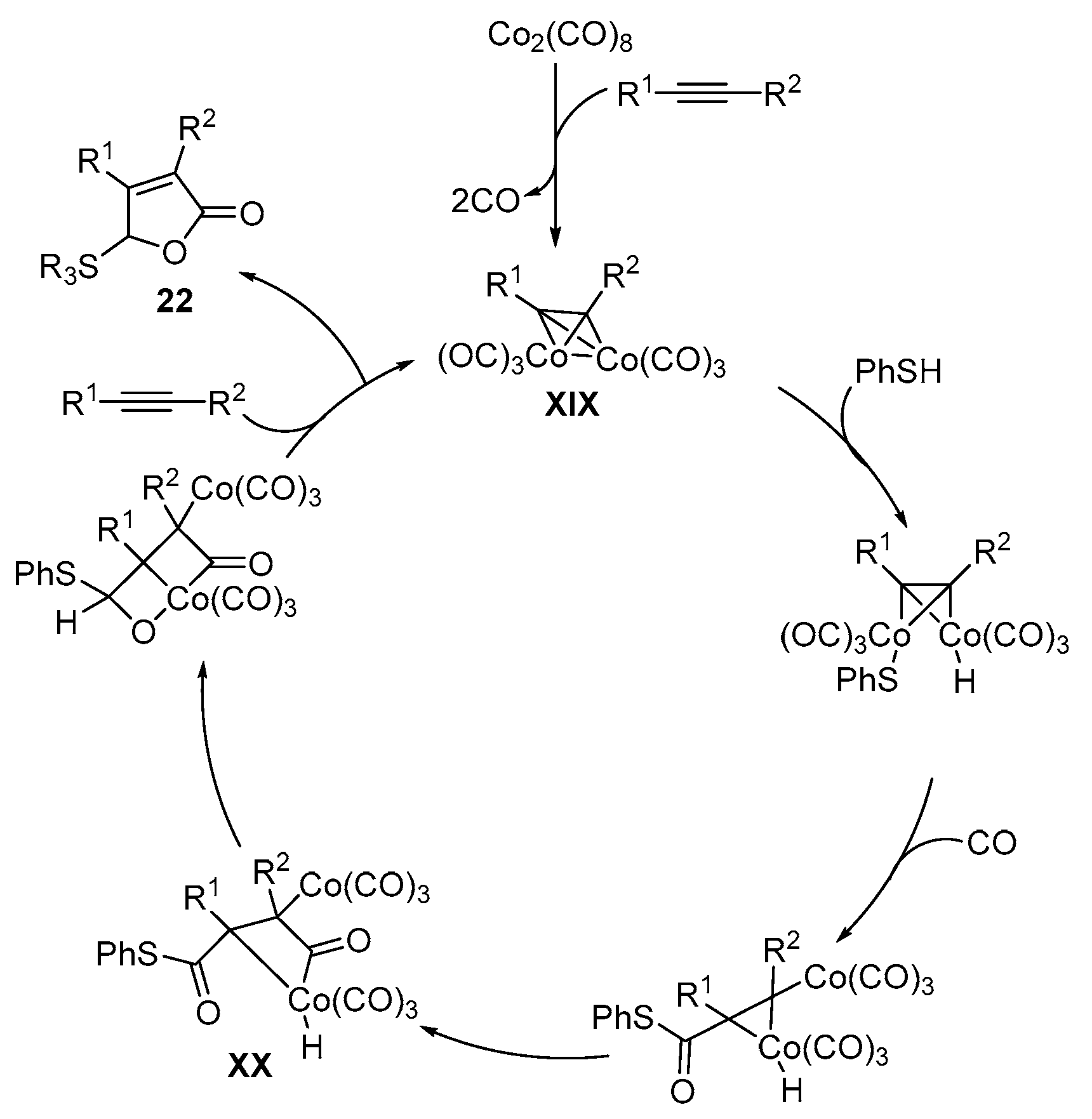
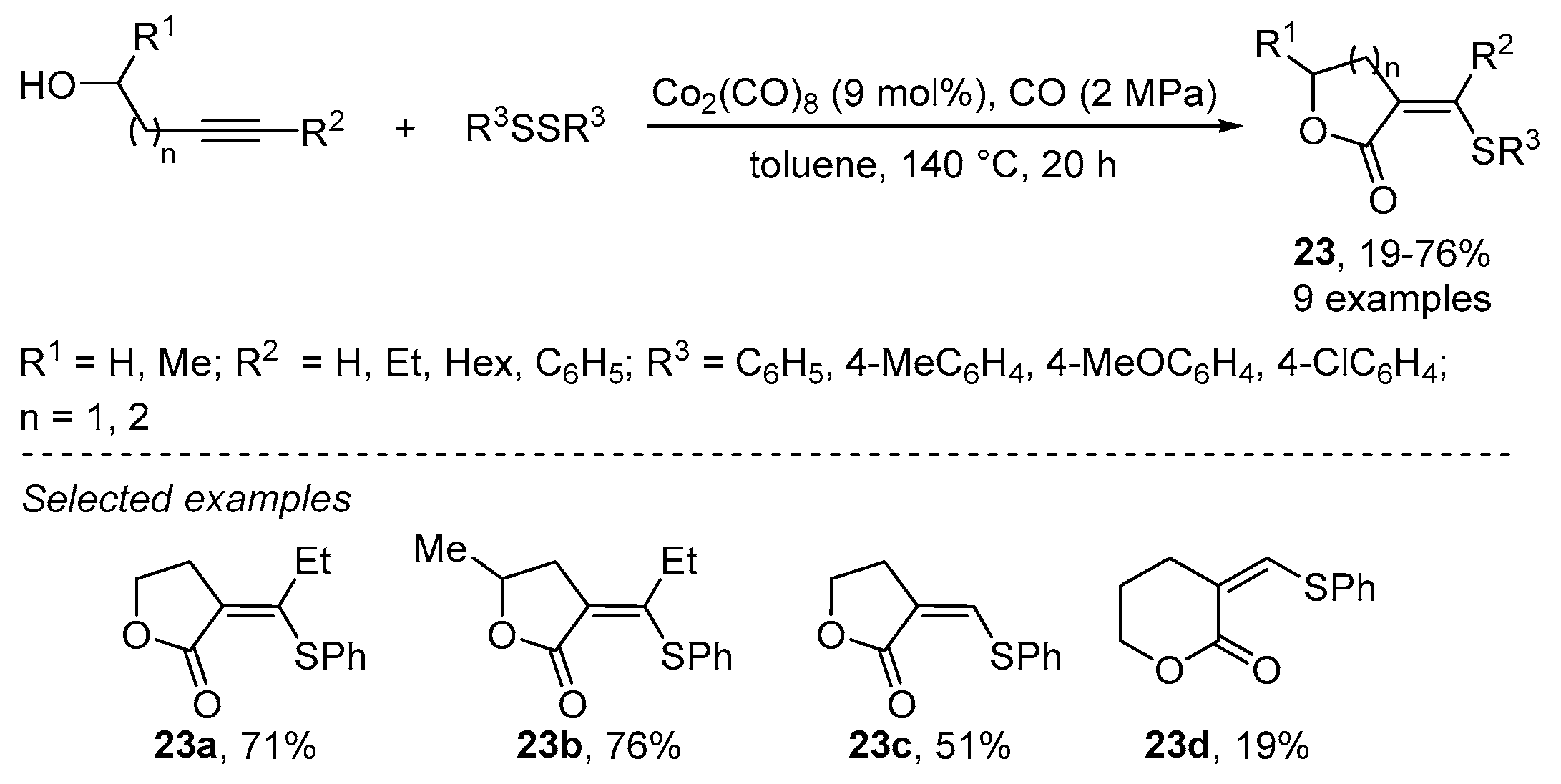
2.2. Using Cobalt as Catalyst
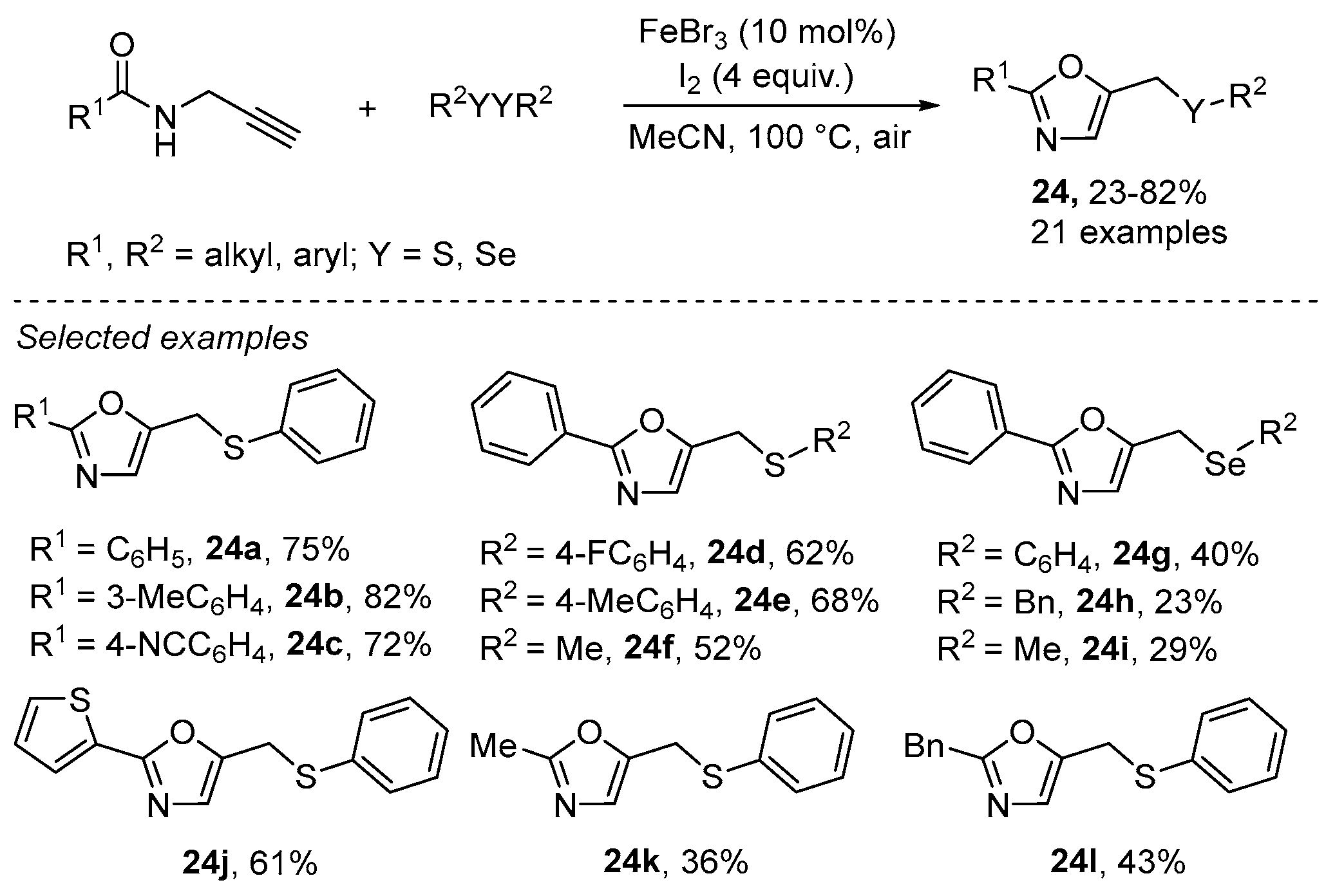
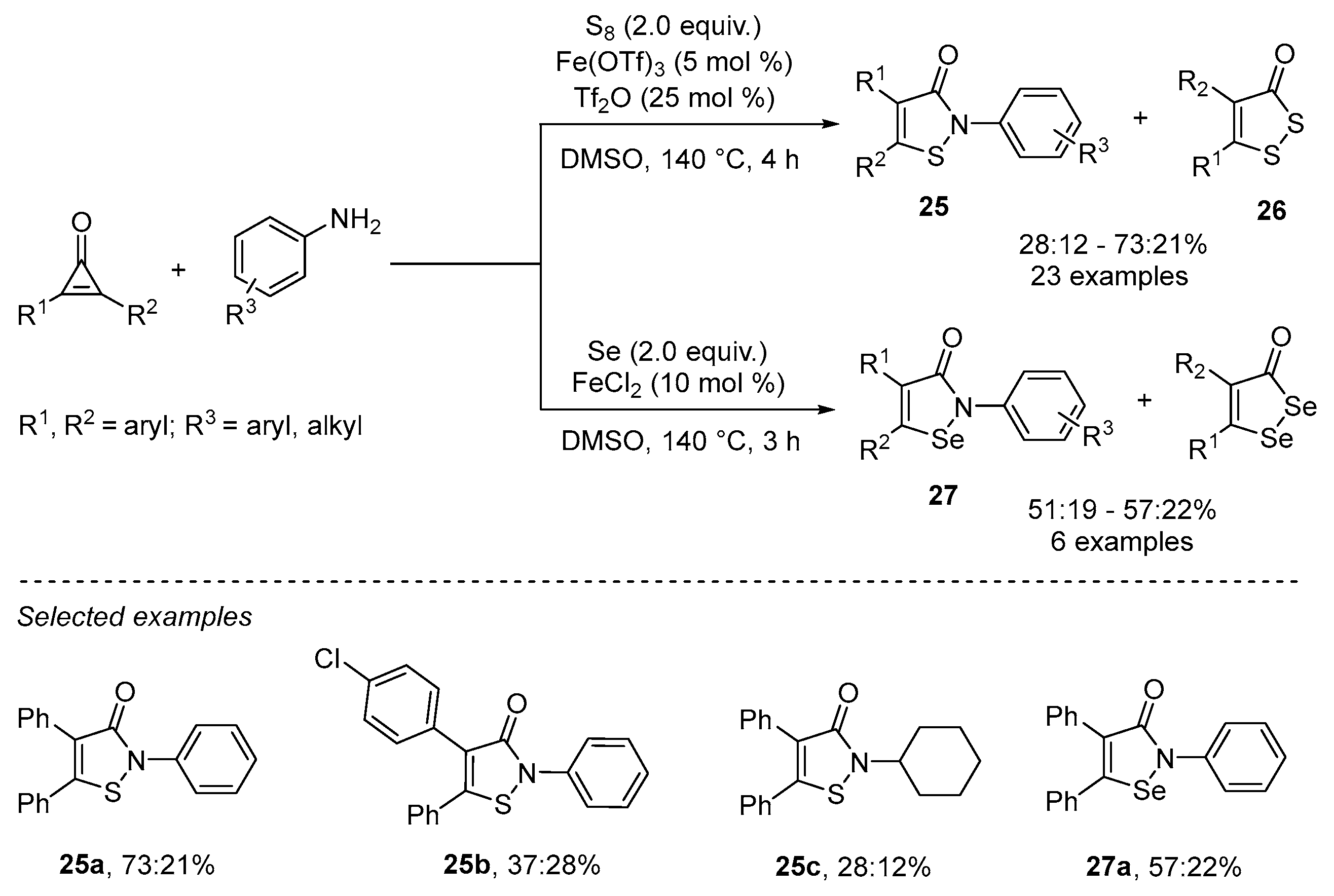
2.3. Using Iron as Catalyst
2.3.1. N-Based Heterocycles
2.3.2. O-Based Heterocycles
2.3.3. Chalcogen-Based Heterocycles
2.3.4. Miscellaneous
2.4. Using Palladium as Catalyst
2.5. Using Silver as Catalyst
2.5.1. N-Based Heterocycles
2.5.2. O-Based Heterocycles
2.6. Using Indium as Catalyst
2.7. Using Nickel as Catalyst
3. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Font, M.; Gulías, M.; Mascareñas, J.L. Transition-Metal-Catalyzed Annulations Involving the Activation of C(sp3)−H Bonds. Angew. Chem. Int. Ed. 2022, 61, e202112848. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of Heterocycles via Palladium π-Olefin and π-Alkyne Chemistry. Chem. Rev. 2004, 104, 2285–2310. [Google Scholar] [CrossRef]
- Penteado, F.; Peglow, T.J.; Silva, M.S.; Perin, G.; Lenardão, E.J. Greening the synthesis of selenium-containing heterocycles: Recent efforts and advances. Curr. Opin. Green Sustain. Chem. 2020, 26, 100372. [Google Scholar] [CrossRef]
- Aronica, L.A.; Albano, G. Supported Metal Catalysts for the Synthesis of N-Heterocycles. Catalysts 2022, 12, 68. [Google Scholar] [CrossRef]
- Achar, T.K.; Al-Thabaiti, S.A.; Mokhtar, M.; Maiti, D. Enantioselective annulation reactions through C(sp2)–H activation with chiral CpxMIII catalysts. Chem. Catal. 2023, 3, 100575. [Google Scholar] [CrossRef]
- Fanourakis, A.; Docherty, P.J.; Chuentragool, P.; Phipps, R.J. Recent Developments in Enantioselective Transition Metal Catalysis Featuring Attractive Noncovalent Interactions between Ligand and Substrate. ACS Catal. 2020, 10, 10672–10714. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.G.; Wang, S.-G.; Oliveira, C.C.; Chamer, N. Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev. 2017, 117, 8908–8976. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.K.; Sharma, D.; Kumari, S.; Chatterjee, R.; Khatravath, M.; Dandela, R. Recent Advances in Transition Metal-Catalyzed Domino-Cyclization Strategies for Functionalized Heterocyclic/Carbocyclic Compounds. ChemistrySelect 2022, 7, e202201408. [Google Scholar] [CrossRef]
- Sindhe, H.; Chaudhary, B.; Chowdhury, N.; Kamble, A.; Kumar, V.; Lad, A.; Sharma, S. Recent advances in transition-metal catalyzed directed C–H functionalization with fluorinated building blocks. Org. Chem. Front. 2022, 9, 1742–1775. [Google Scholar] [CrossRef]
