
Enzymatic Desymmetrisation of Prochiral Phosphines and Phosphine P-Sulfides as a Route to P-Chiral Catalysts


Division of Organic Chemistry, Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Łódź, Poland
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(2), 171; https://doi.org/10.3390/catal12020171
Submission received: 20 December 2021
/
Revised: 18 January 2022
/
Accepted: 26 January 2022
/
Published: 28 January 2022
(This article belongs to the Special Issue 10th Anniversary of Catalysts: Biocatalysis in Analysis and Synthesis—Past, Present and Future)
Abstract
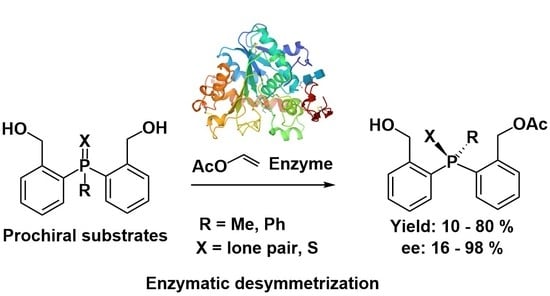
:The enzyme-catalyzed monoacetylation of prochiral bis (2-hydroxymethylphenyl)methylphosphine and bis (2-hydroxymethylphenyl)phenylphosphine and their P-sulfides gave, in one single step, as a result of desymmetrisation, the corresponding monoacetates in moderate yields and with an enantiomeric excess of 16 to 98%, depending on the substrate structure and enzyme applied. The absolute configurations of the selected products were determined by a chemical correlation. This led to the conclusion that, in the case of phosphines, phosphine oxides and phosphine sulfides enzymes preferentially produce compounds of the same spatial arrangement. The new compounds obtained will be transformed into chiral catalysts/ligands.
1. Introduction
Since the pioneering times of the mid-1970s, when the first practical and generally applicable methods in asymmetric synthesis [1] were developed, there has been a tremendous growth in this research field.
One of the fundamental research goals in modern chemistry is the development of efficient and selective procedures to access organic compounds. Among all the methodologies developed to date, catalysis offers an efficient and economical approach to enantiomerically pure substances. In particular, organocatalysis, transition metal catalysis and enzymes, and biotechnology methods are in high demand due to their application in stereoselective carbon–hydrogen, carbon–carbon or carbon–heteroatom bond formations.
Among the commonly used catalysts, there is a substantial number of hetero-organic compounds, especially organic phosphorus and sulfur compounds. Concerning the former ones, there are mainly phosphines and phosphine oxides [2], whose synthesis requires the application of various methodologies. Among them, the desymmetrisation of prochiral phosphorus substrates seems particularly interesting. Such transformations were performed mainly in a chemical manner, using chiral catalysts (for some recent results see [3,4]). There are only a few examples of the use of enzymes as catalysts, such as desymmetrisation [5,6,7,8], three of which came from our laboratory [5,7,8] (vide infra).
The chiral sulfur derivatives that were applied in asymmetric catalysis are variously functionalized sulfinyl compounds, sulfoximines and others (for a recent overview on the use of S-chiral sulfur ligands/catalysts in asymmetric synthesis, see [9]).
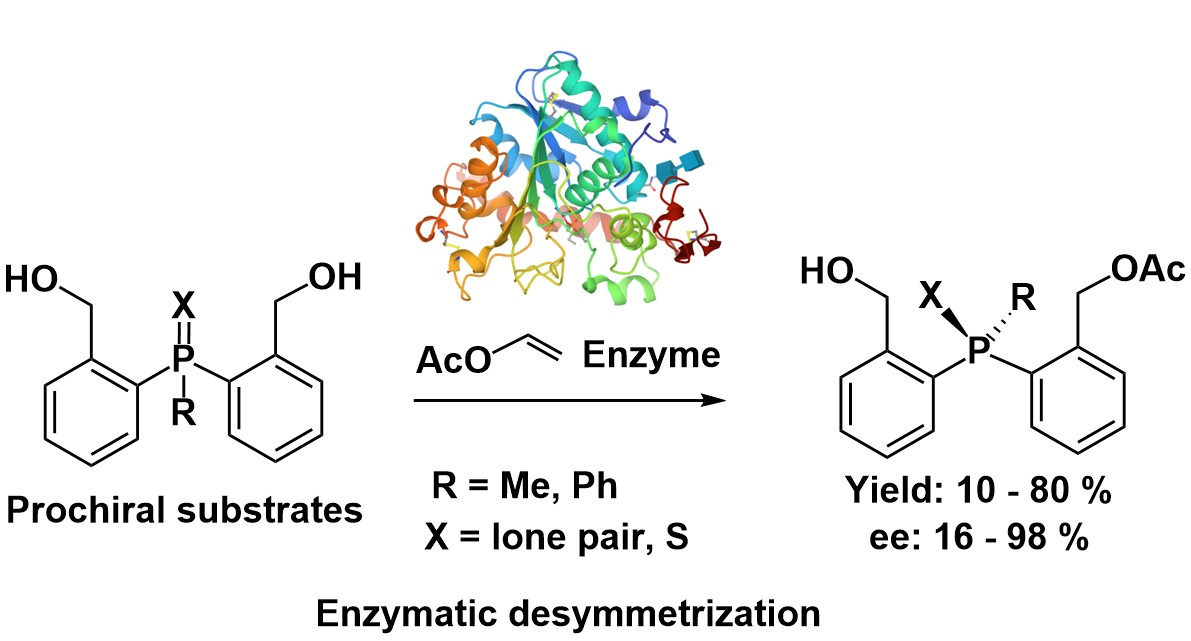
Searching for new chiral catalysts, we synthesized some time ago a series of organosulfur chiral compounds 3, containing a stereogenic sulfinyl moiety, an enantiomeric amine fragment and the hydroxyl group (Scheme 1) [10,11]. They proved to be excellent catalysts in a variety of reactions of asymmetric synthesis [9,12].
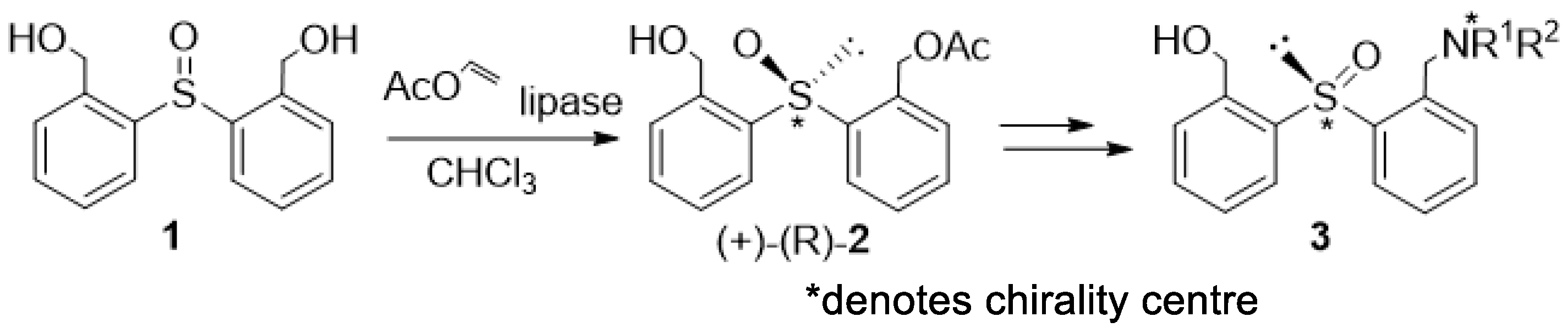
Following these positive results, we decided to synthesize catalysts in which the stereogenic sulfinyl moiety was replaced by a stereogenic phosphorus-containing group. In the first approach, attempts were made at the synthesis of the phosphinyl (P = O) analogues. We synthesized the enantiomerically pure (2-acetoxymethylphenyl)(2′-hydroxy-methylphenyl)phosphine oxide 5, which was planned to be a precursor of the desired ligands 6 (Scheme 2).
The initial steps involving the enzymatic desymmetrisation of 4 (Scheme 2) were fully successful [8]. In all cases, the catalyst precursor 5 was obtained in high yields and with ees up to 99.5%. Unexpectedly, attempts to replace the hydroxyl group with an enantiomerically pure chiral amine moiety via the mesylation of enantiomerically pure 5 always led to mixtures of diastereomers (which was not the case for the corresponding sulfinyl analogs 2), which means that racemization must have taken place at one of the stages of the synthesis. The partial racemization of enantiomeric precursor 5 ultimately led to the formation of inseparable diastereomeric mixtures of the desired ligands 6 [13]. Moreover, the latter proved to be totally ineffective as catalysts for the several reactions of asymmetric synthesis [14].
2. Results and Discussion
2.1. Synthesis of Bis (2-hydroxymethylphenyl)phosphines 12 and 13
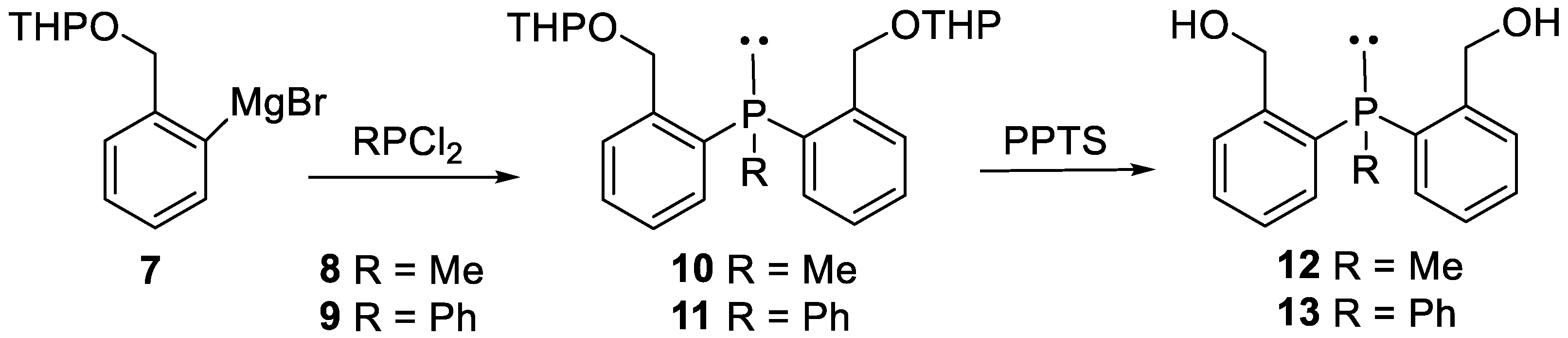
The syntheses of starting phosphines 12 and 13 were based on the reactions of o-tetrahydropyranyloxymethylphenylmagnesium bromide (Mg-THPBB) 7 with the appropriate dichlorophosphines 8 and 9. Thus, the synthesis of bis (2-hydroxymethylphenyl)methylphosphine 12 was achieved by the reaction of dichloro (methyl) phosphine 8 with Mg-THPBB 7 to give 10, which was then treated with pyridinium p-toluenesulfonate (PPTS) to remove the THP protecting group. In turn, the synthesis of bis (2-hydroxymethylphenyl)phenylphosphine 13 was accomplished in a similar way, starting from dichloro (phenyl) phosphine and treatment of the resulting 11 with PPTS [8] (Scheme 3).