- Mancuso, R.; Dalpozzo, R. Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts 2018, 8, 458. [Google Scholar] [CrossRef]
- Cintas, P.; Palmisano, G.; Cravotto, G. Power ultrasound in metal-assisted synthesis: From classical Barbier-like reactions to click chemistry. Ultrason. Sonochem. 2011, 18, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.G.; Jensen, K.F. The role of flow in green chemistry and engineering. Green Chem. 2013, 15, 1456–1472. [Google Scholar] [CrossRef]
- Hou, W.; Xu, H. Incorporating Selenium into Heterocycles and Natural Products—From Chemical Properties to Pharmacological Activities. J. Med. Chem. 2022, 65, 4436–4456. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Hernández, M.; Alcolea, V.; Pérez-Silanes, S. Potential of sulfur-selenium isosteric replacement as a strategy for the development of new anti-chagasic drugs. Acta Trop. 2022, 233, 106547. [Google Scholar] [CrossRef]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Shaaben, S.; Sasse, F.; Burkholz, T.; Jacob, C. Sulfur, selenium and tellurium pseudopeptides: Synthesis and biological evaluation. Bioorg. Med. Chem. 2014, 22, 3610–3619. [Google Scholar] [CrossRef]
- Orian, L.; Toppo, S. Organochalcogen peroxidase mimetics as potential drugs: A long story of a promise still unfulfilled. Free Radic. Biol. Med. 2014, 66, 65–74. [Google Scholar] [CrossRef]
- Bandeira, P.T.; Dalmolin, M.C.; de Oliveira, M.M.; Nunes, K.C.; Garcia, F.P.; Nakamura, C.V.; de Oliveira, A.R.M.; Piovan, L. Synthesis, Antioxidant Activity and Cytotoxicity of N-Functionalized Organotellurides. Bioorg. Med. Chem. 2019, 27, 410–415. [Google Scholar] [CrossRef]
- Sachdeva, H.; Khaturia, S.; Saquib, M.; Khatik, N.; Khandelwal, A.R.; Meena, R.; Sharma, K. Oxygen- and Sulphur-Containing Heterocyclic Compounds as Potential Anticancer Agents. Appl. Biochem. Biotechnol. 2022, 194, 6438–6467. [Google Scholar] [CrossRef]
- Pang, Y.; An, B.; Lou, L.; Zhang, J.; Yan, J.; Huang, L.; Li, X.; Yin, S. Design, Synthesis, and Biological Evaluation of Novel Selenium-Containing Isocombretastatins and Phenstatins as Antitumor Agents. J. Med. Chem. 2017, 60, 7300–7314. [Google Scholar] [CrossRef]
- de Souza, D.; Mariano, D.O.C.; Nedel, F.; Schultze, E.; Campos, V.F.; Seixas, F.; da Silva, R.S.; Munchen, T.S.; Ilha, V.; Dornelles, L.; et al. New Organochalcogen Multitarget Drug: Synthesis and Antioxidant and Antitumoral Activities of Chalcogenozidovudine Derivatives. J. Med. Chem. 2015, 58, 3329–3339. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.G.; Zugman, T.; Bandeira, P.T.; Dalmolin, M.C.; Scariot, D.B.; Garcia, F.P.; de Oliveira, A.R.M.; Nakamurab, C.V.; Piovan, L. Complementary Performance of Organoselenides and Organotellurides as Antimicrobials Agents. J. Braz. Chem. Soc. 2021, 32, 462–475. [Google Scholar] [CrossRef]
- Cervo, R.; Lopes, T.R.R.; de Vasconcelos, A.R.; Cargnelutti, J.F.; Schumacher, R.F.; Tirloni, B.; dos Santos, S.S.; Abram, U.; Lang, E.; Cargnelutti, R. Coordination compounds containing 2-pyridylselenium ligands: Synthesis, structural characterization, and antibacterial evaluation. New J. Chem. 2021, 45, 12863–12870. [Google Scholar] [CrossRef]
- Reis, A.S.; Vogt, A.G.; Pinza, M.P.; Voss, G.T.; da Fonseca, C.A.R.; Paltian, J.J.; Peglow, T.J.; Vaucher, R.A.; Echenique, J.V.Z.; Soares, M.P.; et al. Modulation of COX-2, INF-ɣ, glutamatergic and opioid systems contributes to antinociceptive, anti-inflammatory and anti-hyperalgesic effects of bis(3-amino-2-pyridine) diselenide. Chem.-Biol. Interact. 2019, 311, 108790. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Njardarson, J.T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top. Curr. Chem. 2019, 376, 1–34. [Google Scholar] [CrossRef]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals to Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Breder, A.; Ortgies, S. Recent developments in sulfur- and selenium-catalyzed oxidative and isohypsic functionalization reactions of alkenes. Tetrahedron Lett. 2015, 56, 2843–2852. [Google Scholar] [CrossRef]
- Cargnelutti, R.; Schumacher, R.F.; Belladona, A.L.; Kazmierczak, J.C. Coordination chemistry and synthetic approaches of pyridyl-selenium ligands: A decade update. Coord. Chem. Rev. 2021, 426, 213537. [Google Scholar] [CrossRef]
- Singh, A.; Kaushik, A.; Dhau, J.S.; Kumar, R. Exploring coordination preferences and biological applications of pyridyl-based organochalcogen (Se, Te) ligands. Coord. Chem. Rev. 2022, 450, 214254. [Google Scholar] [CrossRef]
- Gabriele, B.; Mancuso, R.; Larock, R.C. Recent Advances in the Synthesis of Iodoheterocycles via Iodocyclization of Functionalized Alkynes. Curr. Org. Chem. 2014, 18, 341–358. [Google Scholar] [CrossRef]