2.2. Desymmetrisation of Bis (2-hydroxymethylphenyl)phosphines 12 and 13
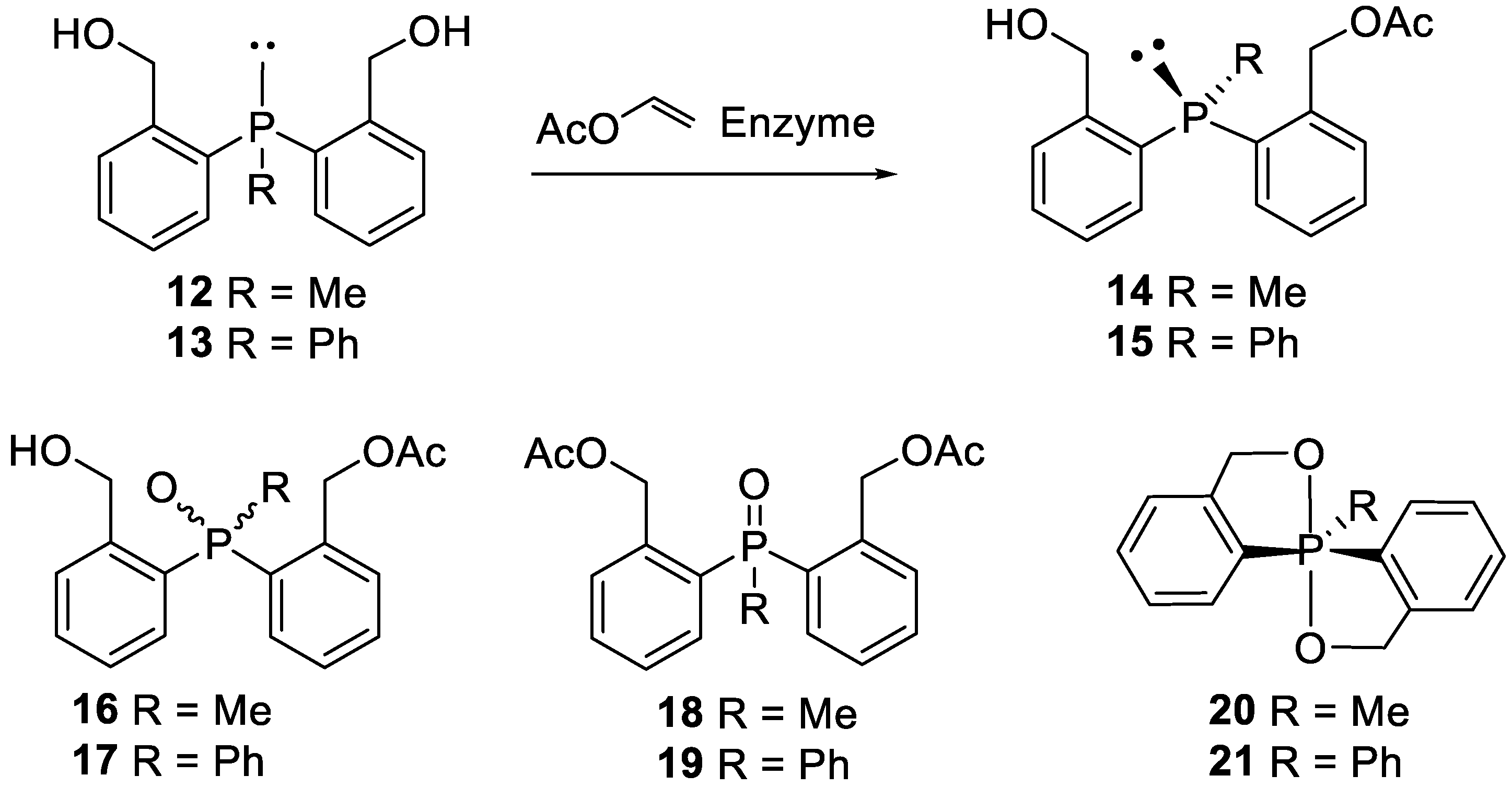
Due to the fact that in our laboratory we have successfully used enzymes for the synthesis of non-racemic hetero-organic compounds, particularly those containing stereogenic phosphorus and sulfur atoms, we decided to use also in this case such a procedure for desymmetrisation of the appropriate substrates. Thus, prochiral phosphines 12 and 13 were subjected to asymmetric acetylation with an excess of vinyl acetate in various solvents at 30 °C, using selected enzymes as biocatalysts (Scheme 4).
The reactions were monitored by 31P NMR. After completion, the enzymes were filtered off, the solvent and excess of vinyl acetate were evaporated to leave the residue, which, in addition to the desired enantiomerically enriched monoacetates 14 and 15, contained some byproducts. These were the corresponding oxidized derivatives 16 and 17, diacetates 18 and 19 and phosphoranes 20 and 21, whose formation proved inevitable in spite of all the undertaken protection from air (as shown in Scheme 4)
The residue was separated via column chromatography and the results are collected in Table 1. The inspection of Table 1 clearly shows that the products exhibiting the highest enantiomeric excess were obtained using CAL-B as the biocatalyst and toluene as the solvent in acetylation of diol 12 and CAL-B as the biocatalyst and t-butyl methyl ether as the solvent in acetylation of diol 13. The relatively low yield of the desired monoacetates may be due to the formation of byproducts (vide supra). Some attempts to improve the results, using increased amounts of enzymes, did not lead to the expected better outcome.
The comparison of the above results with those obtained in our previous paper devoted to the desymmetrisation of the P = O analogs (vide supra and Scheme 2) [8] clearly shows that the replacement of the phosphinyl (P = O) moiety by the phosphino (P-:) one results in a lower stereoselectivity of the reaction and lower yields of the products.
2.3. Desymmetrisation of Bis (2-hydroxymethylphenyl)phosphine Sulfides
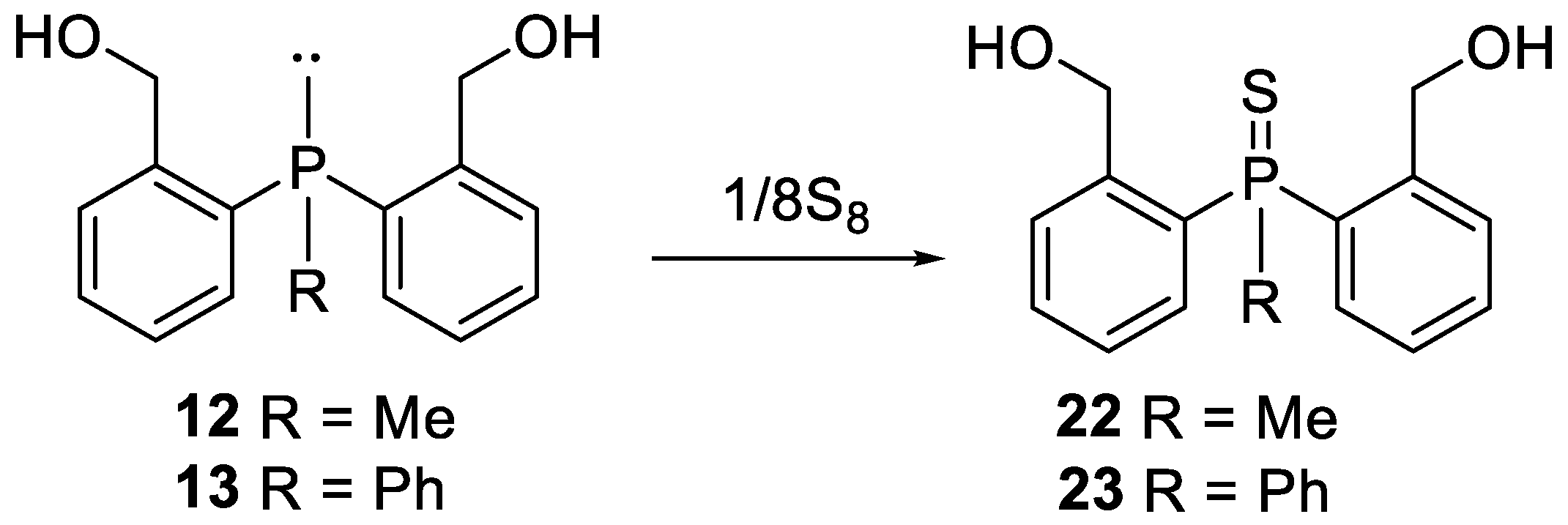
Because the results obtained during desymmetrisation of bis (2-hydroxymethyl-phenyl)phosphines 12 and 13 were acceptable, but not very satisfactory and, moreover, some byproducts arising from oxidation phosphines were formed, we decided to synthesize appropriate phosphine sulfides 22 and 23. Prochiral bis (2-hydroxymethylphenyl)phosphines 12 and 13 were treated with elemental sulfur to give the appropriate phosphine sulfides 22 and 23 (Scheme 5).
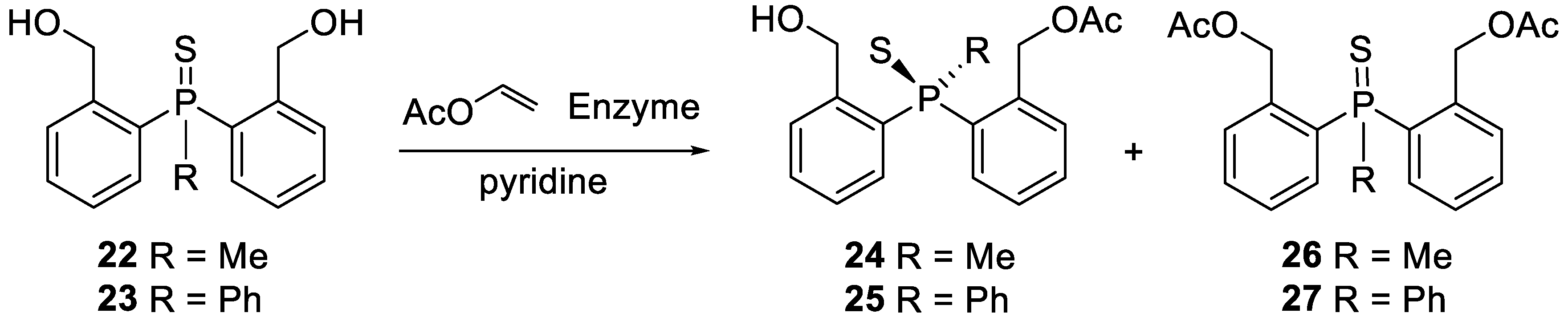
The latter were subjected to asymmetric acetylation with an excess of vinyl acetate in various solvents at 30 °C, using a number of lipases (Scheme 6).
The reactions were monitored by 31P NMR. After completion, the enzymes were filtered off, the solvent and excess of vinyl acetate were evaporated, and the residue was separated via column chromatography. Interestingly, also in this case, the formation of achiral diacetates 26 and 27 was observed, which, in some cases, may be responsible for a decreased yield of monoacetates. The results are collected in Table 2.
The inspection of Table 2 clearly shows that the reaction time, yields and enantioselectivity strongly depended on the lipase and the solvent used. Monoacetate 24 was obtained in good yield, but with moderate enantioselectivity. In turn, monoacetate 25 was formed in good yield and enantioselectivity. The best results were obtained using PFL as the biocatalyst and t-butyl methyl ether as the solvent. Again, attempts to use increased amounts of enzymes did not lead to higher outcomes. Nevertheless, it should be stressed that in both cases presented above, the stereoselective enzymatic transformation was achieved using the substrates in which the prostereogenic phosphorus atom and the reacting hydroxy oxygen were distant from each other by four bonds.
2.4. Determination of the Absolute Configuration of Monoacetates 14 and 24
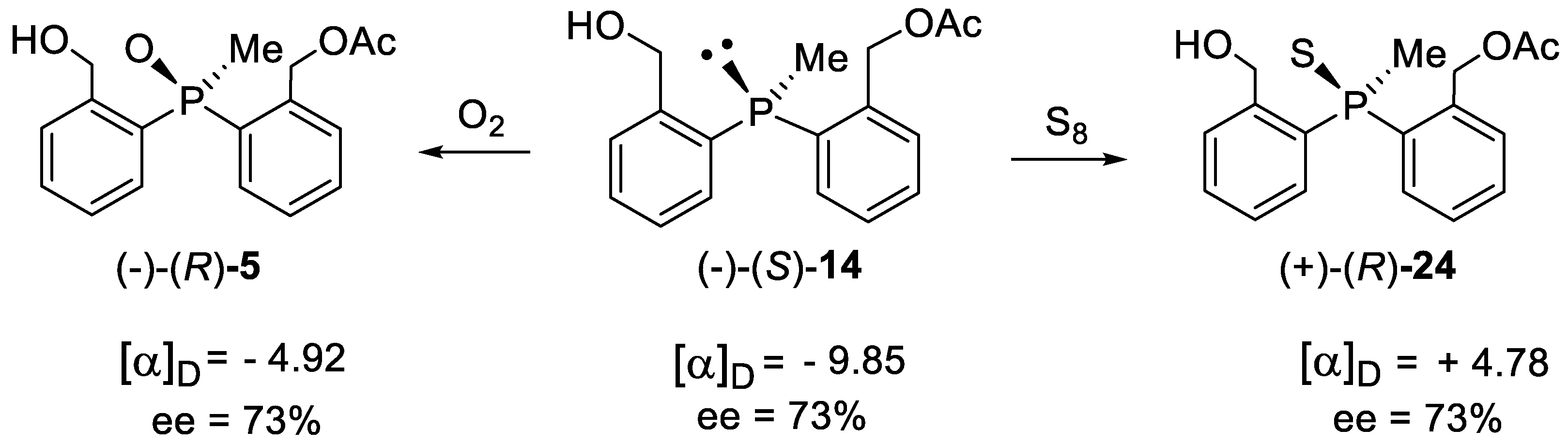
The absolute configuration of the newly obtained enantiomerically enriched monoacetates was determined as follows: phosphine 14 (Table 1, entry 4) was oxidized with air to give phosphine oxide (-)-(R)-5, whose absolute configuration and optical rotation are known from our previous work [8]. Since oxidation with air proceeds with the retention of the configuration at phosphorus [15], (S) configuration was ascribed to (-)-phosphine 14 obtained. In turn, the reaction of phosphine 14 with elemental sulfur, proceeding also with retention of configuration at phosphorus [15], gave phosphine sulfide 24. Hence, the absolute configuration of the latter is (+)-(R) (Scheme 7), that is, the same as the one obtained in the enzymatic desymmetrisation.