- Godoi, B.; Schumacher, R.F.; Zeni, G. Synthesis of Heterocycles via Electrophilic Cyclization of Alkynes Containing Heteroatom. Chem. Rev. 2011, 111, 2937–2980. [Google Scholar] [CrossRef]
- Castellanos, A.; Flecther, S.P. Current Methods for Asymmetric Halogenation of Olefins. Chem. Eur. J. 2011, 17, 5766–5776. [Google Scholar] [CrossRef]
- Hennecke, U. New Catalytic Approaches towards the Enantioselective Halogenation of Alkenes. Chem.-Asian J. 2012, 7, 456–465. [Google Scholar] [CrossRef]
- Sun, K.; Wang, X.; Li, C.; Wang, H.; Li, L. Recent advances in tandem selenocyclization and tellurocyclization with alkenes and alkynes. Org. Chem. Front. 2020, 7, 3100–3119. [Google Scholar] [CrossRef]
- Jurinic, C.K.; Belladona, A.L.; Schumacher, R.F.; Godoi, B. Diorganyl Dichalcogenides and Copper/Iron Salts: Versatile Cyclization System To Achieve Carbo- and Heterocycles from Alkynes. Synthesis 2021, 53, 2545–2558. [Google Scholar] [CrossRef]
- Zeni, G.; Godoi, B.; Jurinic, C.K.; Belladona, A.L.; Schumacher, R.F. Transition Metal-Free Synthesis of Carbo- and Heterocycles via Reaction of Alkynes with Organylchalcogenides. Chem. Rec. 2021, 21, 2880–2895. [Google Scholar] [CrossRef]
- Bartz, R.H.; Dapper, L.H.; Kazmierczak, J.C.; Schumacher, R.F.; Perin, G.; Thurow, S.; Penteado, F.; Lenardão, E.J. Lighting Up the Organochalcogen Synthesis: A Concise Update of Recent Photocatalyzed Approaches. Catalysts 2023, 13, 520. [Google Scholar] [CrossRef]
- Qian, P.-C.; Liu, Y.; Song, R.-J.; Xiang, J.-N.; Li, J.-H. Copper-Catalyzed Oxidative ipso-Cyclization of N-(p-Methoxyaryl)propiolamides with Disulfides and Water Leading to 3-(Arylthio)-1-azaspiro [4.5]deca-3,6,9-triene-2,8-diones. Synlett 2015, 26, 1213–1216. [Google Scholar] [CrossRef]
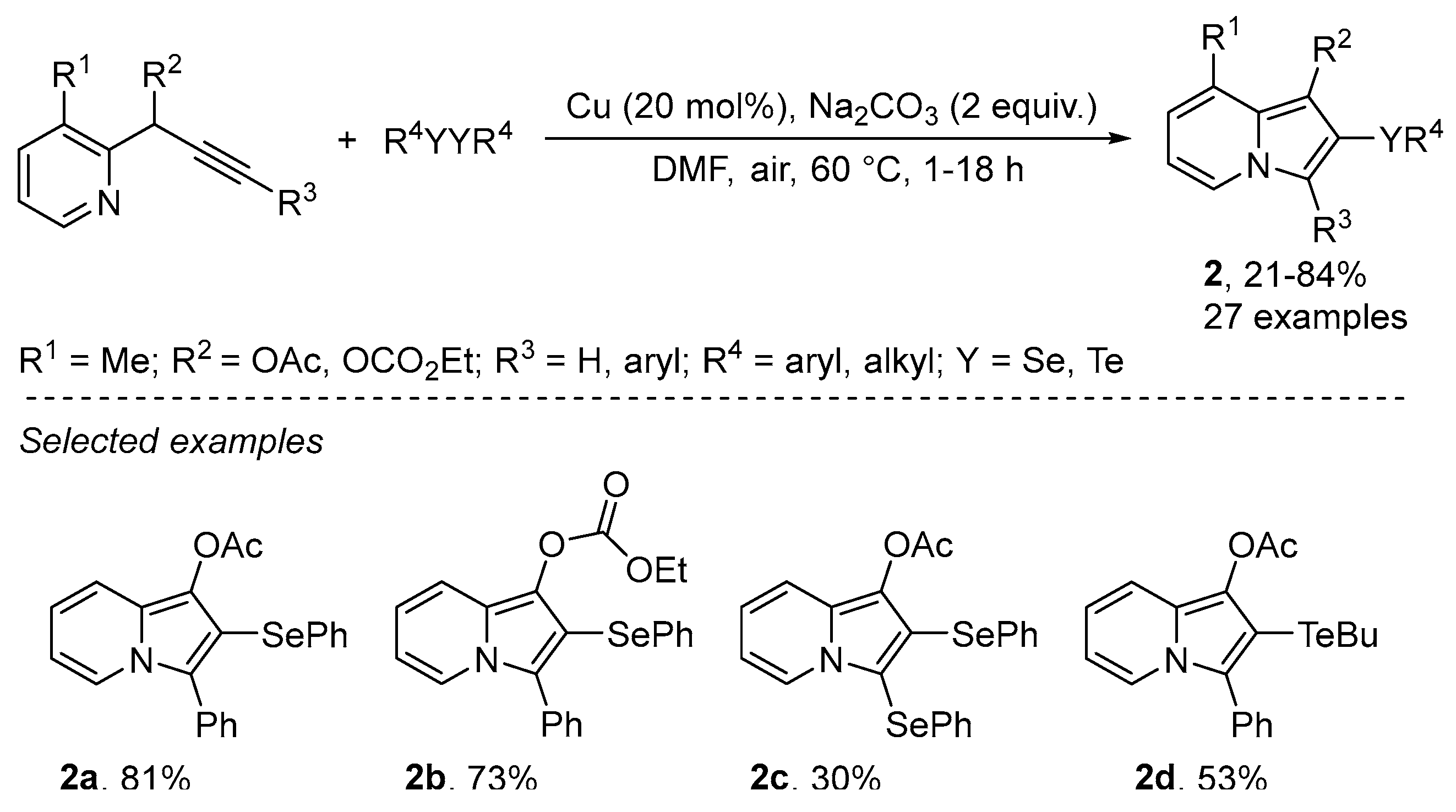
- Goulart, T.A.C.; Back, D.F.; Zeni, G. Copper-Catalyzed Carbon-Nitrogen/Carbon-Selenium Bonds Formation: Synthesis of 2-(Organochalcogenyl)-indolizines. Adv. Synth. Catal. 2017, 359, 1901–1911. [Google Scholar] [CrossRef]
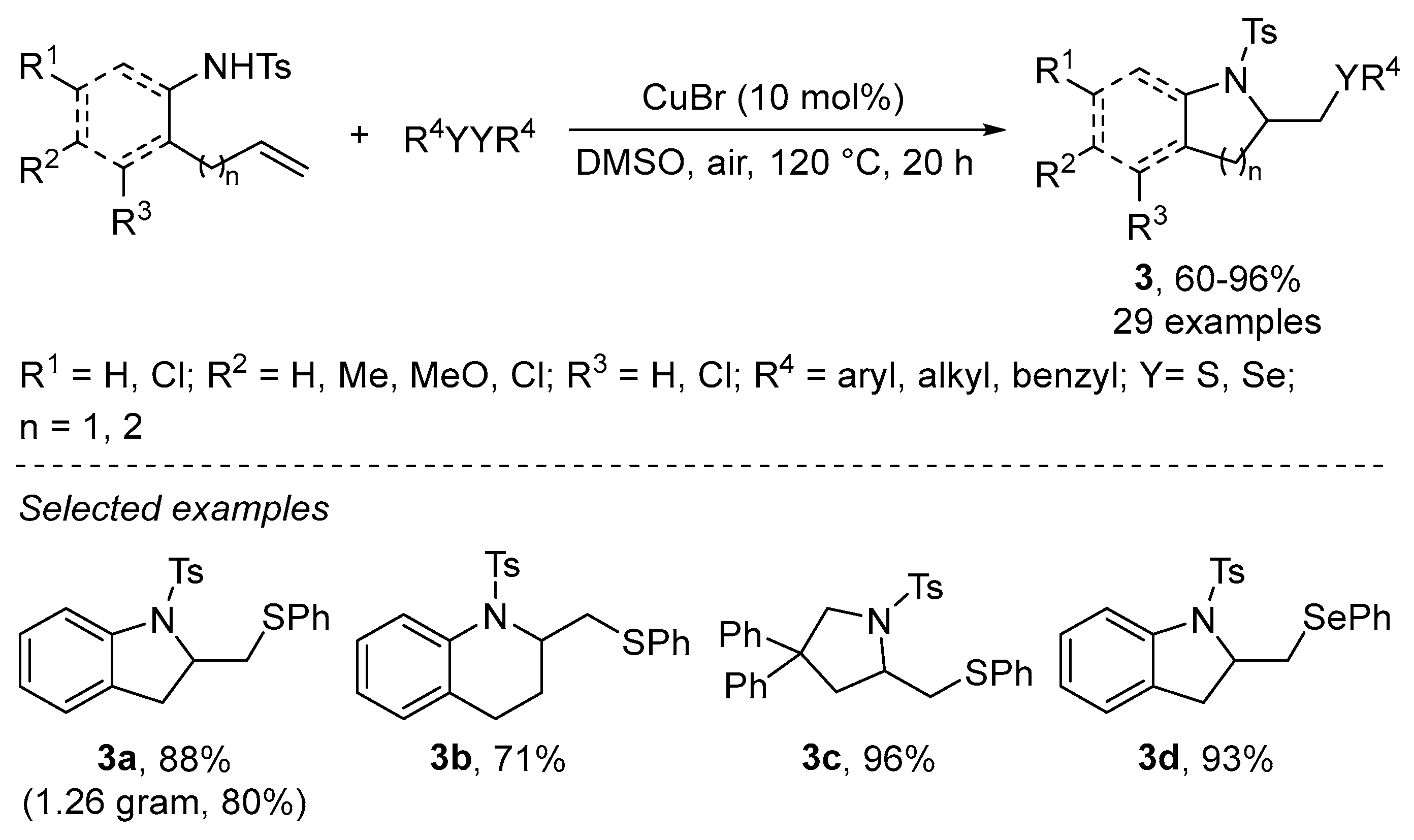
- Ni, Y.; Zuo, H.; Li, Y.; Wu, Y.; Zhong, F. Copper-Catalyzed Regioselective Intramolecular Electrophilic Sulfenoamination via Lewis Acid Activation of Disulfides under Aerobic Conditions. Org. Lett. 2018, 20, 4350–4353. [Google Scholar] [CrossRef]
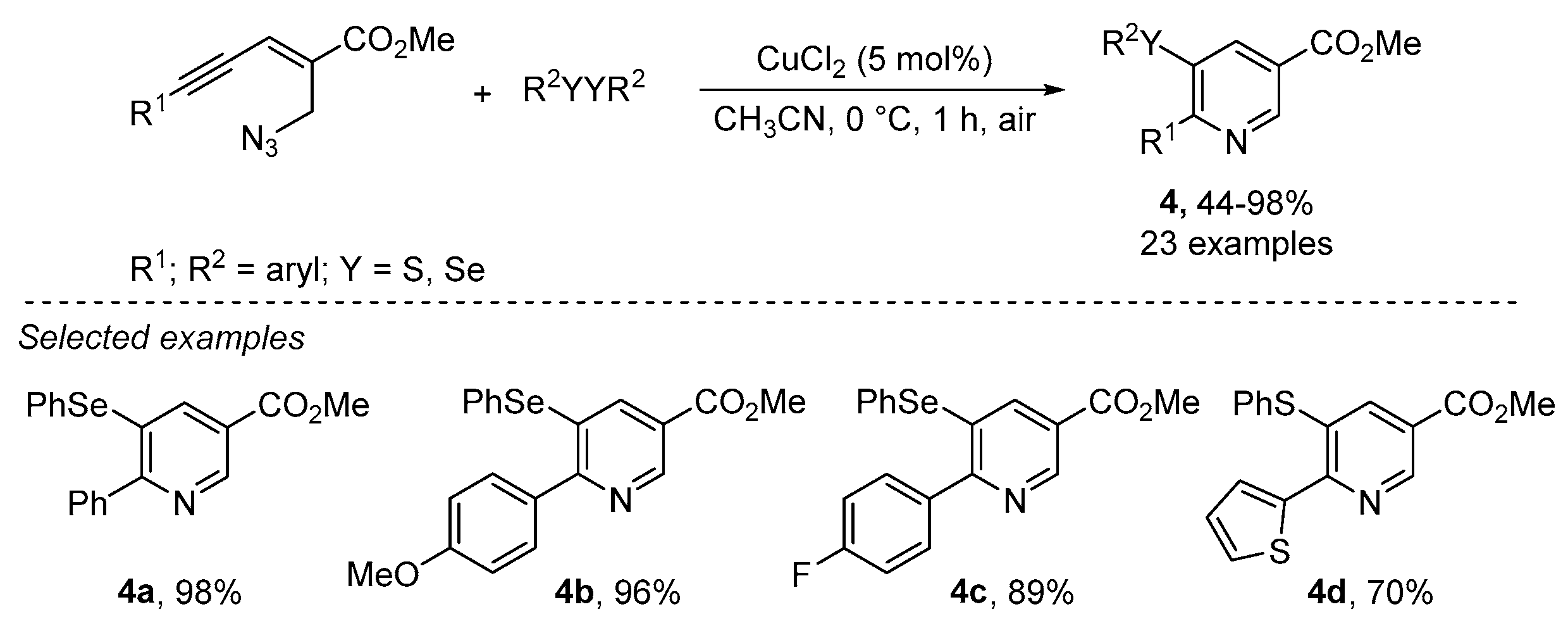
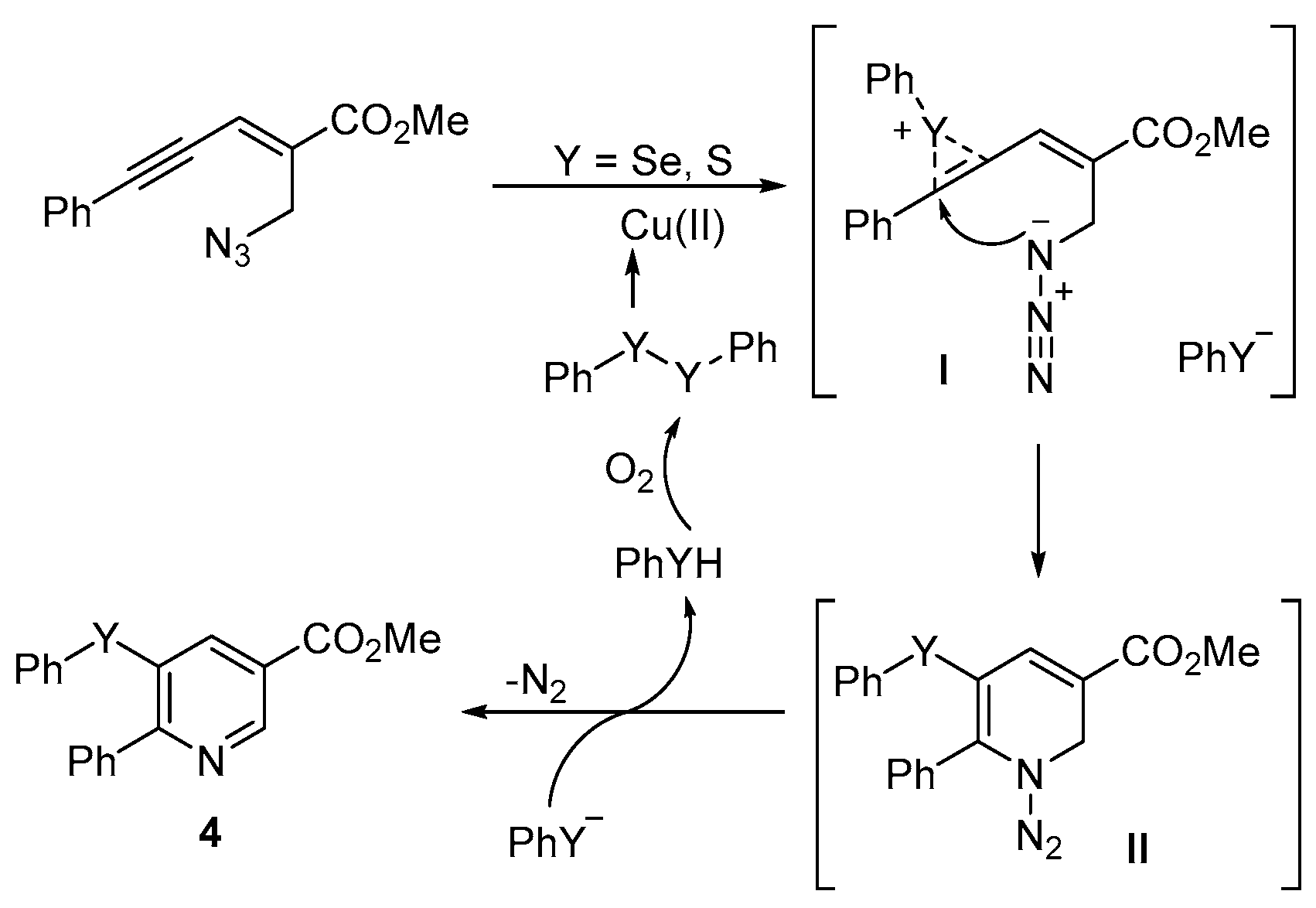