Unfortunately, no such correlation could be made for phosphine monoacetate 15 and phosphine sulfide acetate 25 due to the lack of the corresponding phosphine oxides of known absolute configuration. Moreover, all the chiral products were oils, which excluded X-ray analysis.
3. Materials and Methods
3.1. General Information
The synthesized products were purified by column chromatography on Merck 60 silica gel (0.063–0.200 mm) or preparative plate chromatography using Merck 60 F254 silica gel plate (2.5 mm). TLC was performed on Merck 60 F254 silica gel plate (0.25 mm). Solvents were dried using general procedures and distilled prior to use. The NMR spectra were recorded in CDCl3 with a Bruker AV 200 spectrometer. The chemical shifts (δ) are expressed in ppm, the coupling constants (J) are given in Hz. Mass spectra were recorded with a Finnigan MAT95 Voyager Elite spectrometer or Synapt G2-Si mass spectrometer (Waters) equipped with an ESI source and quadrupole-time-of-Flight mass analyser. Optical rotations were measured on a Perkin-Elmer 241 MC polarimeter. HPLC analysis was made using Varian Pro Star 210 instrument using column with chiral filling Chiralcel OD or Chiralpak AS. The enzymes were purchased from AMANO or SIGMA. Enzymes: CAL-B (Novozym 435)–lipase acrylic resin from Candida antarctica (E.C. 3.1.1.3), SIGMA-ALDRICH; PFL–lipase from Pseudomonas fluorescens (E.C. 3.1.1.3), SIGMA-ALDRICH; PS–lipase from Pseudomonas species (E.C. 3.1.1.3), AMANO, CR–lipase from Candida rugosa (E.C. 3.1.1.3), SIGMA-ALDRICH; LPL–Lipoprotein lipase (E.C. 3.1.1.34), SIGMA-ALDRICH. All the NMR spectra are collected in the “Supplementary Materials”.
3.2. Synthesis of Bis (2-hydroxymethylphenyl)phosphines 12 and 13
3.2.1. Synthesis of Bis [2-(2′-tetrahydropyranyloxy)methylphenyl]phosphine 10 and 11
To magnesium (0.264 g, 0.01 mol) under nitrogen was added a solution of 2′-(2-tetrahydropyranyloxymethyl)bromobenzene 7, obtained according to the known procedure [8] (3 g, 0.01 mol) in THF (8 mL) followed by a small crystal of iodine. The mixture was gently heated to initiate the Grignard reagent formation. After the magnesium completely dissolved, appropriate dichlorophosphine 8 or 9 (0.649 g for 8 or 0.985 g for 9, 0.0055 mol) dissolved in THF (5 mL) was added and the solution was stirred for 3 h. THF was evaporated, saturated aqueous solution of NH4Cl (10 mL) was added to the residue and the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anhydrous MgSO4 and the solvent was removed to give crude phosphine 10 or 11.
- Bis [2-(2′-tetrahydropyranyloxy)methylphenyl]methylphosphine 10
- Crude yield: 1.970 g, 90%
- 31P NMR (CDCl3): δ = −48.3
- Bis [2-(2′-tetrahydropyranyloxy)methylphenyl]phenylphosphine 11
- Crude yield: 2.535 g, 94%
- 31P NMR (CDCl3): δ = −25.7
3.2.2. Synthesis of Bis (2-hydroxymethylphenyl)phosphines 12 and 13
To a solution of crude phosphine 10 or 11 (0.0054 mol) in EtOH (40 mL) pyridinium p-toluenesulfonate (PPTS) (0.2 eq., 0.27 g, 0.0011 mol) was added and the mixture was stirred at 55 ºC for 7 h. EtOH was evaporated, to the residue saturated aqueous solution of NaHCO3 (10 mL) was added and the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anhydrous MgSO4. After the evaporation of the solvent, the crude products were purified by column chromatography using dichloromethane-acetone in a gradient from 5:1 to 1:1 as eluent to give products 12 and 13, respectively.
- Bis (2-hydroxymethylphenyl)methylphosphine 12
- Oil, isolated yield: 0.439 g, 25%
- 31P NMR (CDCl3): δ = −50.8;
- 1H NMR (CDCl3): δ = 1.59 (d, J = 3.8 Hz, 3H), 4.67–4.90 (m, 4H), 7.27–7.39 (m, 8H);
- MS (+ESI): m/z = 261 (M + H);
- HRMS (+ESI): m/z = 261.1049, calcd for C15H18PO2 (M + H), 261.1044.
- Bis (2-hydroxymethylphenyl)phenylphosphine 13
- Oil, isolated yield: 0.098 g, 56%
- 31P NMR (CDCl3): δ = −27.5;
- 1H NMR (CDCl3): δ = 4.75–5.04 (m, 4H), 6.94–7.52 (m, 13H);
- MS (+ESI): m/z = 323 (M + H);
- HRMS (+ESI): m/z = 323.1208, calcd for C20H20PO2 (M + H), 323.1201.
3.3. Synthesis of Bis (2-hydroxymethylphenyl)phosphine Sulfides 22 and 23
To obtain bis(2-hydroxymethylphenyl)phosphine sulfides 22 and 23, to the solution of phosphine 12 and 13 (0.180 g, 0.692 mmol for 12 and 0.134 g, 0.416 mmol for 13) in dichloromethane (20 mL) under nitrogen elemental sulfur (1 eq., 0.022 g, 0.692 mmol for 12 and 0.014 g, 0.416 mmol for 13) was added. The mixture was refluxed until the substrate disappeared, which was found by 31P NMR. Then, the reaction mixture was filtered through celite and the solvent was evaporated. The crude reaction mixture was purified by column chromatography using dichloromethane-acetone 6:1 with the addition of triethylamine (0.03% vol.) to give pure 22 and 23.
- Bis (2-hydroxymethylphenyl)methylphosphine sulfide 22
- Yellowish solid, m. p. 92–94 °C, isolated yield: 0.124 g, 61%
- 31P NMR (CDCl3): δ = 34.2;
- 1H NMR (CDCl3): δ = 2.34 (d, J = 13.2 Hz, 3H), 3.71 (br. s, 1H), 4.39–4.75 (m, 4H), 7.11–7.72 (m, 8H);
- 13C NMR (CDCl3): δ = 23.68 (d, JP-Me = 60.5 Hz), 62.93 (d, JP-CH2OH = 6.0 Hz), 128.15, 131.03, 131.12, 131.73, 132.22, 132.30, 132.36, 132.74, 143.97, 144.04 (Aryl);
- MS (+ESI): m/z = 293 (M + H);
- HRMS (+ESI): m/z = 293.0771, calcd for C15H18PO2S (M + H), 293.0765.
- Bis (2-hydroxymethylphenyl)phenylphosphine sulfide 23
- Yellowish solid, m. p. 132–134 °C, isolated yield: 0.111 g, 76%
- 31P NMR (CDCl3): δ = 40.7;
- 1H NMR (CDCl3): δ = 3.85 (br. s, 1H), 4.67–4.82 (m, 4H), 5.29–5.49 (m, 2H), 6.78–7.72 (m, 13H);
- MS (CI): m/z = 355 (M + H);
- HRMS (CI): m/z = 354.0845, calcd for C20H19PO2S (M + H), 354.0843.
3.4. General Procedure for the Enzymatic Desymmetrization of Bis (2-hydroxymethylphenyl)phosphines 12 and 13 and Bis (2-hydroxymethylphenyl)phosphine Sulfides 22 and 23
To a solution of the prochiral phosphine diol (12 or 13, 0.1 mmol) or prochiral P-sulfide diol (22 or 23, 0.1 mmol) in a solvent (5 mL) pyridine (3 eq., 0.024 mL, 0.3 mmol), vinyl acetate (0.5 mL) and an enzyme (20 mg) were added. In the case of phosphines 12 and 13, the reaction was carried out under a nitrogen atmosphere. The whole mixture was stirred at room temperature. The conversion degree was determined by 31P NMR. Then, the enzyme was filtered off and the solvents were evaporated. The crude reaction mixture was separated by column chromatography using dichloromethane-acetone in gradient from 100:1 to 1:1 with the addition of triethylamine (0.03% vol.) as eluent, to give pure enantiomerically enriched phosphine monoacetates 14 and 15 and P-sulfide monoacetates 24 and 25. The results are collected in Table 1 for phosphines 12 and 13 and in Table 2 for sulfides 22 and 23.
- (2-acetoxymethylphenyl)(2′-hydroxymethylphenyl)methylphosphine 14
- 31P NMR (CDCl3): δ = −48.4;
- 1H NMR (CDCl3): δ = 1.59 (d, J = 3.6 Hz, 3H), 1.86 (s, 3H), 4.69–4.89 (m, 4H), 7.19–7.48 (m, 8H);
- MS (CI): m/z = 303 (M + H);
- HRMS (+ESI): m/z = 303.1158, calcd for C17H20PO3 (M + H), 303.1150.
- (2-acetoxymethylphenyl)(2′-hydroxymethylphenyl)phenylphosphine 15
- 31P NMR (CDCl3): δ = −26.3;
- 1H NMR (CDCl3): δ = 1.62 (s, 3H), 4.71–4.82 (m, 2H), 5.29–5.49 (m, 2H), 6.81–7.44 (m, 13H);
- MS (FAB): m/z = 365 (M + H);
- HRMS (FAB): m/z = 365.1317, calcd for C22H22PO3 (M + H), 365.1306.
- (2-acetoxymethylphenyl)(2′-hydroxymethylphenyl)methylphosphine sulfide 24
- 31P NMR (CDCl3): δ = 33.7;
- 1H NMR (CDCl3): δ = 1.89 (s, 3H), 2.38 (d, J = 13.2 Hz, 3H), 4.31–4.75 (m, 2H), 4.94–5.09 (m, 2H), 7.36–8.15 (m, 8H);
- MS (+ESI): m/z = 335 (M + H);
- HRMS (+ESI): m/z = 335.0876, calcd for C17H20PO3S (M + H), 335.0871.
- (2-acetoxymethylphenyl)(2′-hydroxymethylphenyl)phenylphosphine sulfide 25
- 31P NMR (CDCl3): δ = 41.2;
- 1H NMR (CDCl3): δ = 1.84 (s, 3H), 4.01 (br. s, 1H), 4.60–4.82 (m, 2H), 5.38–5.55 (m, 2H), 6.89–7.73 (m, 13H);
- MS (CI): m/z = 397 (M + H);
- HRMS (+ESI): m/z = 397.1023, calcd for C22H22PO3S (M + H), 397.1027.
4. Conclusions
Prochiral bis (2-hydroxymethylphenyl) phosphines and their sulfides could be successfully transformed into enantiomerically enriched monoacetyl derivatives by a desymmetrisation procedure using an enzymatic acetylation reaction. The use of enzymes proved to be useful in obtaining the desired products with high stereoselectivity in one step. The determination of their absolute configuration proved that, in the case of phosphines, phosphine oxides and phosphine sulfides enzymes preferentially produce compounds of the same spatial arrangement. The new compounds obtained will be transformed into chiral catalysts/ligands. The appropriate investigations are underway.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12020171/s1: NMR spectra of all the new compounds.