- Reddy, C.R.; Ranjan, R.; Prajapti, S.K. Copper-catalyzed intramolecular chalcogenoamination of enynyl azides: Synthesis of 5-selenyl/sulfenyl nicotinates. Org. Lett. 2019, 21, 623–626. [Google Scholar] [CrossRef] [PubMed]
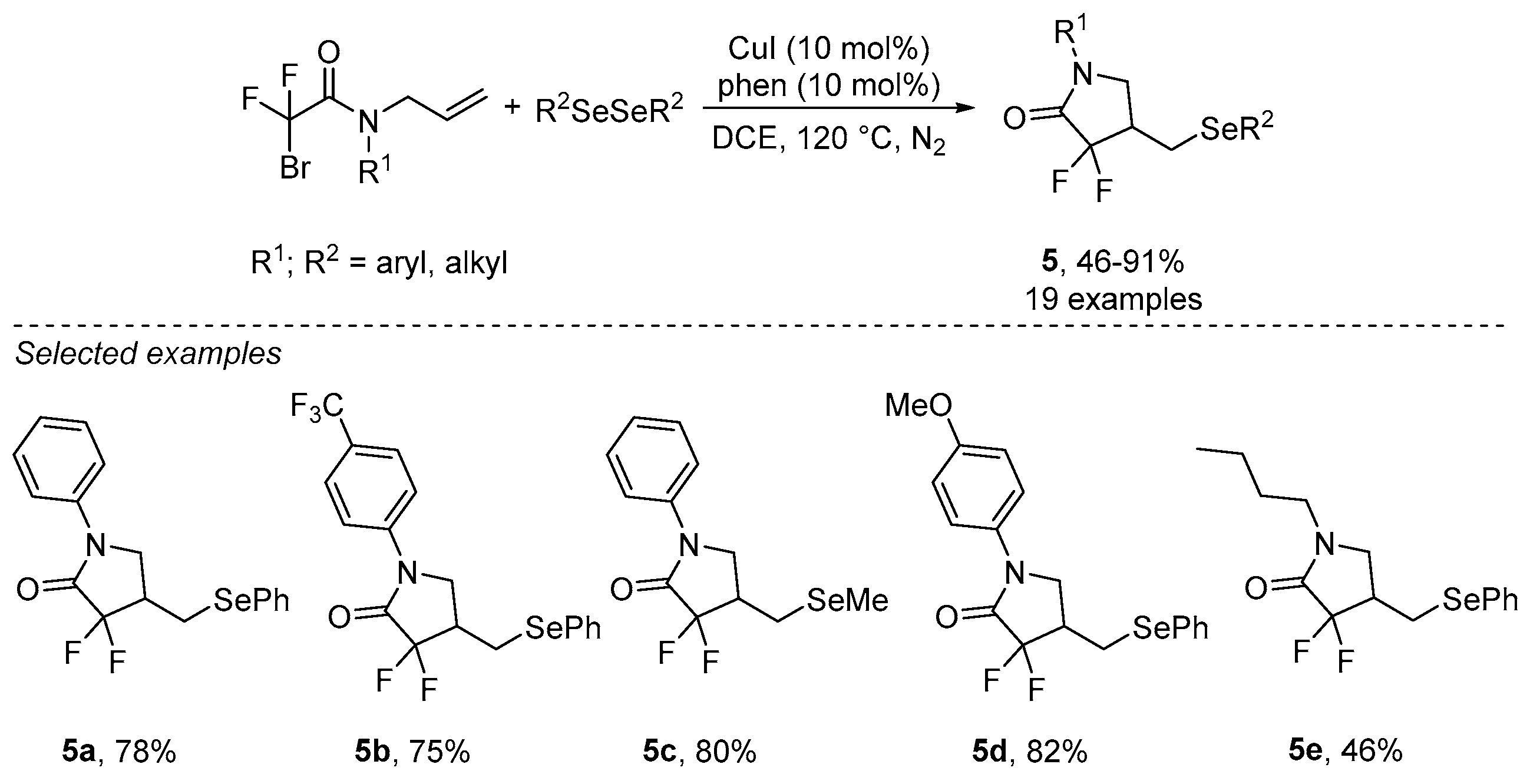
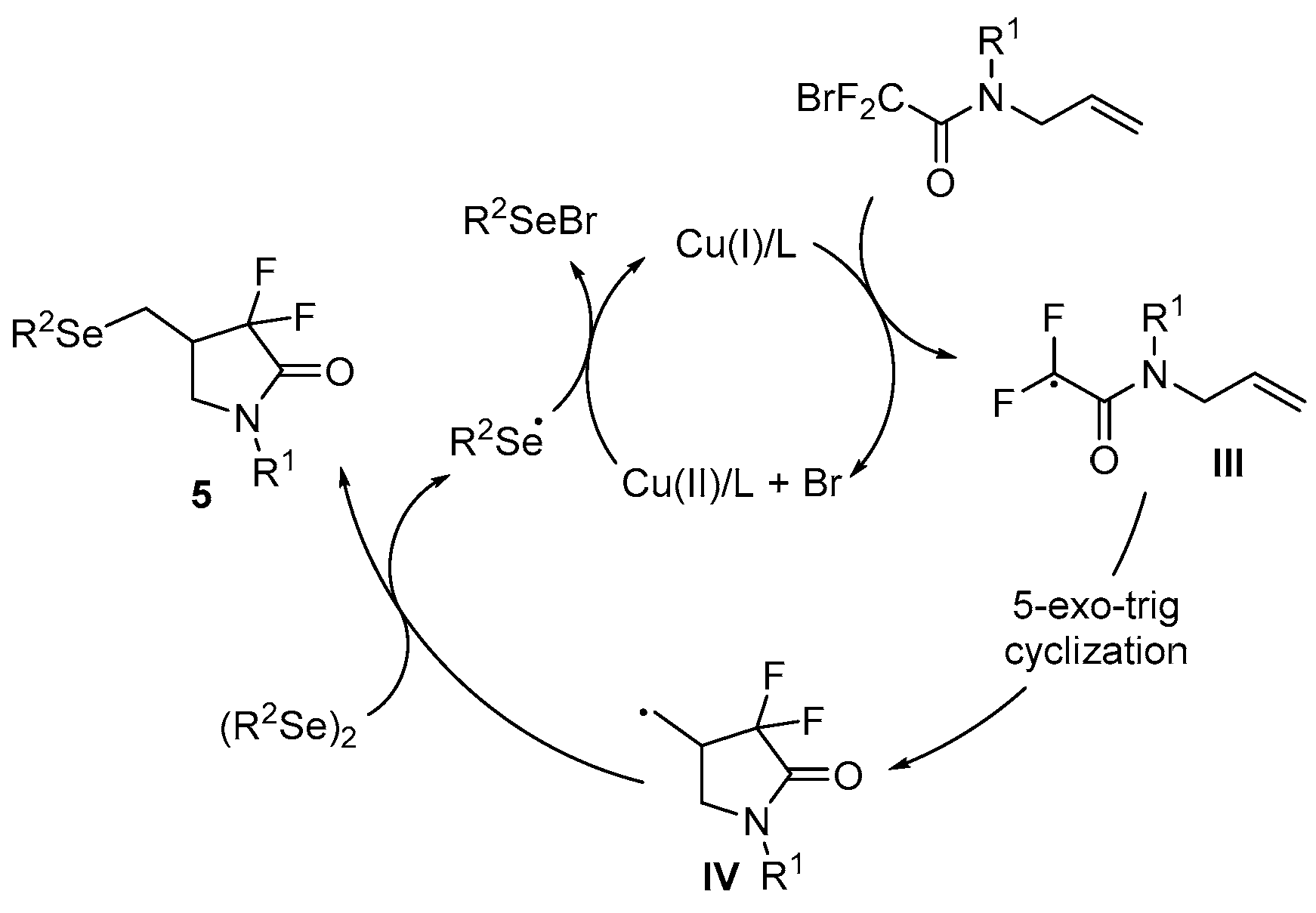
- Sun, K.; Wang, S.; Feng, R.; Zhang, Y.; Wang, X.; Zhang, Z.; Zhang, B. Copper-catalyzed radical selenodifluoromethylation of alkenes: Access to CF2-containing γ-lactams. Org. Lett. 2019, 21, 2052–2055. [Google Scholar] [CrossRef] [PubMed]
- Gou, R.; Zhang, Y.; Wu, S.W.; Liu, F. Synthesis of Polysubstituted 3-Chalcogenated Indoles through Copper (I) Iodide-Catalyzed Three-Component Domino Reactions. Synlett 2019, 30, 207–212. [Google Scholar] [CrossRef]
- Sahoo, S.R.; Sarkar, D.; Henkel, F.; Reuter, H. Copper (I)-Catalyzed Synthesis of Functionalized Indolizinones from Substituted Pyridine Homologated Ynones. J. Org. Chem. 2020, 85, 902–911. [Google Scholar] [CrossRef] [PubMed]
- He, F.S.; Yao, Y.; Tang, Z.; Xie, W.; Wu, J. Copper-catalyzed regio-and chemoselective selenosulfonylation of 1,6-enynes from sulfur dioxide. Org. Chem. Front. 2021, 8, 6119–6124. [Google Scholar] [CrossRef]
- Liu, W.; Chen, W.; Wang, L. Synthesis of 2-selenyl(sulfenyl)benzofurans via Cu-catalyzed tandem reactions of 2-(gemdibromovinyl) phenols with diorganyl diselenides(disulfides). RSC Adv. 2013, 3, 4723–4730. [Google Scholar] [CrossRef]
- Zhu, R.; Buchwald, S.L. Versatile Enantioselective Synthesis of Functionalized Lactones via Copper-Catalyzed Radical Oxyfunctionalization of Alkenes. J. Am. Chem. Soc. 2015, 137, 8069–8077. [Google Scholar] [CrossRef]
- Gao, Y.; Gao, Y.; Tanh, X.; Peng, J.; Hu, M.; Wu, W.; Jiang, H. Copper-Catalyzed Oxysulfenylation of Enolates with Sodium Sulfinates: A Strategy To Construct Sulfenylated Cyclic Ethers. Org. Lett. 2016, 18, 1158–1161. [Google Scholar] [CrossRef]
- Sahoo, S.R.; Sarkar, D.; Henkel, F.; Reuter, H. Copper (i) catalyzed synthesis of selanyl methylene 4-chromanol and aurone derivatives. Org. Biomol. Chem. 2020, 18, 4619–4627. [Google Scholar] [CrossRef]
- Maity, P.; Kundu, D.; Roy, R.; Ranu, B.C. A Direct Synthesis of Selenophenes by Cu-Catalyzed One-Pot Addition of a Selenium Moiety to (E,E)-1,3-Dienyl Bromides and Subsequent Nucleophilic Cyclization. Org. Lett. 2014, 16, 4122–4125. [Google Scholar] [CrossRef]