Author Contributions
Conceptualization, M.K. and P.K.; methodology, M.K.; investigation, L.M.; writing—original draft preparation, L.M.; writing—review and editing, M.K.; supervision, P.K.; funding acquisition, L.M. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support by the National Research Centre (NCN), Poland, grant PRELUDIUM: UMO-2014/13/N/ST5/03481 for L. M., is gratefully acknowledged.
Data Availability Statement
Some or all data, models, or code that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Enders, D.; Hoffmann, R.W. Asymmetrische Synthese. Chem. Unserer Zeit 1985, 19, 177–199. [Google Scholar] [CrossRef]
- Börner, A. (Ed.) Phosphorus Ligands in Asymmetric Catalysis; Wiley-VCH: Karlsruhe, Germany, 2008. [Google Scholar]
- Gammon, J.J.; Viktoria, H.; Gessner, V.H.; Barker, G.R.; Granander, J.; Whitwood, A.C.; Strohmann, C.; O’Brien, P.; Brian, K.B. Synthesis of P-Stereogenic Compounds via Kinetic Deprotonation and Dynamic Thermodynamic Resolution of Phosphine Sulfides: Opposite Sense of Induction Using (-)-Sparteine. J. Am. Chem. Soc. 2010, 132, 13922–13927. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.-H.; Zhou, Q.-Y.; Yang, C.; Chen, L.; Cheng, J.-P.; Li, X. Access to P-stereogenic compounds via desymmetrizing enantioselective bromination. Chem. Sci. 2021, 12, 4582. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasiński, P.; Żurawiński, R.; Albrycht, M.; Mikołajczyk, M. The first enzymatic desymmetrizations of prochiral phosphine oxides. Tetrahedron Asymmetry 2003, 14, 3379–3384. [Google Scholar] [CrossRef]
- Wiktelius, D.; Johansson, M.J.; Luthmann, K.; Kann, N. A Biocatalytic Route to P-Chirogenic Compounds by Lipase-Catalyzed Desymmetrization of a Prochiral Phosphine−Borane. Org. Lett. 2005, 7, 4991–4994. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasiński, P.; Rachwalski, M.; Kwiatkowska, M.; Mikołajczyk, M.; Wieczorek, W.M.; Szyrej, M.; Sieroń, L.; Rutjes, F.P.J.T. Enzyme-promoted desymmetrisation of prochiral bis(cyanomethyl)phenylphosphine oxide. Tetrahedron Asymmetry 2007, 18, 2108–2112. [Google Scholar] [CrossRef]
- Kaczmarczyk, S.; Kwiatkowska, M.; Madalińska, L.; Barbachowska, A.; Rachwalski, M.; Błaszczyk, J.; Sieroń, L.; Kiełbasiński, P. Enzymatic Synthesis of Enantiopure Precursors of Chiral Bidentate and Tridentate Phosphorus Catalysts. Adv. Synth. Catal. 2011, 353, 2446–2454. [Google Scholar] [CrossRef]
- Otocka, S.; Kwiatkowska, M.; Madalińska, L.; Kiełbasiński, P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef] [PubMed]
- Rachwalski, M.; Kwiatkowska, M.; Drabowicz, J.; Kłos, M.; Wieczorek, W.M.; Szyrej, M.; Sieroń, L.; Kiełbasiński, P. Enzyme-promoted Desymmetrization of Bis(2-hydroxymethylphenyl)sulfoxide as a Route to Tridentate Chiral Catalysts. Tetrahedron Asymmetry 2008, 19, 2096–2101. [Google Scholar] [CrossRef]
- Leśniak, S.; Rachwalski, M.; Sznajder, E.; Kiełbasiński, P. New Highly Efficient Aziridine-functionalized Tridentate Sulfinyl Catalysts for Enantioselective Diethylzinc Addition to Carbonyl Compounds. Tetrahedron Asymmetry 2009, 20, 2311–2314. [Google Scholar] [CrossRef]
- Kiełbasiński, P.; Kwiatkowska, M.; Cierpiał, T.; Rachwalski, M.; Leśniak, S. The Sulfinyl Group: Its Importance for Asymmetric Synthesis and Biological Activity. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 649–653. [Google Scholar] [CrossRef]
- Kaczmarczyk, S.; Madalińska, L.; Kiełbasiński, P. Unexpected Racemization of 2-Hydroxymethylphenylphosphine Oxides. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 249–253. [Google Scholar] [CrossRef]
- Madalińska, L.; Kwiatkowska, M.; Kaczmarczyk, S.; Kiełbasiński, P.; Division of Organic Chemistry, Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza, Poland. 2022; (unpublished results).
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry 2020, 12, 108. [Google Scholar] [CrossRef] [Green Version]
Scheme 1.
Synthesis of chiral tridentate sulfinyl catalysts.

Scheme 2.
Synthesis of chiral phosphine oxides as substrates for potential tridentate ligands.

Scheme 3.
Synthesis of bis (2-hydroxymethylphenyl)phosphines 12 and 13.

Scheme 4.
Desymmetrisation of bis (2-hydroxiymethylphenyl)phosphines 12 and 13.

Scheme 5.
Synthesis of bis (2-hydroxymethylphenyl)phosphine sulfides 22 and 23.

Scheme 6.
Desymmetrisation of bis (2-hydroxymethylphenyl)phosphine sulfides 22 and 23.

Scheme 7.
Chemical correlation of the absolute configuration of 14 and 24.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Monoacetates 14 and 15 via Desymmetrisation of bis (2-hydroxymethylphenyl)phosphines 12 and 13.
Table 1.