- Casola, K.K.; Gomes, M.R.; Back, D.F.; Zeni, G. Electrophilic Cyclization Involving Carbon−Selenium/Carbon−Halide Bond Formation: Synthesis of 3-Substituted Selenophenes. J. Org. Chem. 2018, 83, 6706–6718. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Li, M.; Jin, Y.; Jiang, H.; Wu, W. Synthesis of 2,5-disubstituted selenophenes via a copper-catalyzed regioselective [2+2+1] cyclization of terminal alkynes and selenium. Chem. Commun. 2022, 58, 6522–6525. [Google Scholar] [CrossRef] [PubMed]
- Bilheri, F.N.; Pistoia, R.P.; Back, D.F.; Zeni, G. Copper/Palladium-Catalyzed Cyclization/Cross-Coupling Cascade Reaction of 2-gem-Dibromovinyl Aryl Selenides: Synthesis of 2-Substituted Benzo[b]selenophenes. Adv. Synth. Catal. 2017, 359, 4208–4216. [Google Scholar] [CrossRef]
- Higuchi, Y.; Higashimae, S.; Tamai, T.; Ogawa, A. A highly selective cobalt-catalyzed carbonylative cyclization of internal alkynes with carbon monoxide and organic thiols. Tetrahedron 2013, 69, 11197–11202. [Google Scholar] [CrossRef]
- Higashimae, S.; Tamai, T.; Nomoto, A.; Ogawa, A. Selective Thiolative Lactonization of Internal Alkynes Bearing a Hydroxyl Group with Carbon Monoxide and Organic Disulfides Catalyzed by Transition-Metal Complexes. J. Org. Chem. 2015, 80, 7126–7133. [Google Scholar] [CrossRef]
- Gao, X.-H.; Qian, P.-C.; Zhang, X.-G.; Deng, C.-L. FeBr3-Catalyzed Tandem Reaction of N-Propargylamides with Disulfides or Diselenides for the Synthesis of Oxazole Derivatives. Synlett 2016, 27, 1110–1115. [Google Scholar] [CrossRef]
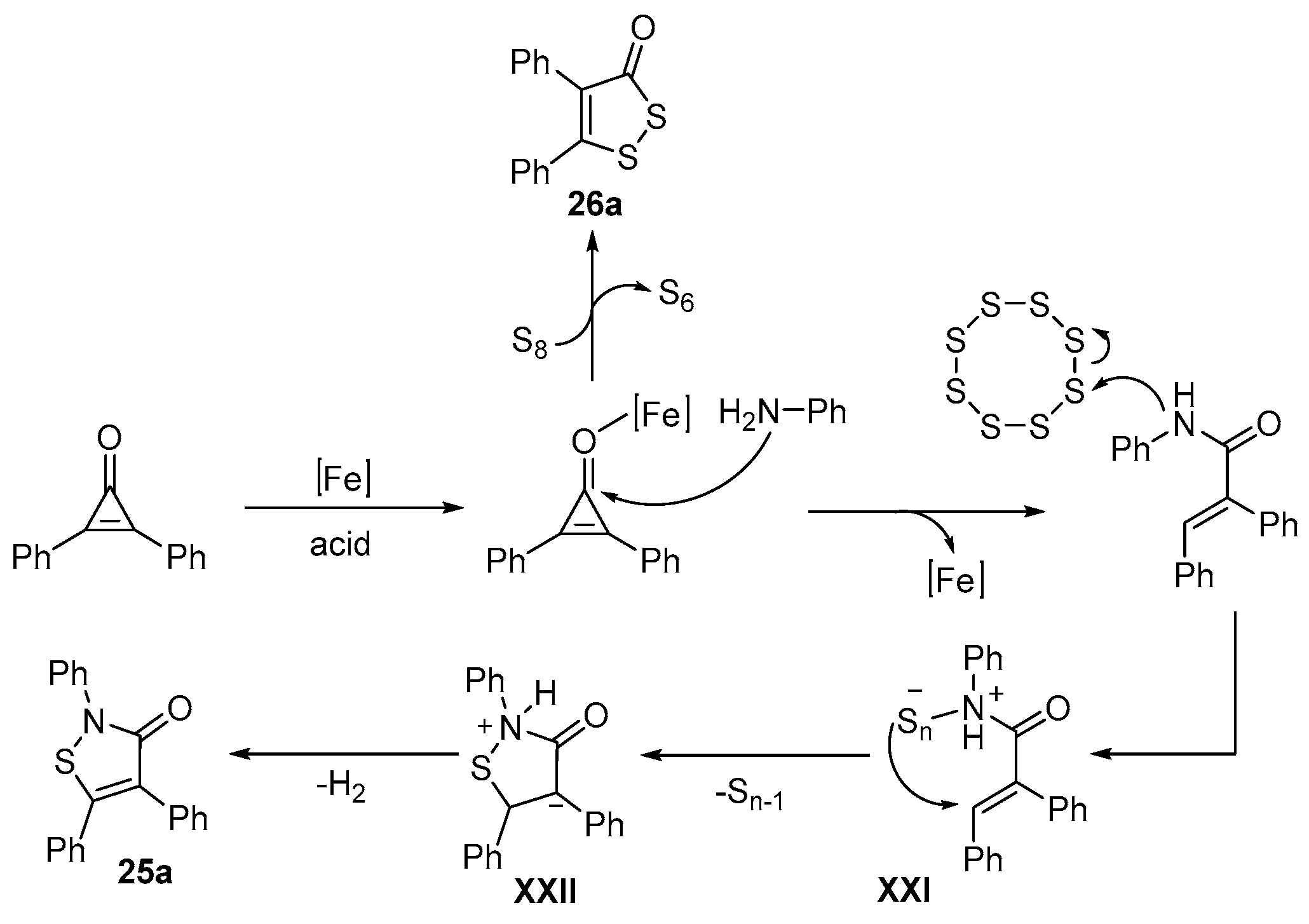
- Wang, H.; Yan, R. Iron-Catalyzed One-Step Synthesis of Isothiazolone/1,2-Selenazolone Derivatives via [3+1+1] Annulation of Cyclopropenones, Anilines, and Elemental Chalcogens. Adv. Synth. Catal. 2022, 364, 715–719. [Google Scholar] [CrossRef]
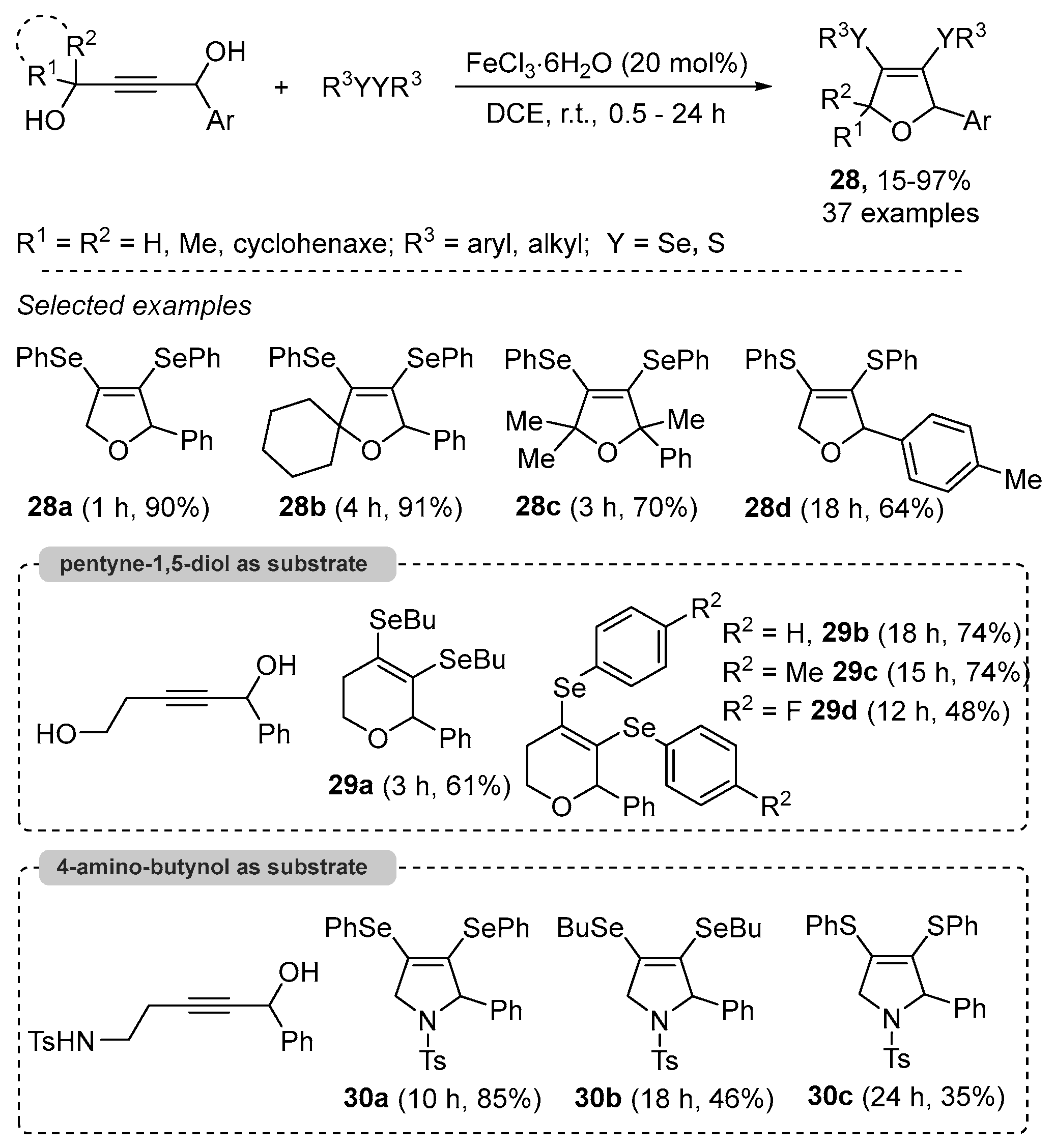
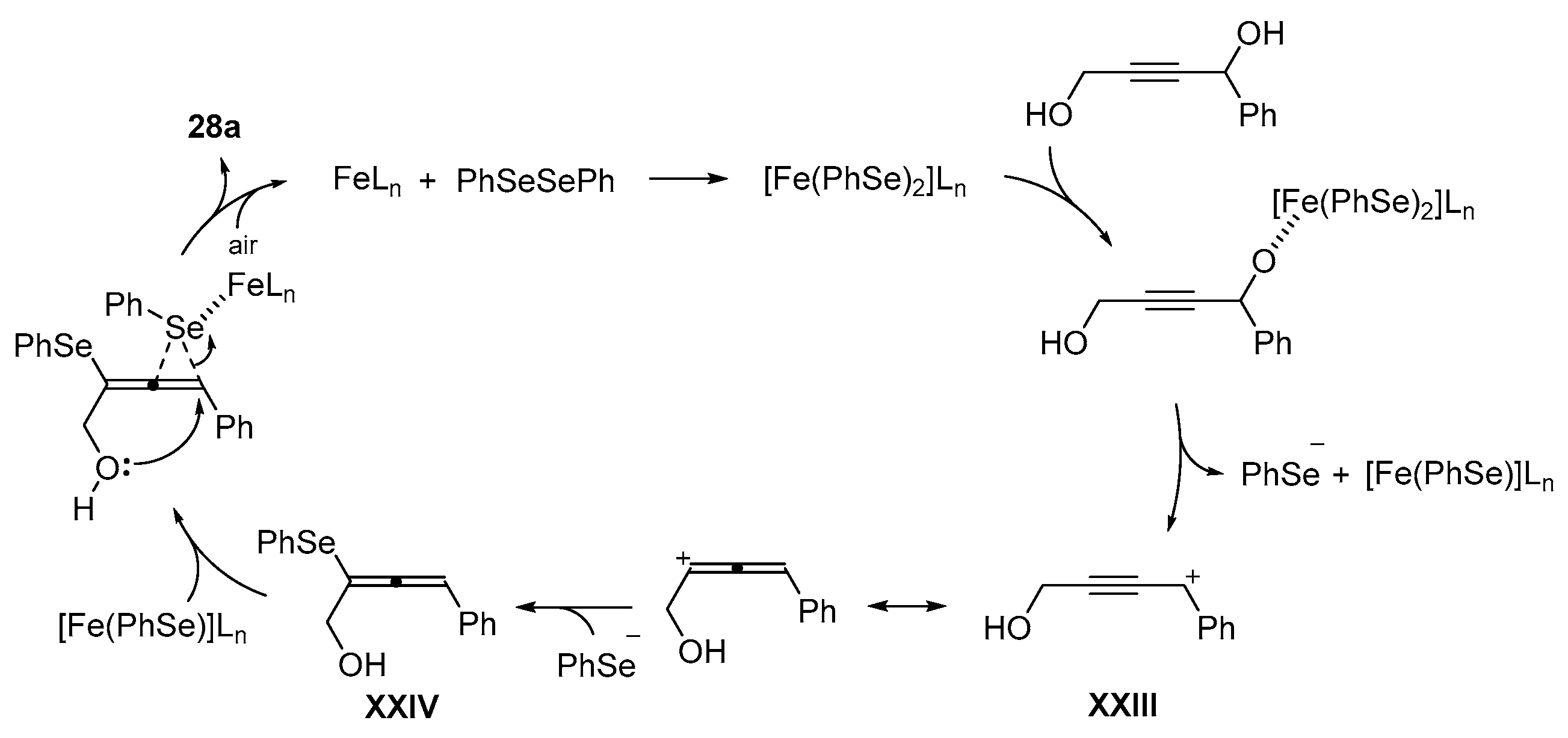
- Casola, K.K.; Back, D.F.; Zeni, G. Iron-Catalyzed Cyclization of Alkynols with Diorganyl Diselenides: Synthesis of 2,5-Dihydrofuran, 3,6-Dihydro-2H-pyran, and 2,5-Dihydro-1H-pyrrole Organoselanyl Derivatives. J. Org. Chem. 2015, 80, 7702–7712. [Google Scholar] [CrossRef]
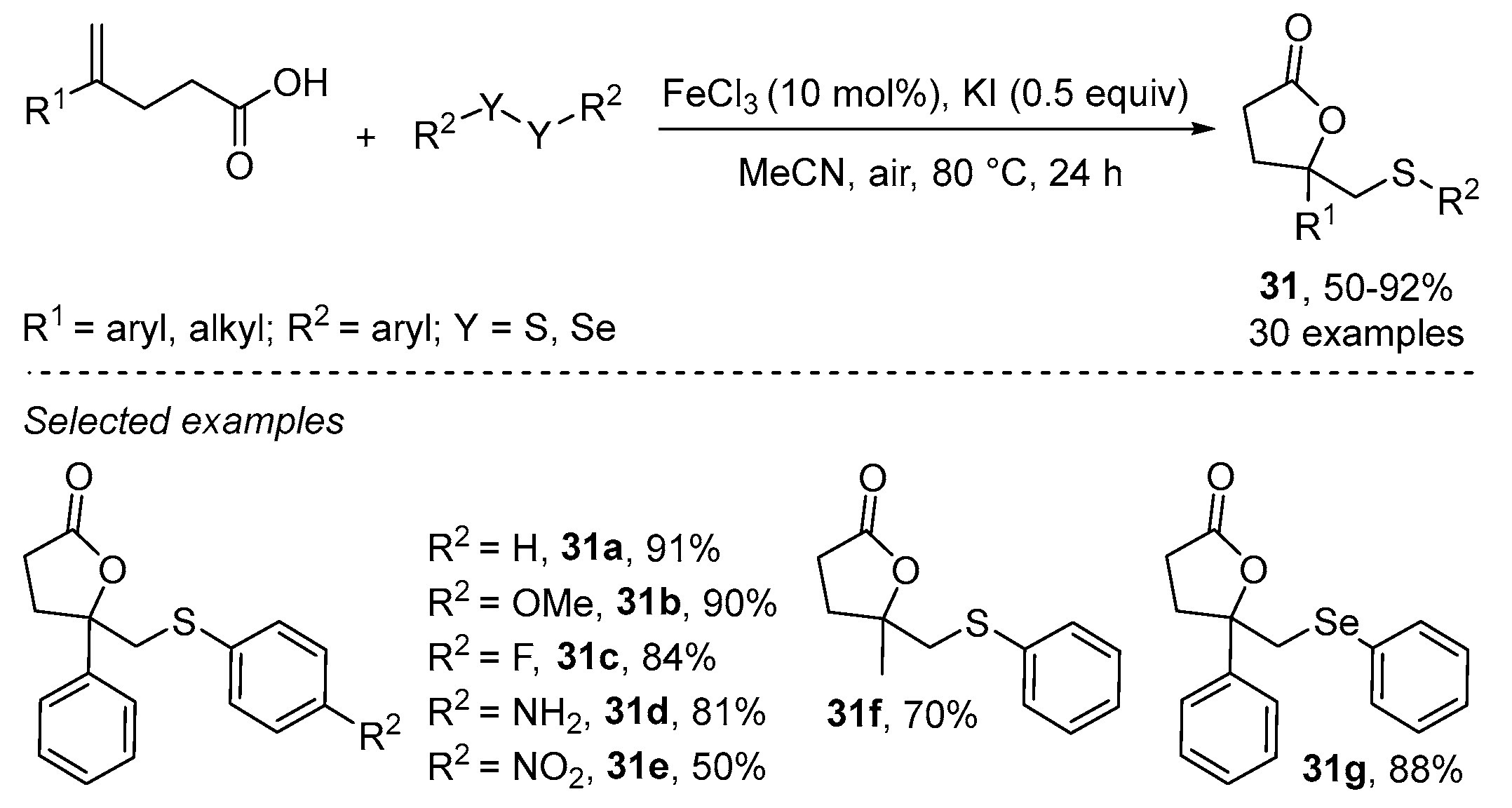
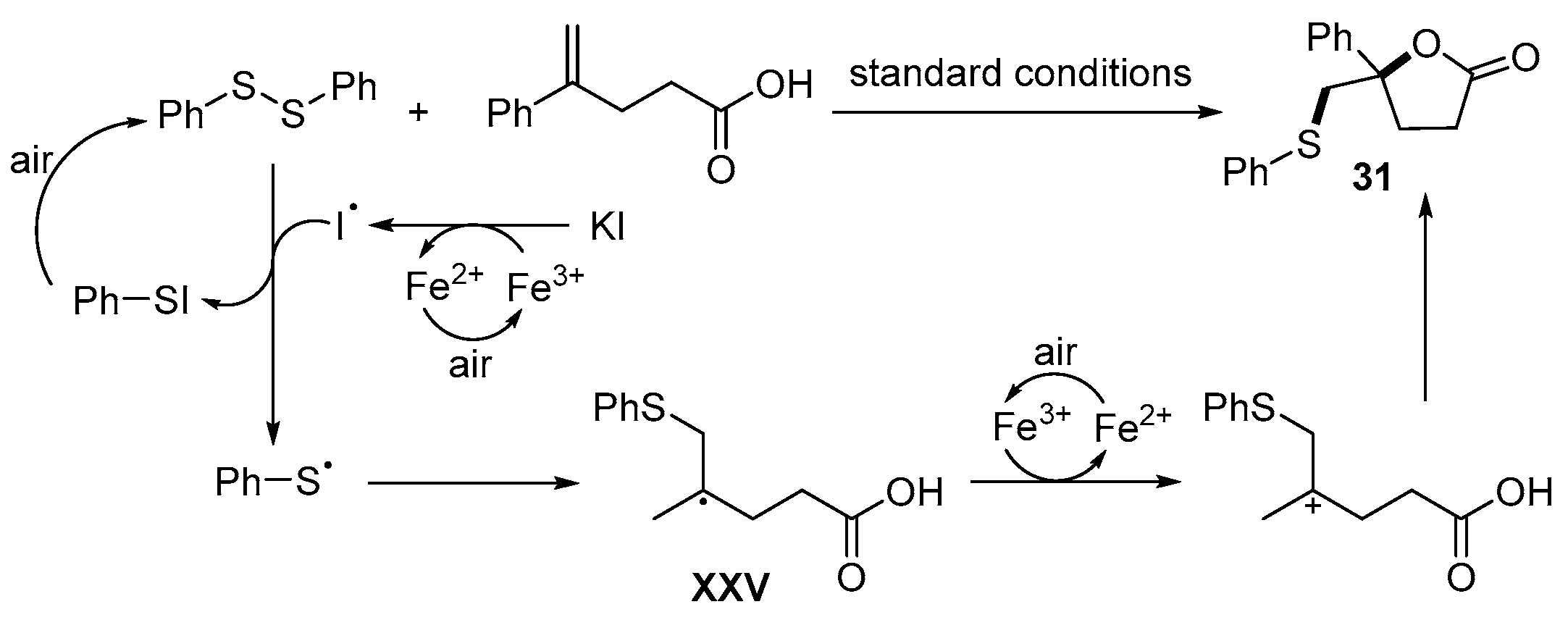
- Cheng, F.; Wang, L.-L.; Mao, Y.-H.; Dong, Y.-X.; Liu, B.; Zhu, G.-F.; Yang, Y.-Y.; Guo, B.; Tang, L.; Zhang, J.-Q. Iron-Catalyzed Radical Annulation of Unsaturated Carboxylic Acids with Disulfides for the Synthesis of γ-Lactones. J. Org. Chem. 2021, 86, 8620–8629. [Google Scholar] [CrossRef]
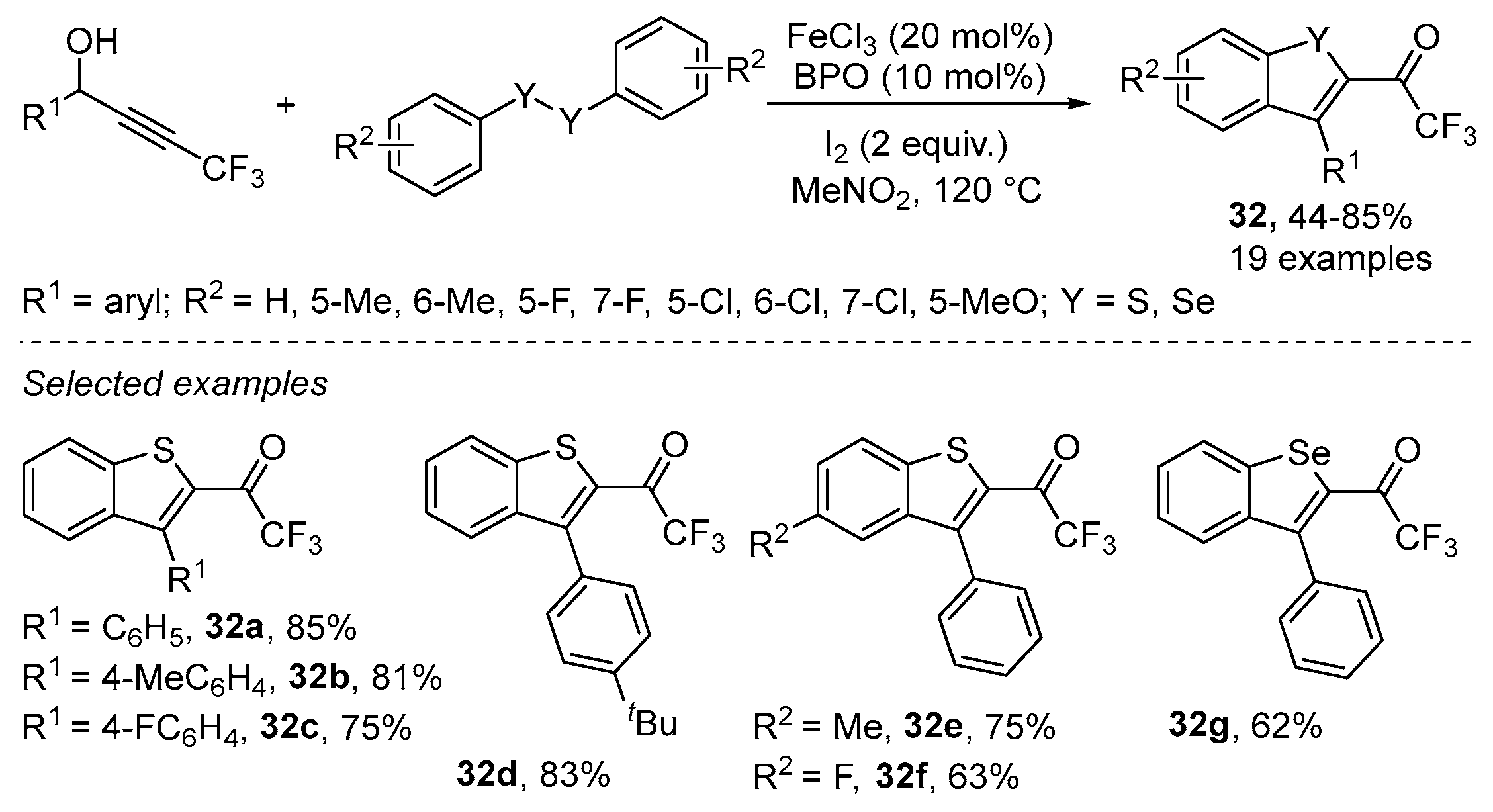
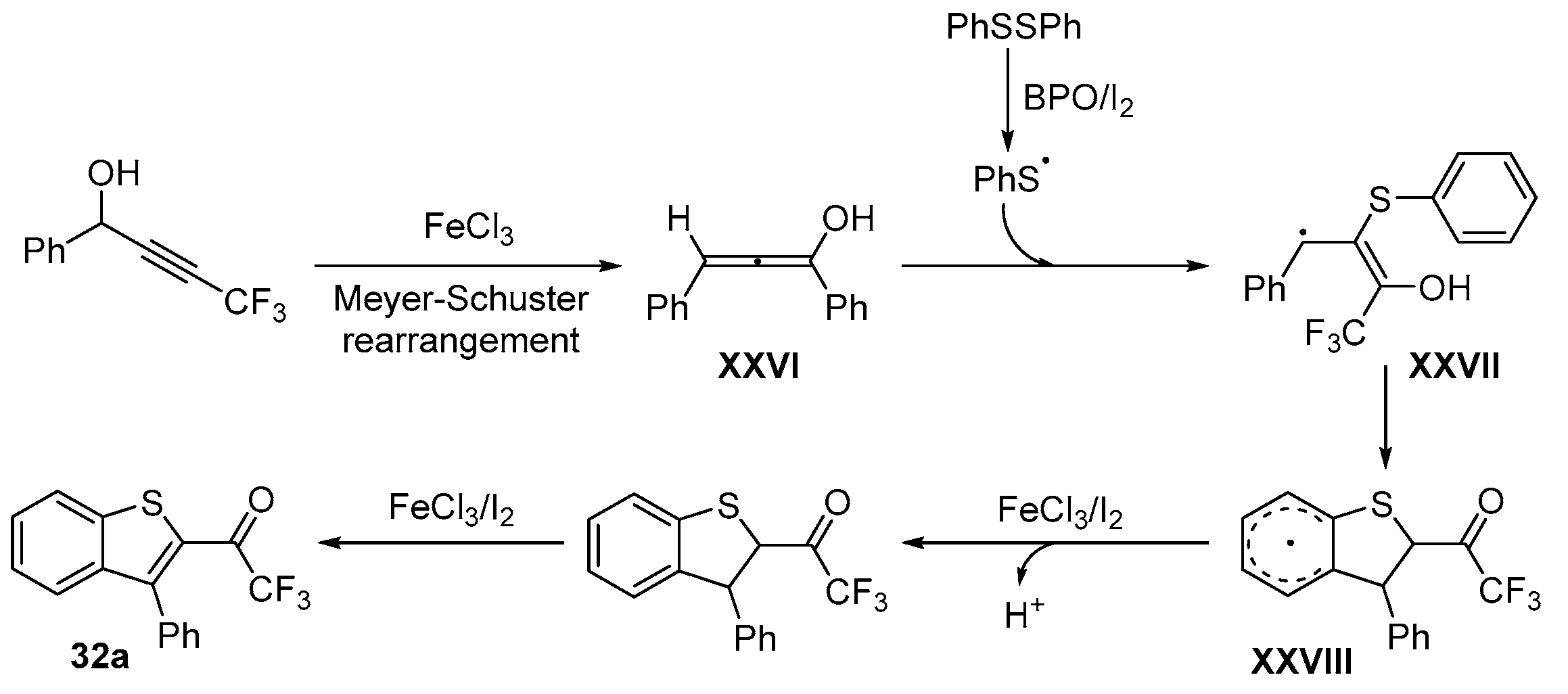
- Lin, Y.-F.; Wang, C.; Hu, B.-L.; Qian, P.-C.; Zhang, X.-G. Iron-Catalyzed Thiocyclization for the Synthesis of Trifluoromethylated Benzothiophenes by C–H Functionalization of Aryl Disulfides. Synlett 2017, 28, 707–712. [Google Scholar] [CrossRef]
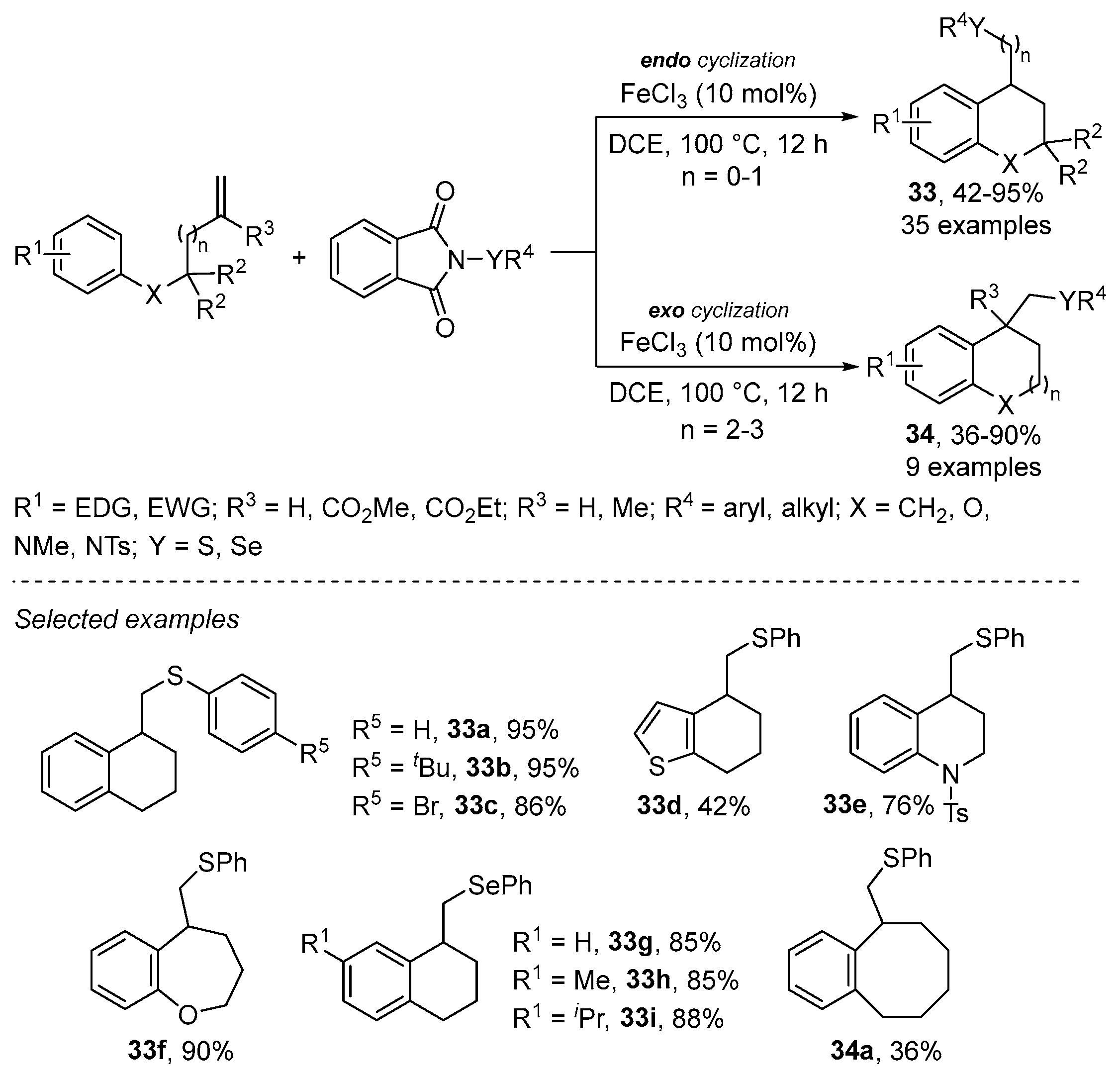
- Lv, L.; Li, Z. FeCl3-Catalyzed Regio-Divergent Carbosulfenylation of Unactivated Alkenes: Construction of a Medium-Sized Ring. J. Org. Chem. 2018, 83, 10985–10994. [Google Scholar] [CrossRef] [PubMed]
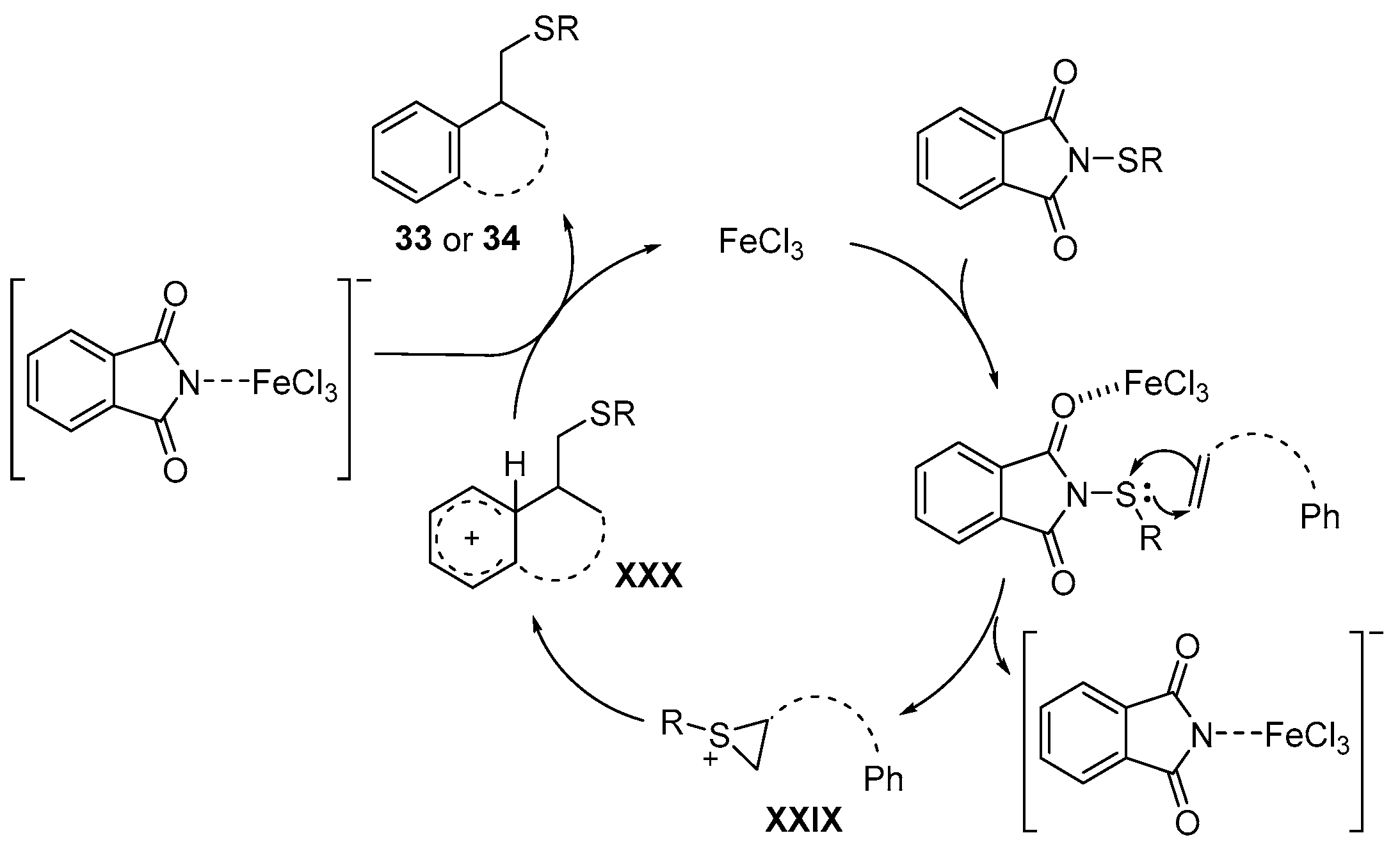
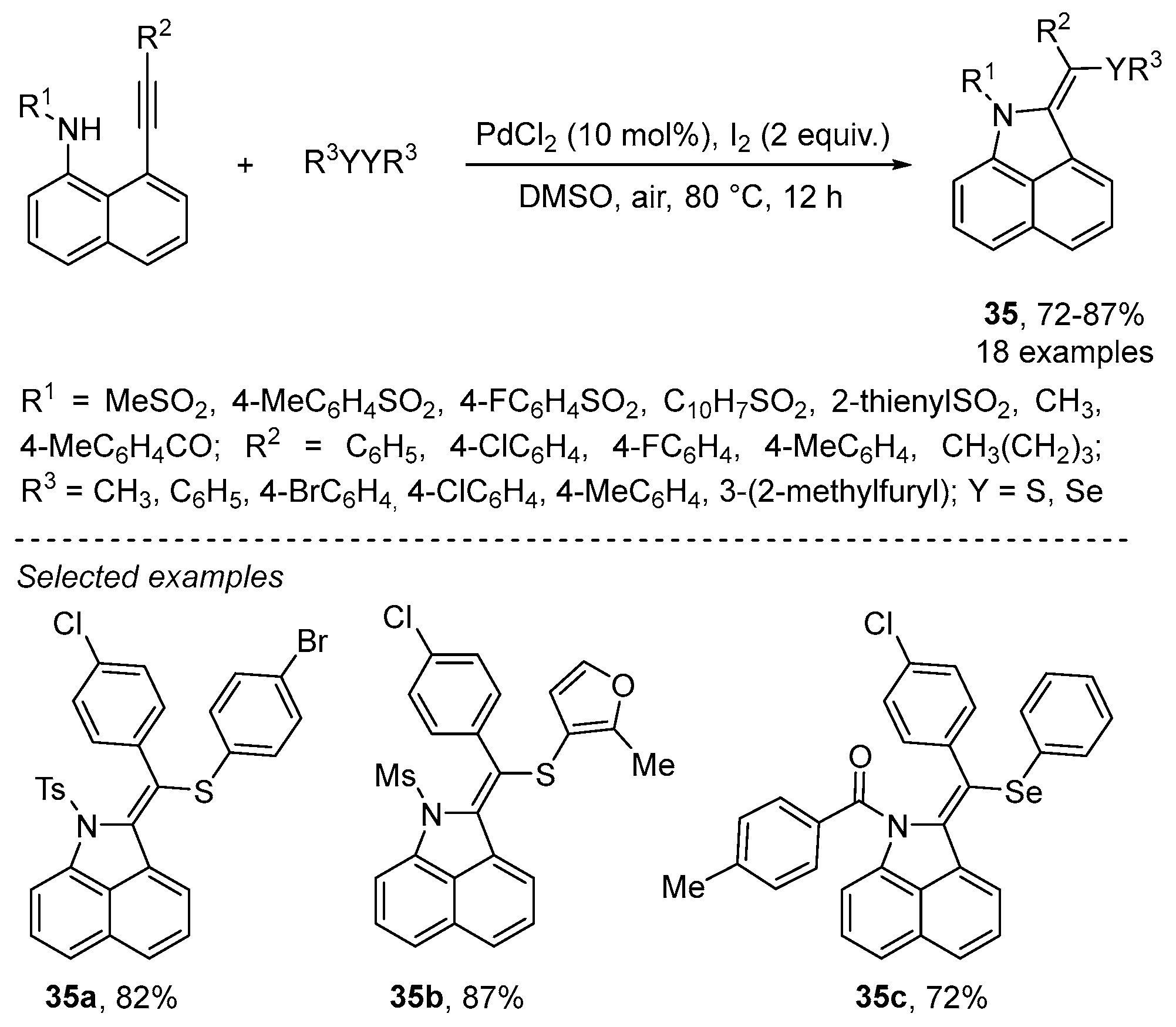
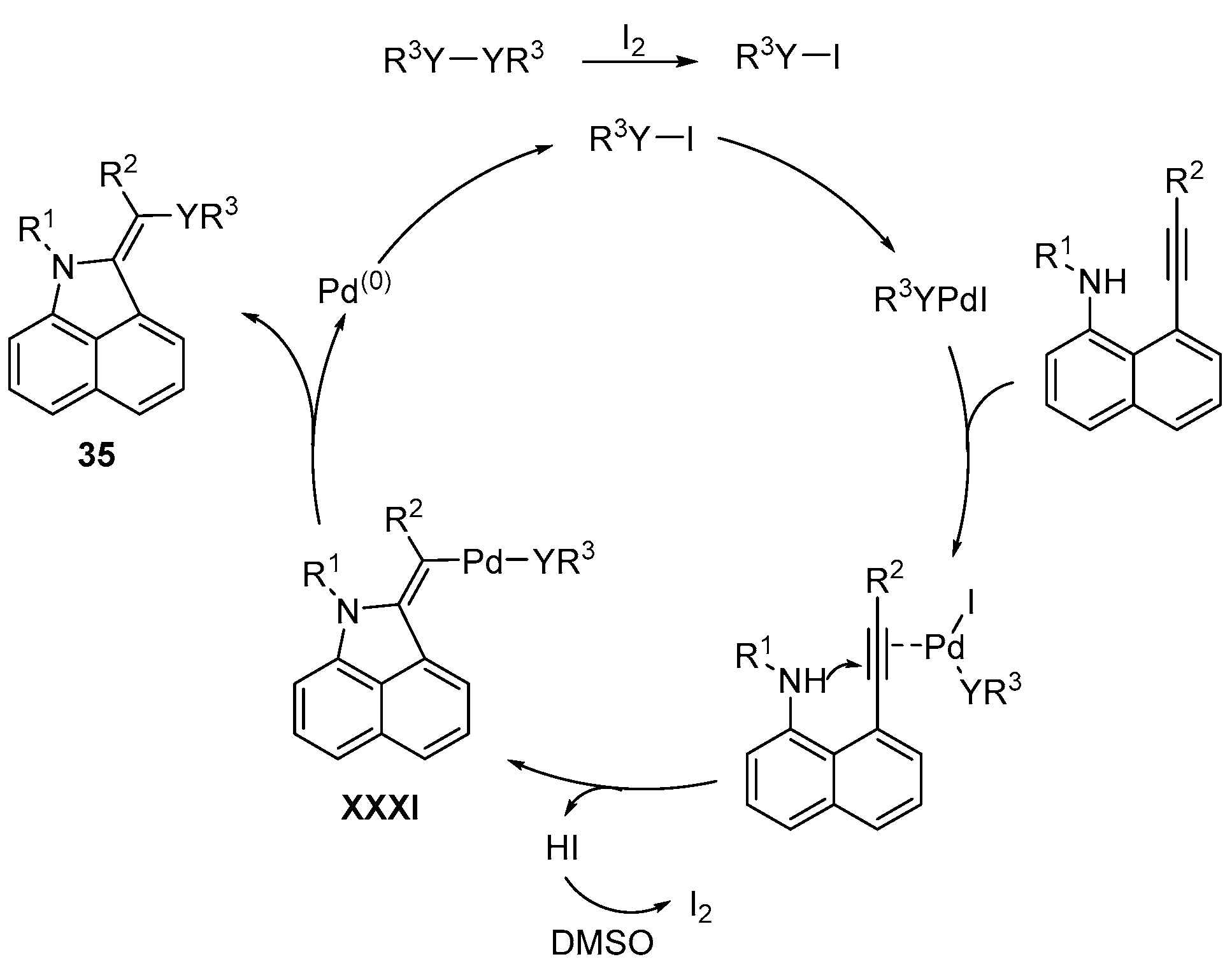
- Bi, C.; Zhang, L.; Qiu, G.; Li, X.; Yao, J.; Zhou, H. Stereoselective and regioselective 5-exo-dig cyclization of 8-alkynylnaphthalen-1-amines for the synthesis of (E)-2-(arylthio)alkylene-1,2-dihydrobenzo[cd]indoles. Org. Biomol. Chem. 2018, 16, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, Y.; Hu, M.; Li, C.; Li, M.; He, D.; Jiang, H. A palladium-catalyzed three-component cascade S-transfer reaction in ionic liquids. Green Chem. 2019, 21, 4084–4089. [Google Scholar] [CrossRef]
- Jin, G.-Q.; Gao, W.-X.; Zhou, Y.-B.; Liu, M.-C.; Wu, H.-Y. Synthesis of selenated isochromenones by AgNO3-catalyzed three-component reaction of alkynylaryl esters, selenium powder and ArB(OH)2. RSC Adv. 2020, 10, 30439–30442. [Google Scholar] [CrossRef] [PubMed]
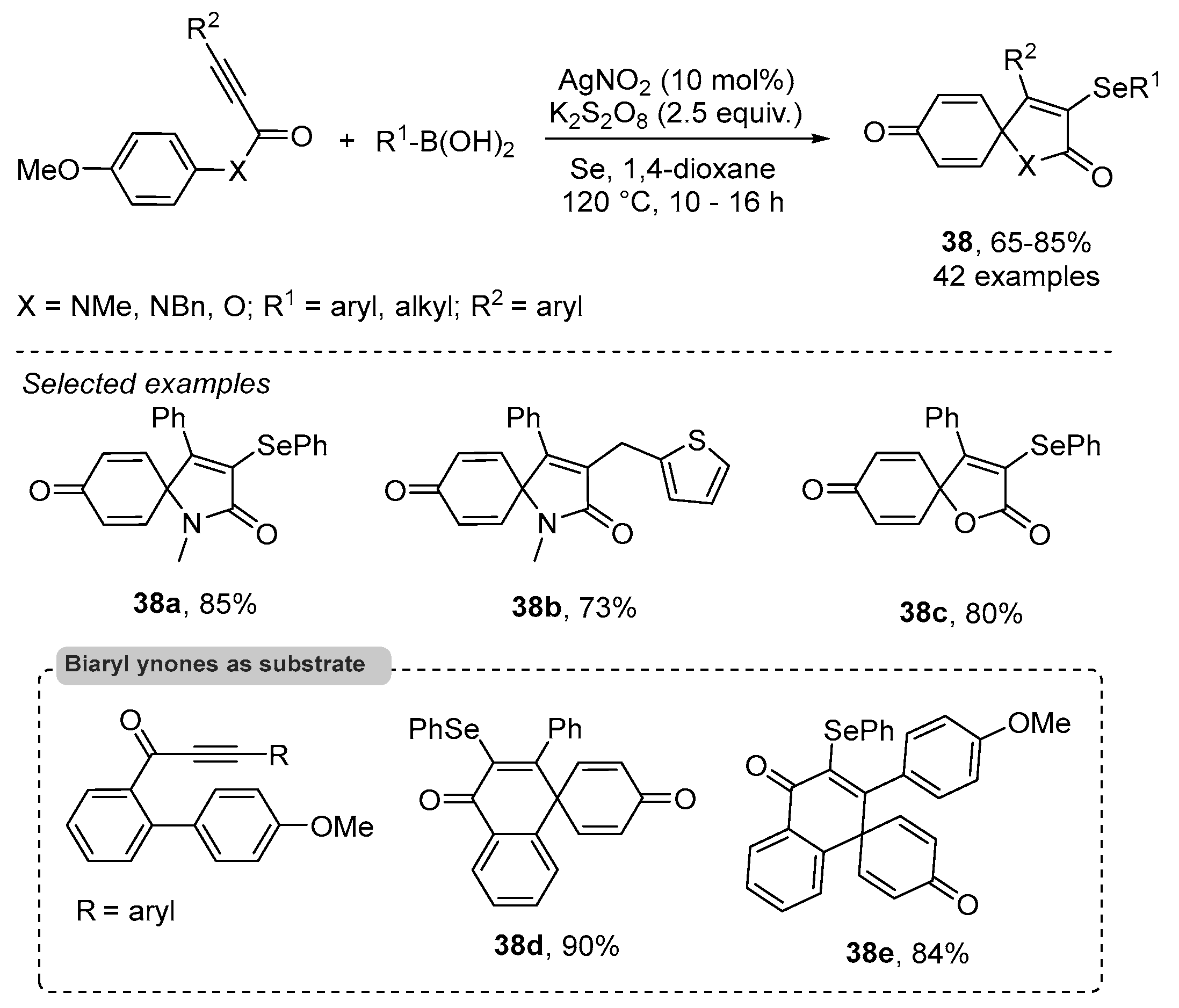
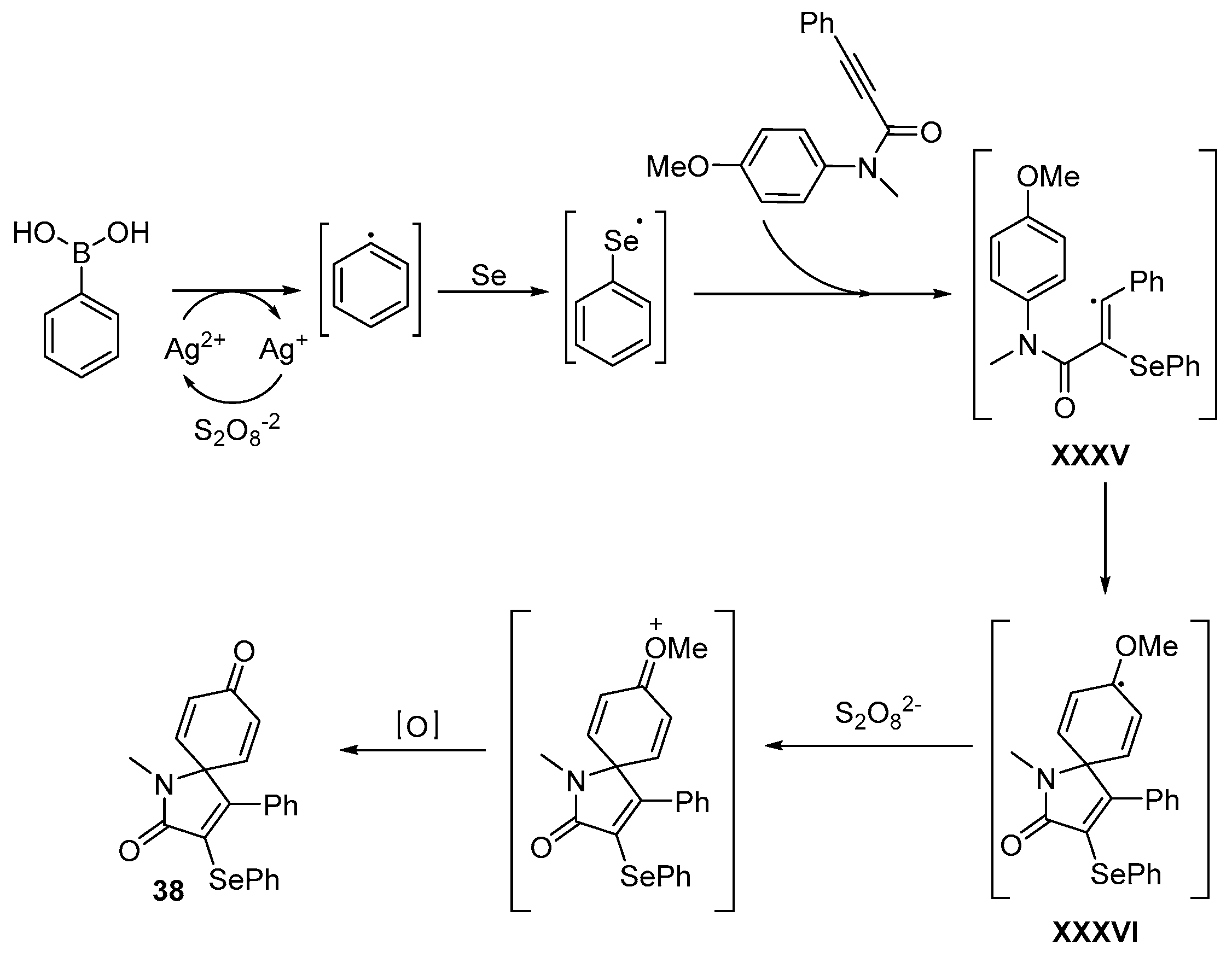
- Reddy, C.R.; Subbarao, M.; Kolgave, D.H.; Ajaykumar, U.; Vinaya, P.P. Access to Diverse Seleno-spirocyclohexadienones via Ag(II)-Catalyzed Selenylative ipso-Annulation with Se and Boronic Acids. ACS Omega 2022, 7, 38045–38052. [Google Scholar] [CrossRef] [PubMed]
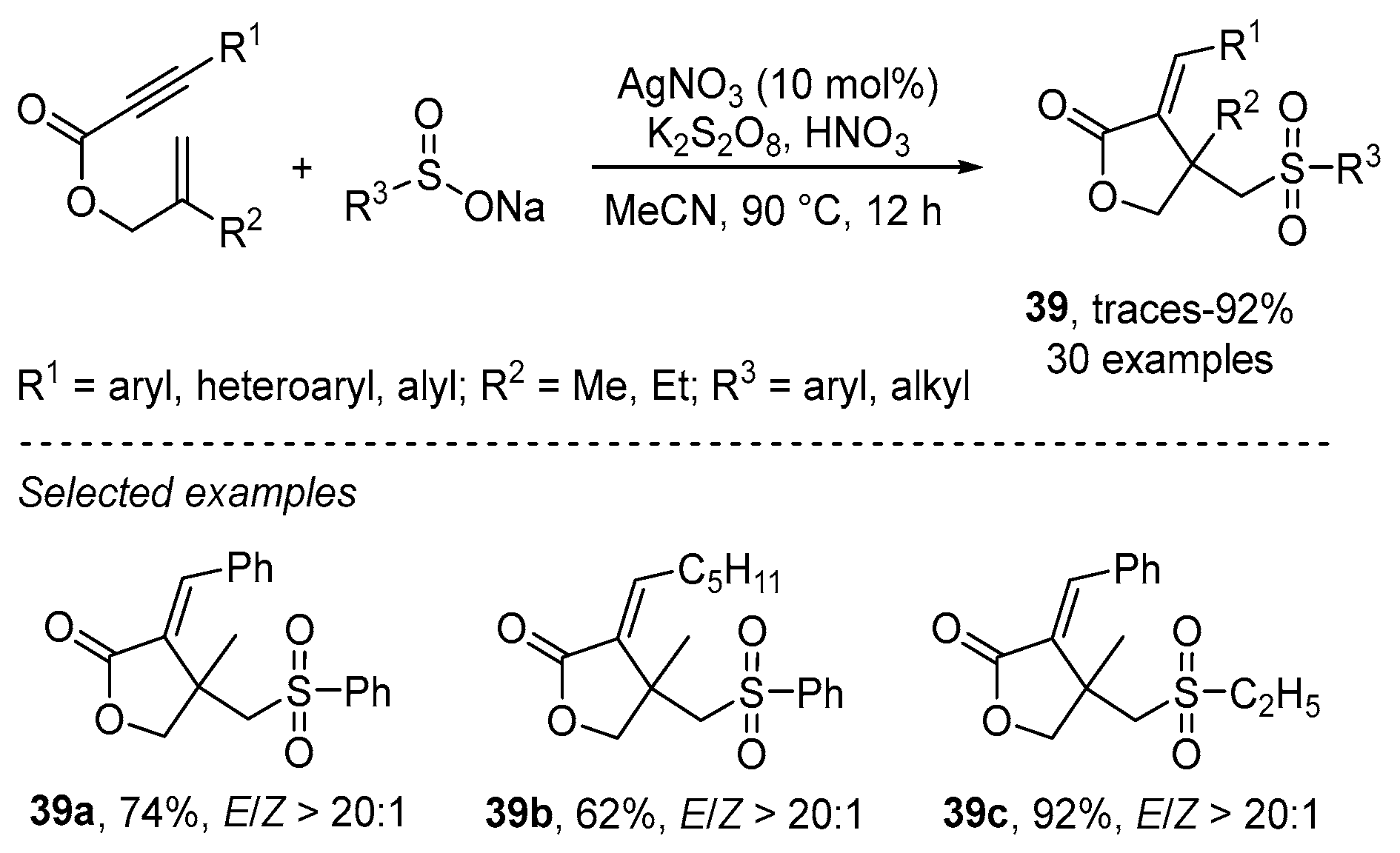
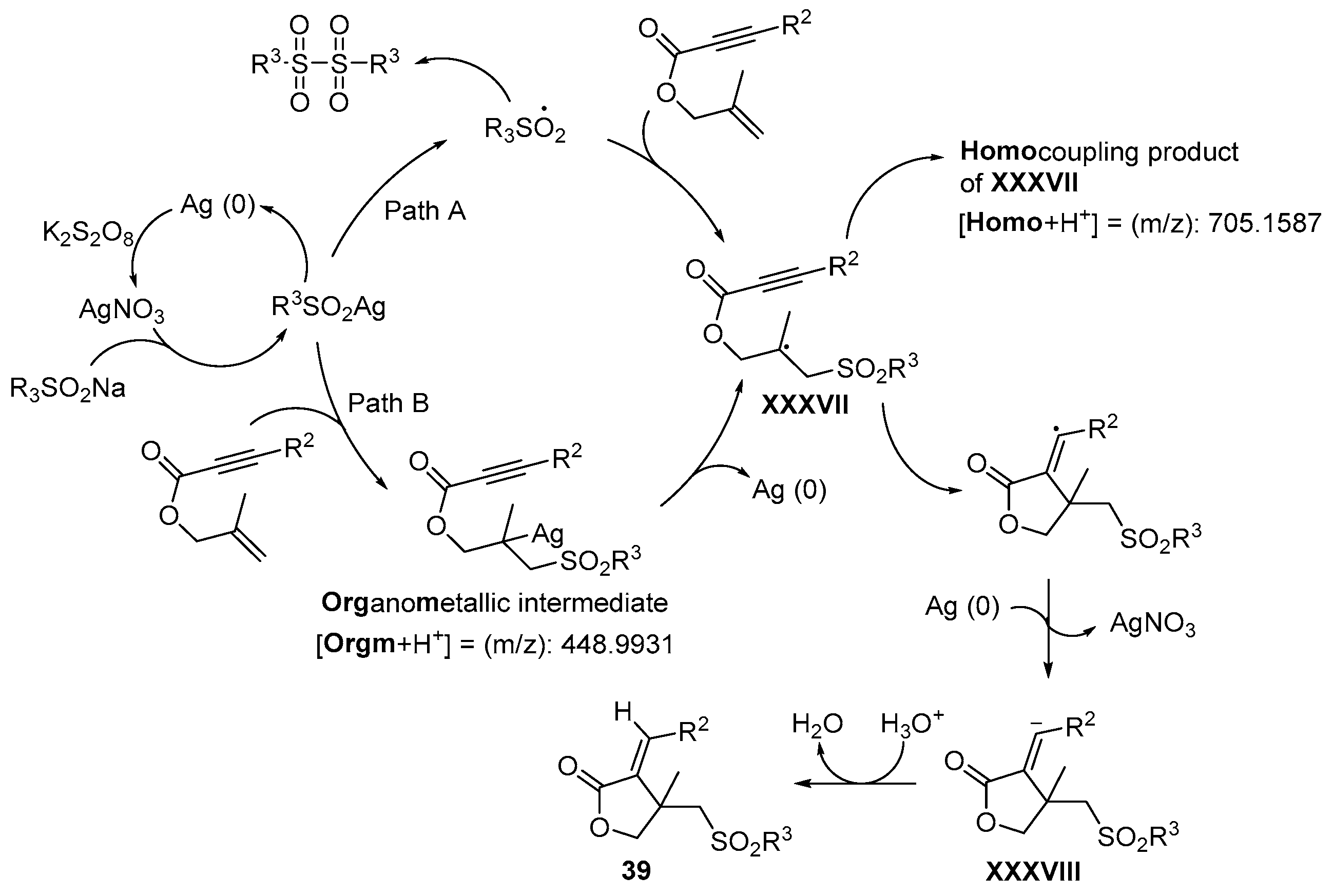
- Wu, W.; Yi, S.; Yu, Y.; Huang, W.; Jiang, H. Synthesis of Sulfonylated Lactones via Ag-Catalyzed Cascade Sulfonylation/Cyclization of 1,6-Enynes with Sodium Sulfinates. J. Org. Chem. 2017, 82, 1224–1230. [Google Scholar] [CrossRef]
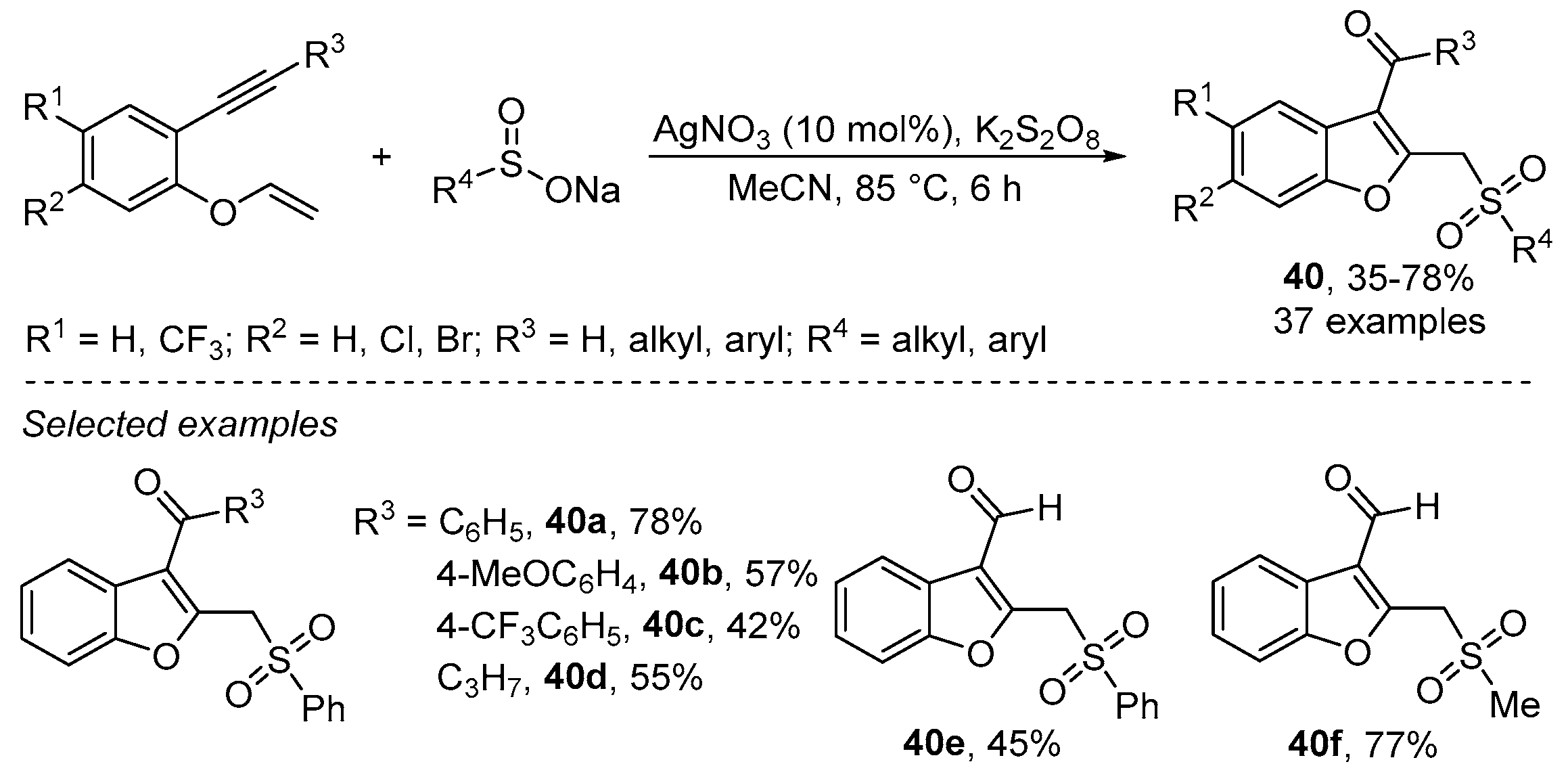
- Wu, W.; Yi, S.; Luo, D.; Jiang, H. Ag-Catalyzed Oxidative Cyclization Reaction of 1,6-Enynes and Sodium Sulfinate: Access to Sulfonylated Benzofurans. Org. Lett. 2017, 19, 2825–2828. [Google Scholar] [CrossRef]