Monoacetates 14 and 15 via Desymmetrisation of bis (2-hydroxymethylphenyl)phosphines 12 and 13.
| Entry | Diol | R | Enzyme (a) | Solvent | Time | Monoacetate | |||
|---|---|---|---|---|---|---|---|---|---|
| [Days] | Symbol | Yield [%] | [α]D (b) | Ee | |||||
| [%] (c) | |||||||||
| 1 | 12 | Me | CAL-B | CH2Cl2 | 8 | 14 | 53 | −5.40 | 40 |
| 2 | 12 | Me | CAL-B | i-Pr2O | 1 | 14 | 52 | −7.10 | 53 |
| 3 | 12 | Me | CAL-B | TBME | 1 | 14 | 49 | −8.70 | 64 |
| 4 | 12 | Me | CAL-B | toluene | 11 | 14 | 25 | −9.85 | 73 |
| 5 | 13 | Ph | CAL-B | TBME + pyridine | 21 | 15 | 10 | 4.49 | 98 |
| 6 | 13 | Ph | CAL-B | i-Pr2O + toluene + pyridine | 23 | 15 | 10 | 4.42 | 95 |
| 7 | 13 | Ph | CAL-B | acetone | 14 | 15 | 52 | 2.74 | 60 |
| 8 | 13 | Ph | CAL-B | toluene + pyridine | 15 | 15 | 38 | 2.74 | 60 |
| 9 | 13 | Ph | CAL-B | toluene | 6 | 15 | 27 | 2.28 | 50 |
| 10 | 13 | Ph | CAL-B | acetonitrile | 22 | 15 | 26 | 1.26 | 28 |
| 11 | 13 | Ph | CAL-B | CHCl3 | 12 | 15 | 16 | −1.50 | 33 |
| 12 | 13 | Ph | CAL-B | CH2Cl2 | 29 | 15 | 5 | −1.40 | 31 |
| 13 | 13 | Ph | PFL | CHCl3 | 12 | 15 | 6 | −2.26 | 49 |
| 14 | 13 | Ph | LPL | toluene | 28 | 15 | <5 | - | - |
| 15 | 13 | Ph | CR | toluene | 33 | 15 | 11 | 2.54 | 56 |
| 16 | 13 | Ph | PS | CHCl3 | 29 | 15 | 10 | - | - |
| 17 | 13 | Ph | - | Toluene | 24 | - | - | - | - |
(a) Enzyme: CAL-B: Candida antarctica lipase B (Novozym 435); PFL: lipase from Pseudomonas fluorescens; CR: lipase from Candida rugosa; PS: Lipase PS (AMANO); LPL: Lipoprotein lipase; (b) in chloroform (c = 1); (c) the ee values were determined by chiral HPLC: OD, n-Hexane: (i-PrOH: EtOH 4:1) 98%: 2%, Fl. 0.5 mL/min.
Table 2.
Monoacetates 24 and 25 via desymmetrisation of bis (2-hydroxymethylphenyl)phosphine sulfides 22 and 23.
Table 2.
Monoacetates 24 and 25 via desymmetrisation of bis (2-hydroxymethylphenyl)phosphine sulfides 22 and 23.
| Entry | Diol | R | Enzyme (a) | Solvent + Pyridine | Time | Monoacetate | |||
|---|---|---|---|---|---|---|---|---|---|
| [Days] | Symbol | Yield | [α]D (c) | Ee | |||||
| [%] | [%] (d) | ||||||||
| 1 | 22 | Me | CAL-B | TBME (b) | 4 | 24 | 60 | 4.7 | 72 (e) |
| 2 | 22 | Me | CAL-B | toluene | 4 | 24 | 80 | 3.25 | 50 (e) |
| 3 | 22 | Me | CAL-B | acetone | 4 | 24 | 70 | 1.65 | 25 (e) |
| 4 | 22 | Me | CAL-B | CH2Cl2 | 8 | 24 | 49 | 0 | 0 |
| 5 | 22 | Me | CR | TBME | 11 | 24 | 55 | 0 | 0 |
| 6 | 22 | Me | PFL | TBME | 11 | 24 | 60 | 0 | 0 |
| 7 | 23 | Ph | PFL | TBME | 38 | 25 | 60 | −7.57 | 77 |
| 8 | 23 | Ph | PFL | Et2O | 38 | 25 | 55 | −4.44 | 45 |
| 9 | 23 | Ph | PFL | toluene | 38 | 25 | 66 | −1.56 | 16 |
| 10 | 23 | Ph | PFL | CH2Cl2 | 38 | 25 | 44 | −1.62 | 16 |
| 11 | 23 | Ph | CAL-B | Et2O | 38 | 25 | 44 | 3.25 | 54 |
| 12 | 23 | Ph | CAL-B | i-Pr2O | 38 | 25 | 37 | 1.1 | 0 |
| 13 | 23 | Ph | CAL-B | acetone | 38 | 25 | 14 | 2.25 | 23 |
| 14 | 23 | Ph | CAL-B | toluene | 26 | only substrate | |||
| 15 | 23 | Ph | CAL-B | TBME | 16 | 25 | 10 | 2.2 | 23 |
| 16 | 23 | Ph | CR | TBME | 38 | 25 | 23 | 1.8 | 18 |
(a) Enzyme: CAL-B: Candida antarctica lipase B (Novozym 435); PFL: lipase from Pseudomonas fluorescens; CR: lipase from Candida rugose; (b) TBME: t-butyl methyl ether; (c) in chloroform (c = 1); (d) the ee values were determined by chiral HPLC: AS, n-Hexane: (i-PrOH: EtOH 4:1) 96%: 4%, Fl. 0.4 mL/min; (e) the ee values were calculated on the basis of the optical rotation, compared to the one shown in Scheme 7 (vide infra).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Madalińska, L.; Kiełbasiński, P.; Kwiatkowska, M. Enzymatic Desymmetrisation of Prochiral Phosphines and Phosphine P-Sulfides as a Route to P-Chiral Catalysts. Catalysts 2022, 12, 171. https://doi.org/10.3390/catal12020171
AMA Style
Madalińska L, Kiełbasiński P, Kwiatkowska M. Enzymatic Desymmetrisation of Prochiral Phosphines and Phosphine P-Sulfides as a Route to P-Chiral Catalysts. Catalysts. 2022; 12(2):171. https://doi.org/10.3390/catal12020171
Chicago/Turabian StyleMadalińska, Lidia, Piotr Kiełbasiński, and Małgorzata Kwiatkowska. 2022. "Enzymatic Desymmetrisation of Prochiral Phosphines and Phosphine P-Sulfides as a Route to P-Chiral Catalysts" Catalysts 12, no. 2: 171. https://doi.org/10.3390/catal12020171
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.