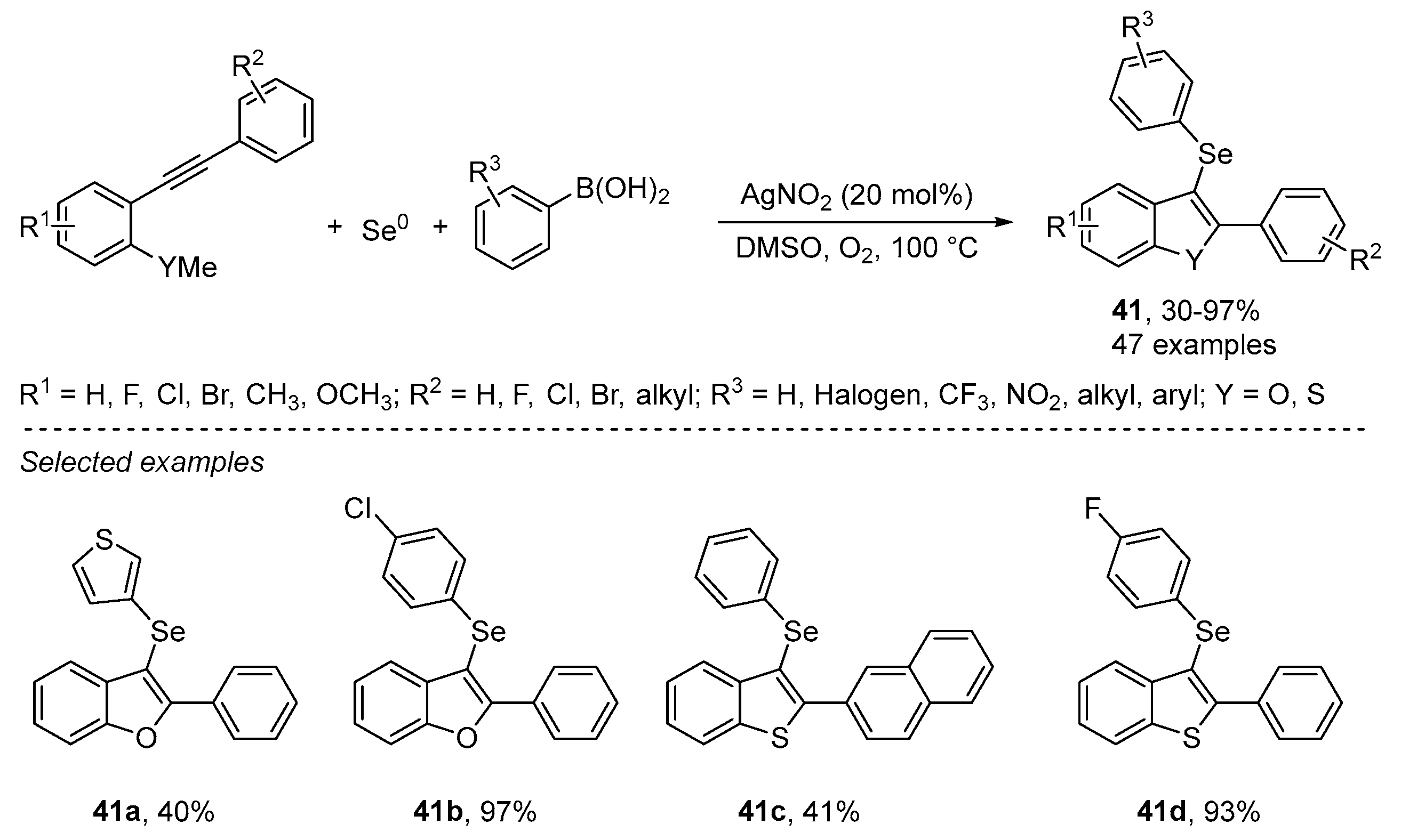
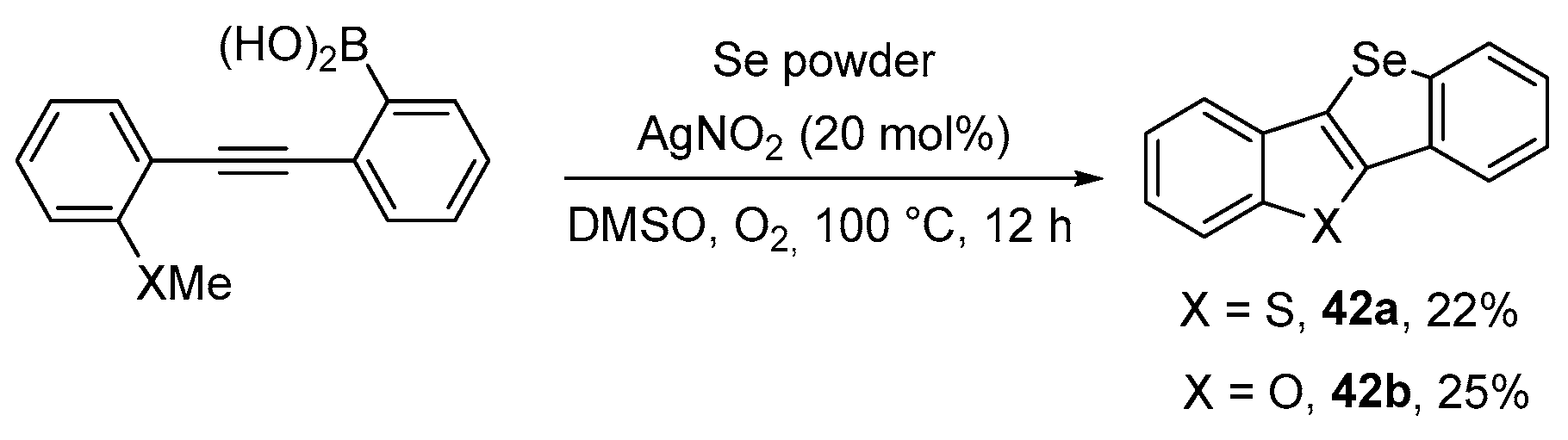
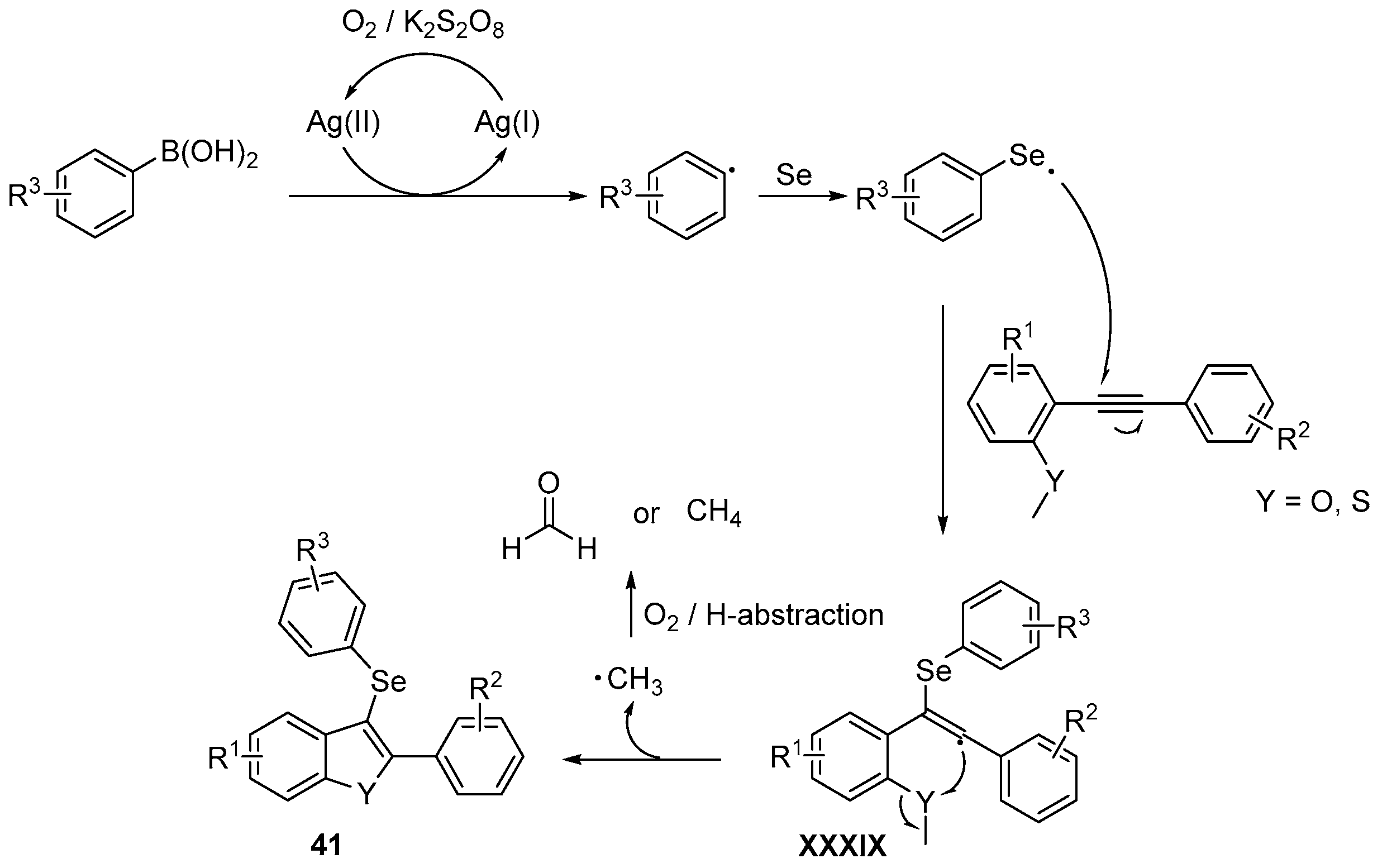
- An, C.; Li, C.-Y.; Huang, X.-B.; Gao, W.-X.; Zhou, Y.-B.; Liu, M.-C.; Wu, H.-Y. Selenium Radical Mediated Cascade Cyclization: Concise Synthesis of Selenated Benzofurans (Benzothiophenes). Org. Lett. 2019, 21, 6710–6714. [Google Scholar] [CrossRef]
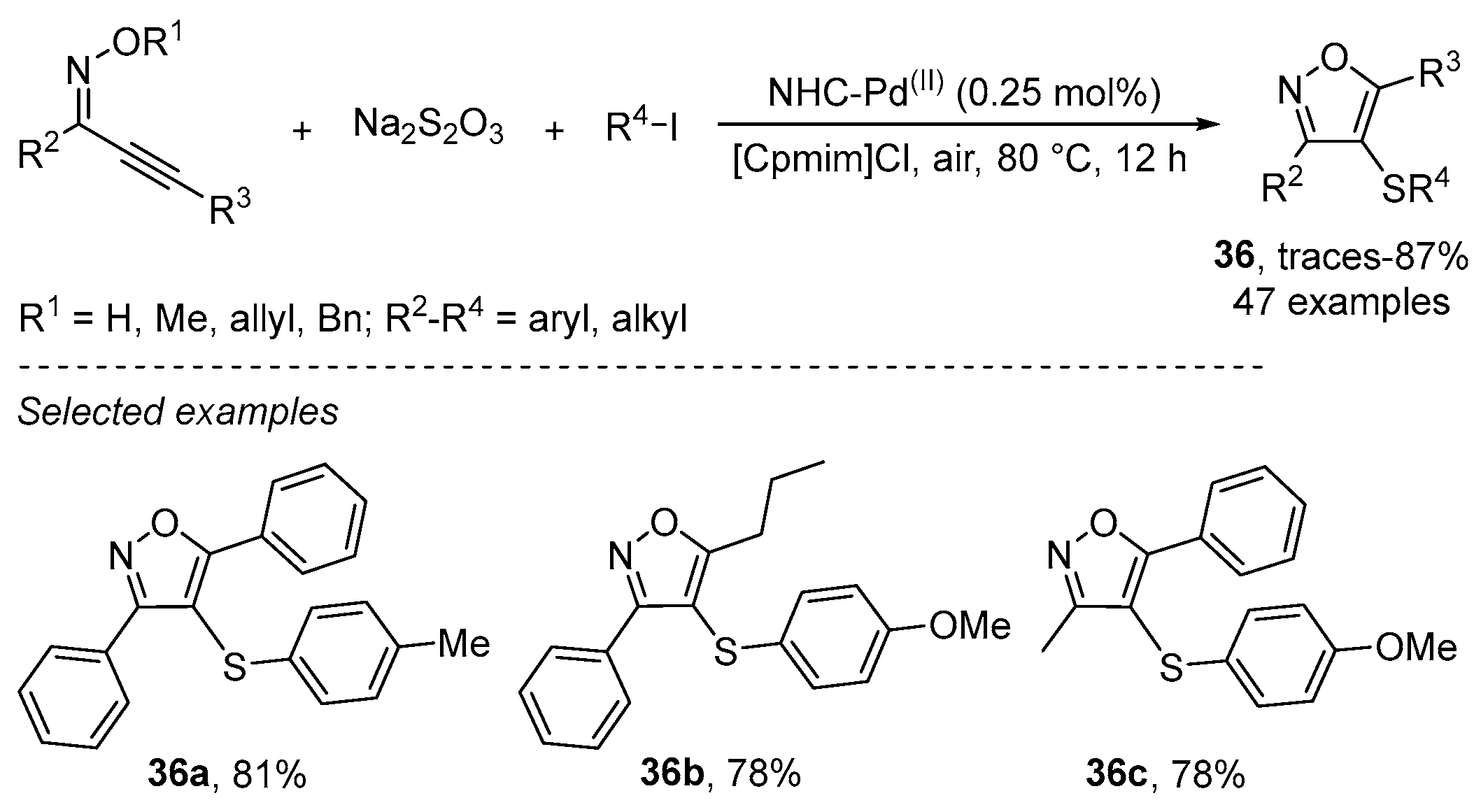
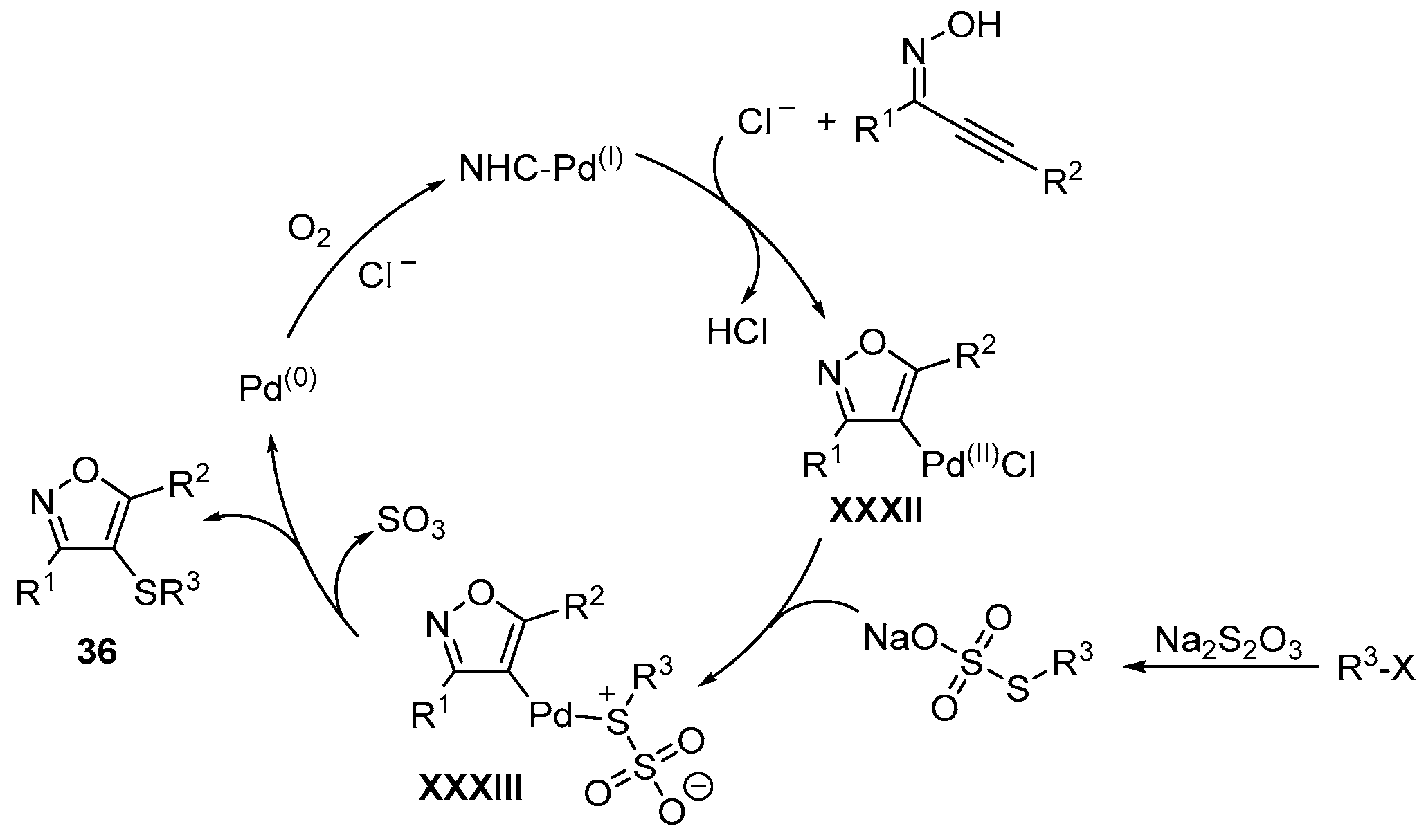
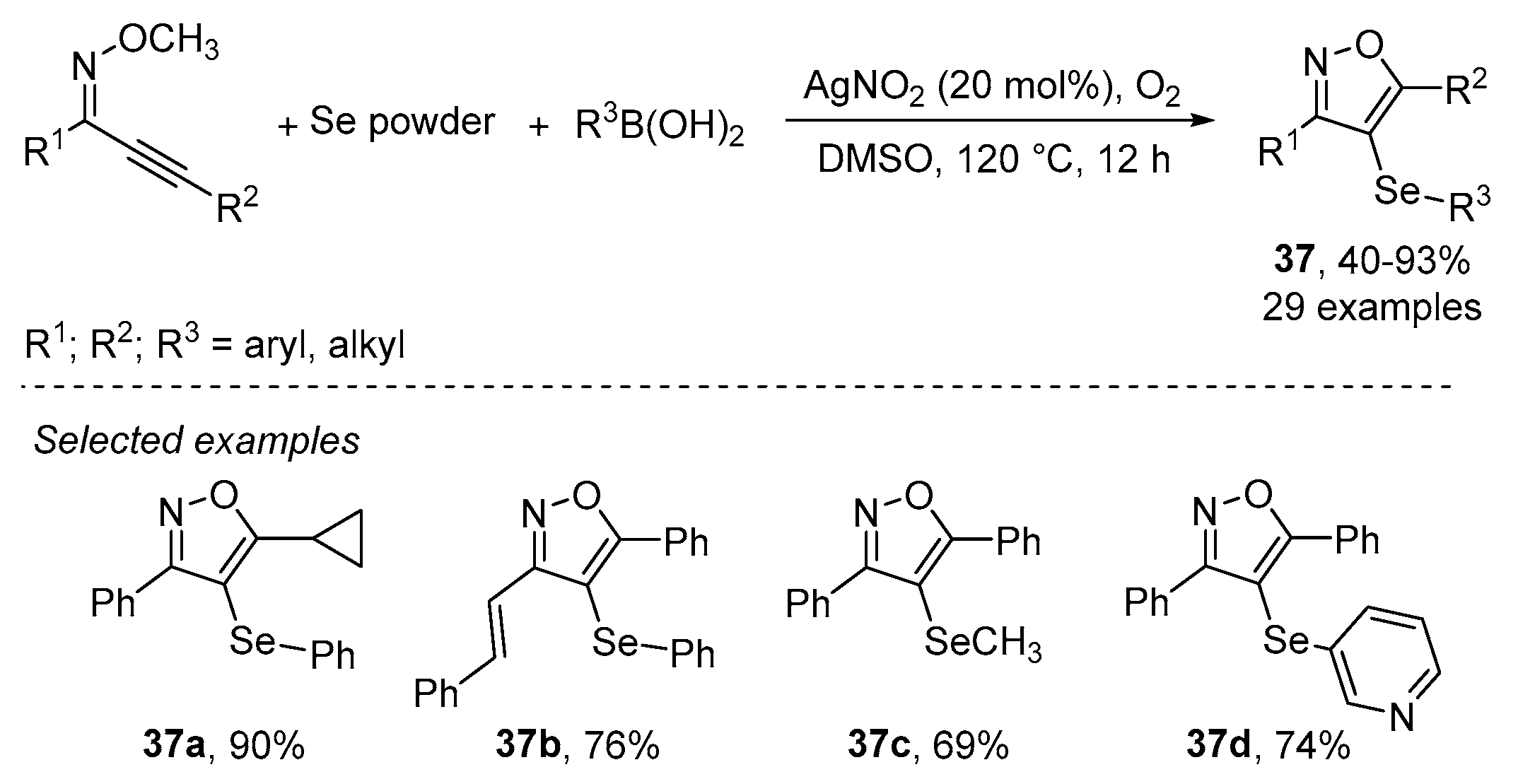
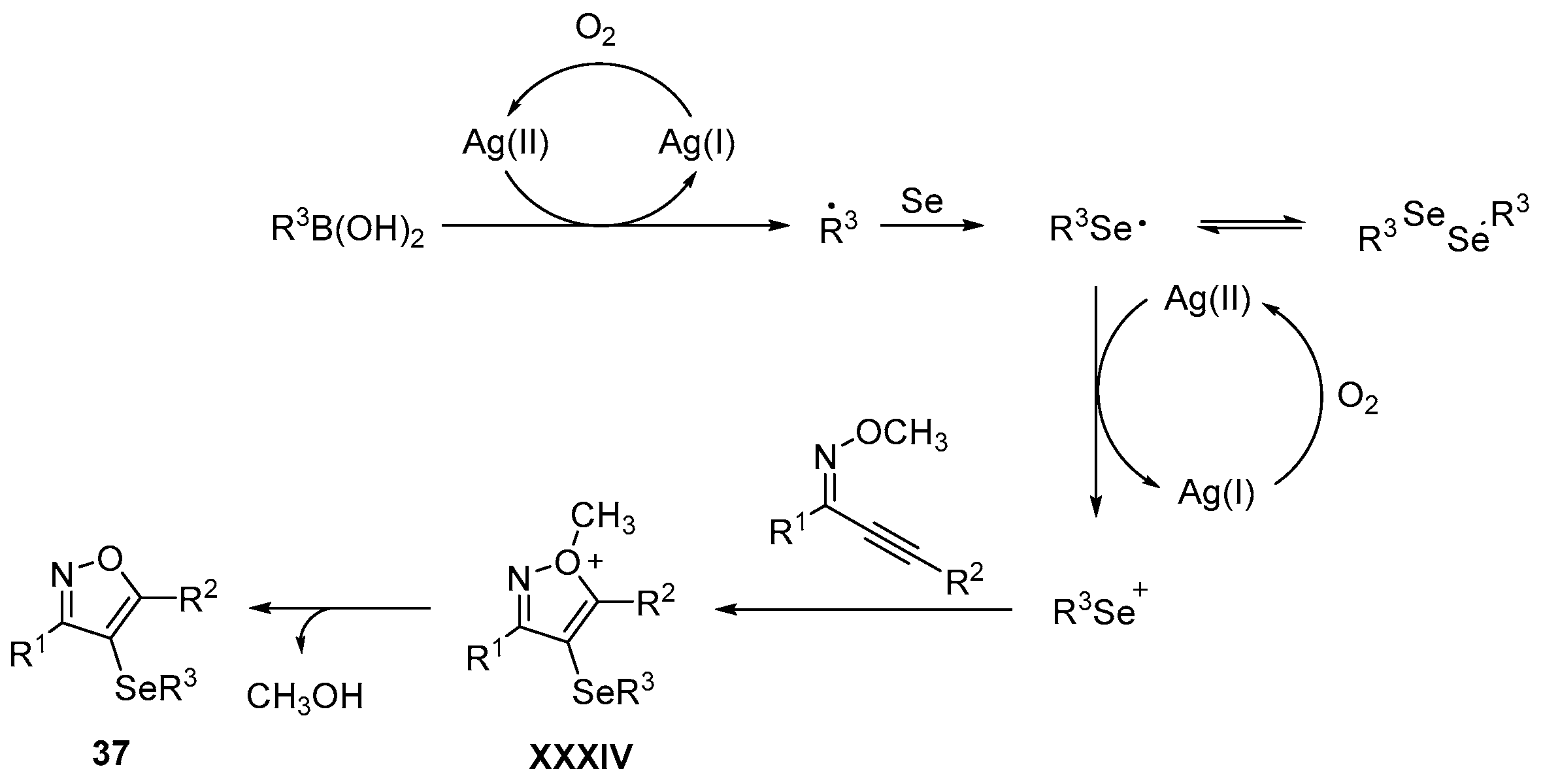
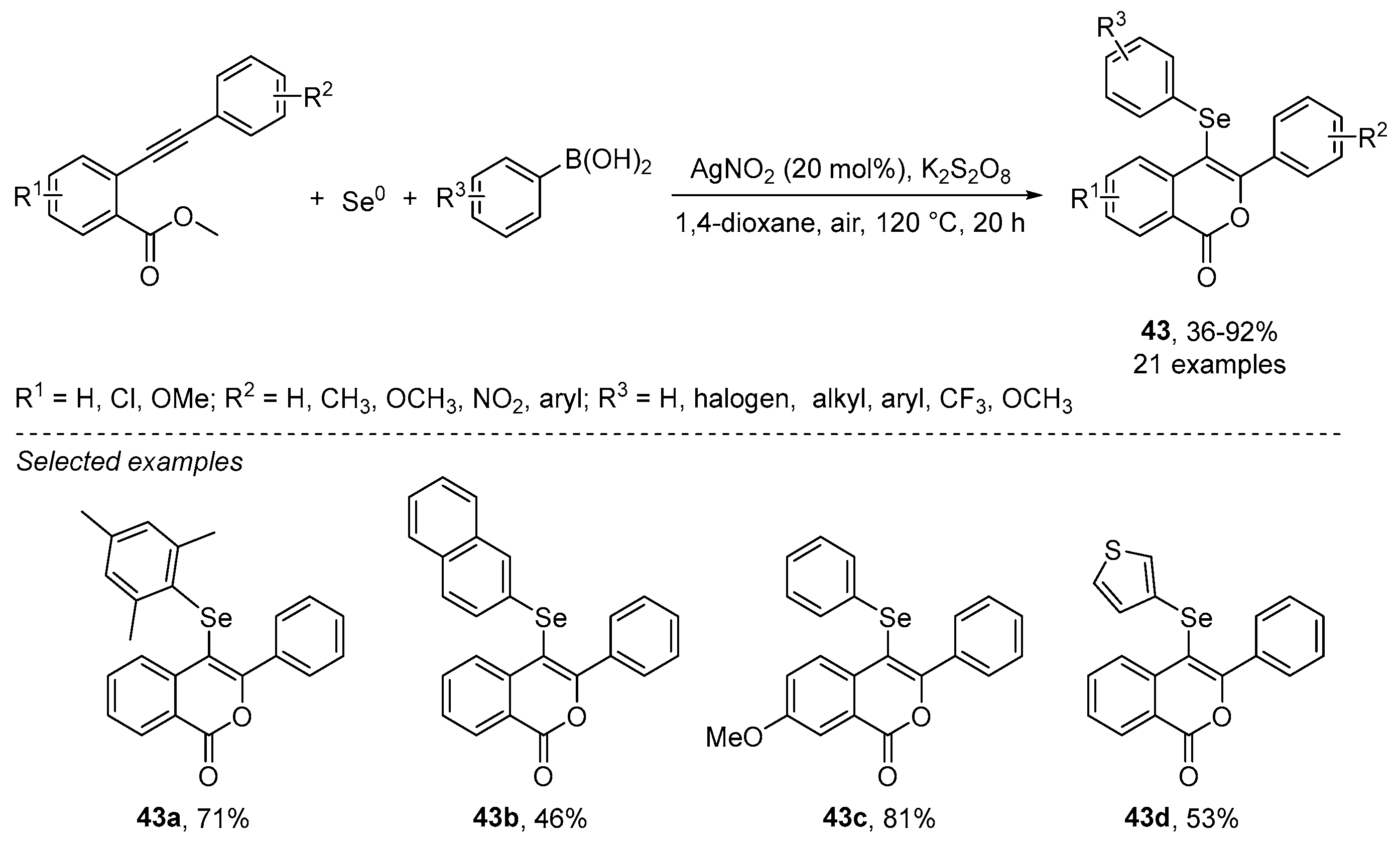
- Wu, J.; Yang, Y.-F.; Huang, X.-B.; Gao, W.-X.; Zhou, Y.-B.; Liu, M.-C.; Wu, H.-Y. Three-Component Reactions of Alkynone o-Methyloximes, Element Selenium, and Boronic Acids Leading to 4-Organoselenylisoxazoles. ACS Omega 2020, 5, 23358–23363. [Google Scholar] [CrossRef]
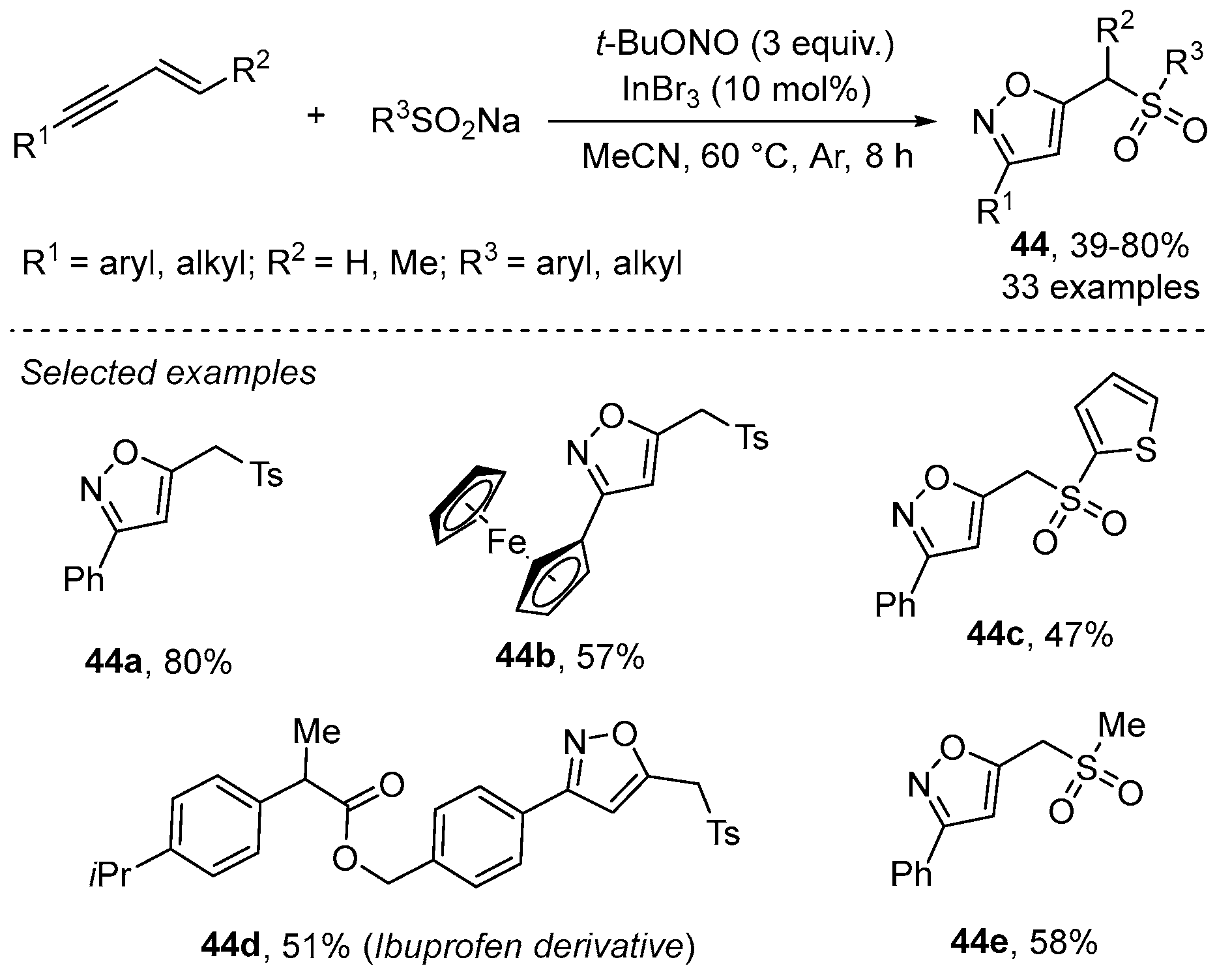
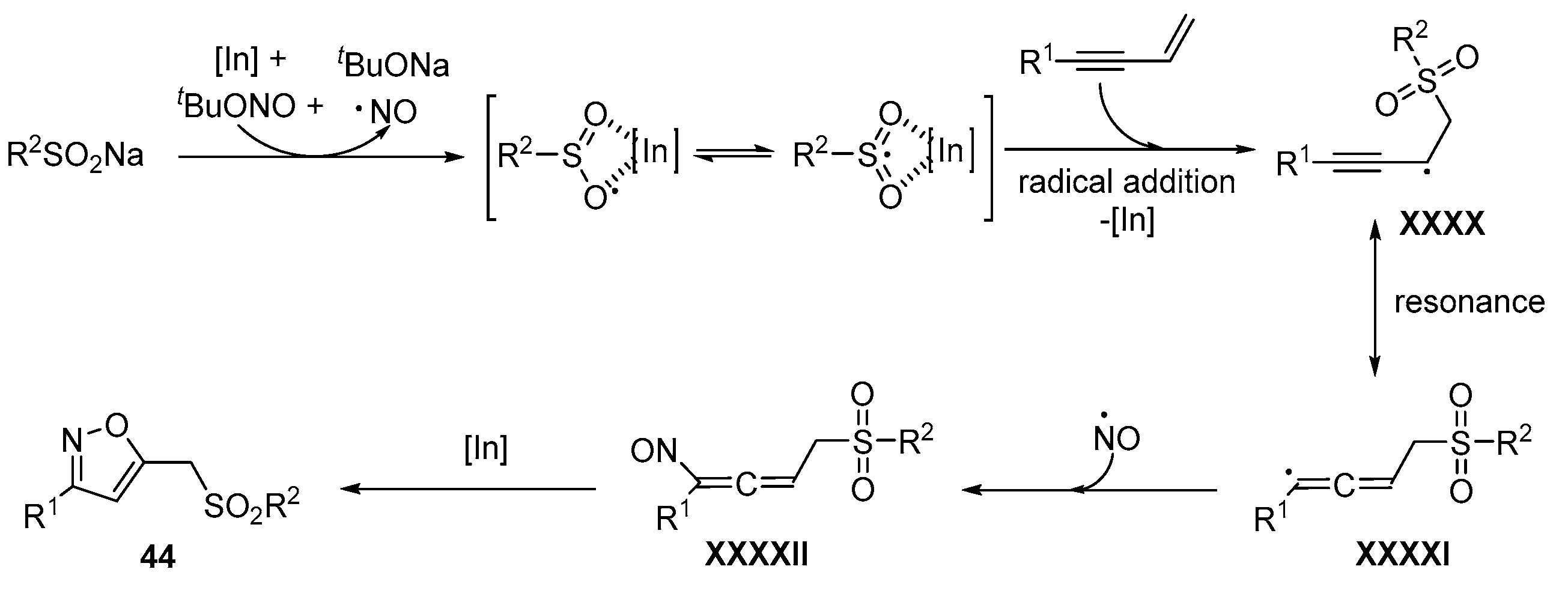
- Yue, X.; Hu, M.; He, X.; Wu, S.; Li, J.-H. A radical-mediated 1,3,4-trifunctionalization cascade of 1,3-enynes with sulfinates and tertbutyl nitrite: Facile access to sulfonyl isoxazoles. Chem. Commun. 2020, 56, 6253–6256. [Google Scholar] [CrossRef]
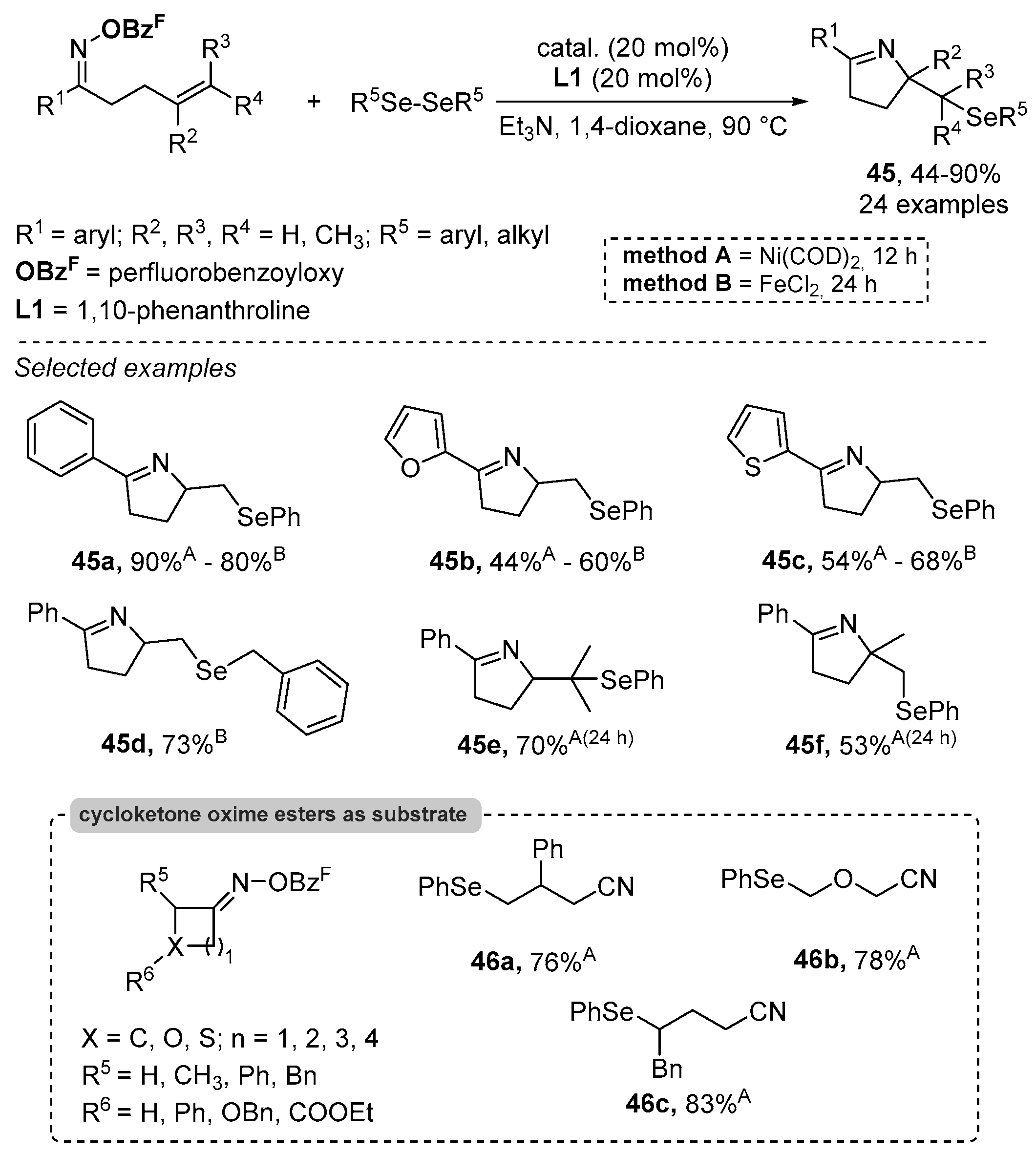
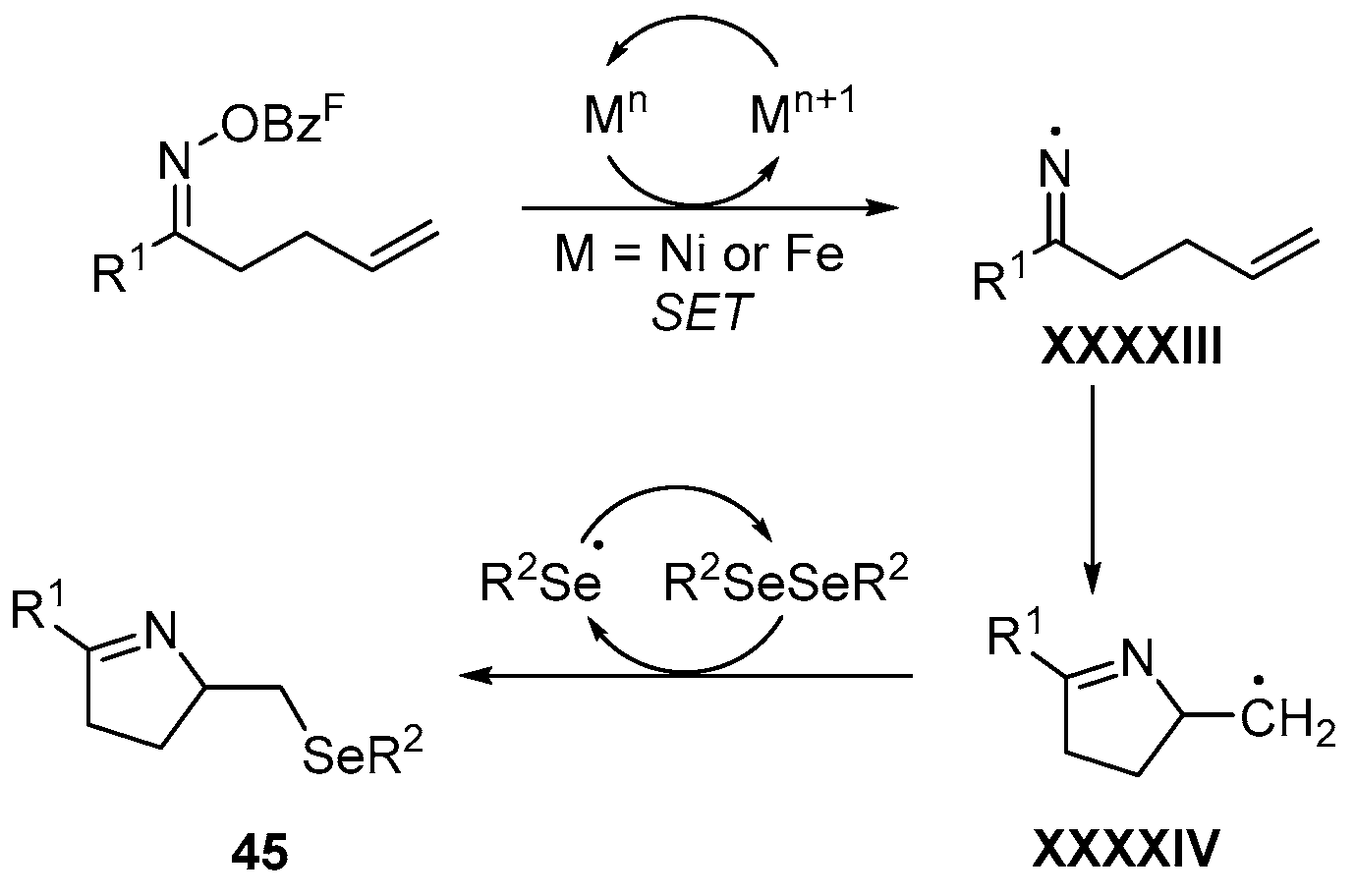
- Liu, L.; Jian, Y.; Hu, W.; Zhao, S.; Shi, Z.-J.; Selander, N.; Zhou, T. Ni and Fe Catalyzed cascade radical reactions of oxime esters with diselenides. Org. Chem. Front. 2022, 9, 3480–3485. [Google Scholar] [CrossRef]




























































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutterres, E.L.; Anjos, T.; Santos, F.B.; Bandeira, P.T.; Penteado, F.; Schumacher, R.F. Recent Approaches in Transition Metal-Catalyzed Chalcogenative Heteroannulation of Alkenes and Alkynes. Catalysts 2023, 13, 1300. https://doi.org/10.3390/catal13091300
Gutterres EL, Anjos T, Santos FB, Bandeira PT, Penteado F, Schumacher RF. Recent Approaches in Transition Metal-Catalyzed Chalcogenative Heteroannulation of Alkenes and Alkynes. Catalysts. 2023; 13(9):1300. https://doi.org/10.3390/catal13091300
Chicago/Turabian StyleGutterres, Elba L., Thiago Anjos, Felipe B. Santos, Pamela T. Bandeira, Filipe Penteado, and Ricardo F. Schumacher. 2023. "Recent Approaches in Transition Metal-Catalyzed Chalcogenative Heteroannulation of Alkenes and Alkynes" Catalysts 13, no. 9: 1300. https://doi.org/10.3390/catal13091300