
Autothermal Reforming of Acetic Acid to Hydrogen and Syngas on Ni and Rh Catalysts


1
School of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
2
Twigg Scientific and Technical Ltd., Cambridge CB25 9AU, UK
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(12), 1504; https://doi.org/10.3390/catal11121504
Submission received: 28 October 2021
/
Revised: 23 November 2021
/
Accepted: 25 November 2021
/
Published: 10 December 2021
(This article belongs to the Special Issue Catalytic Reforming for Syngas and H2 Productions)
Abstract
:The autothermal reforming (ATR) of acetic acid (HAc) as a model bio-oil compound is examined via bench scale experiments and equilibrium modelling to produce hydrogen and syngas. This study compares the performance of nickel (Ni-Al, Ni-CaAl) vs. rhodium (Rh-Al) for particulate packed bed (PPB), and of Rh-Al in PPB vs. Rh with and without Ceria for honeycomb monolith (‘M’) catalysts (R-M and RC-M). All PPB and M catalysts used Al2O3 as main support or washcoat, and when not pre-reduced, exhibited good performance with more than 90% of the HAc converted to C1-gases. The maximum H2 yield (6.5 wt.% of feed HAc) was obtained with both the Rh-Al and Ni-CaAl catalysts used in PPB, compared to the equilibrium limit of 7.2 wt.%, although carbon deposition from Ni-CaAl at 13.9 mg gcat−1 h−1 was significantly larger than Rh-Al’s (5.5 mg gcat−1 h−1); close to maximum H2 yields of 6.2 and 6.3 wt.% were obtained for R-M and RC-M respectively. The overall better performance of the Ni-CaAl catalyst over that of the Ni-Al was attributed to the added CaO reducing the acidity of the Al2O3 support, which provided a superior resistance to persistent coke formation. Unlike Rh-Al, the R-M and RC-M exhibited low steam conversions to H2 and CH4, evidencing little activity in water gas shift and methanation. However, the monolith catalysts showed no significant loss of activity, unlike Ni-Al. Both catalytic PPB (small reactor volumes) and monolith structures (ease of flow, strength, and stability) offer different advantages, thus Rh and Ni catalysts with new supports and structures combining these advantages for their suitability to the scale of local biomass resources could help the future sustainable use of biomasses and their bio-oils as storage friendly and energy dense sources of green hydrogen.
1. Introduction
Autothermal reforming (ATR) of biofuels presents the possibility of using more compact designs than steam reforming, thus incurring lower capital cost [1]. Mid-sized farms could process their own biofuels on-site before transporting the hydrogen produced to a centralised facility or sell directly to private consumers [2], or consume the bio-syngas as green fuel alternative to diesel in gensets at mini- and micro-grids.
Amongst the different biofuels that can be used for hydrogen production, pyrolysis oils or bio-oils have attracted significant attention [3,4,5]. Due to their complex and varying chemical composition, any study involving the use of bio-oil depends to some degree on the pyrolysis method used to produce them and on the biomass feedstock. Unlike direct gasification whose only useful product is syngas and has a large single process heat requirement, converting first the raw biomass via fast pyrolysis has the advantage of producing an energy dense, volatile organic liquid more easily stored and transported, as well as being suitable for a range of catalytic post processing routes to value added products. By careful pyrolysis process controls, the pyrolysis-ATR reforming route should also avoid the heavy tars by-products that are so challenging in biomass gasifier operation, and reduce the energy load over a smaller temperature increment, thus reducing thermodynamic irreversibilities. For an initial experimental study on the ATR of bio-oils, we have decided to choose acetic acid (‘HAc’) as a model compound. Previous studies showed that HAc is a prevalent component of fast pyrolysis of terrestrial biomass, often more than 20% of the total bio-oil composition [6,7]. Even though this is a simple molecule and only represents a fraction of the family of compounds found in actual bio-oil samples, (some heavier and more prone to polymerisation and coking), its use in an autothermal reforming study will be able to contribute to the understanding of the overall performance of an actual bio-oil. Acetic acid is also a significant product of anaerobic digestion used for the production of volatile fatty acids.
The global reaction of complete steam reforming (‘SR’) of HAc, which produces hydrogen and carbon dioxide, is endothermic and generates a stoichiometric maximum of 4 mol H2/mol HAc (or 13.45 wt.% of HAc). However, pressure, temperature and initial reactant concentrations limit the maximum yield through the thermodynamic equilibrium (equilibrium yield).
SR of HAc:
C2H4O2 + 2H2O ⟶ 2CO2 + 4H2 ΔH298 K = 129 kJ molHAc−1
Several authors have used HAc as a model bio-oil model compound in investigating its steam reforming [8,9]. More recent studies have looked at the application of catalysts for autothermal reforming (ATR) of HAc.
The research group of Huang, Wang, Xie and co-workers has been prolific in recent years in successfully investigating Ni, Co, Ni-Mn, Ni-Co based catalysts for ATR of HAc on various novel supports such as perovskites and double layered hydroxides at laboratory scale [10,11,12,13,14,15,16,17]. Steady conversion efficiencies (2.7–3 mol H2/mol HAc.) and lack of deactivation due to coking were observed for feed molar ratio H2O/HAc of 4 (i.e., feed molar steam to carbon ratio ‘S/C’ of 2) at 700 °C for 100 mg of catalyst.
Origins and end of life of spent catalysts, are often overlooked in the literature. Cobalt has been prominent in many recent literature outputs on ATR of acetic acid, despite a significant portion of unethical mining practices in the DRC [18], where 54% of the world’s cobalt is produced and which holds 49% of its reserves [19,20].
This is in a context where more than 50% of Co is used in the booming market of Li-ion batteries. Cobalt features in the EU’s 2017 list of critical raw materials [21] and its demand in the manufacturing industry is increasingly driven by electric vehicles, and rechargeable phones. Recycling of Co in batteries is difficult [22], and in the future will be further strained by the need for battery-based energy storage from intermittent, distributed renewable power generation. There is increasing awareness in the academic community of the need for conducting research on fully recyclable, sustainably sourced catalysts [23]. Industrially manufactured catalysts often already use commercially viable routes for recycling their high value components like Ni, Co and platinum group metals (PGM) using electro/bio-hydrometallurgy or bioleaching, [24,25,26]. Meanwhile, Ni and Co have finite reserves and other markets needs such as those of the steel industry [27]. It is therefore paramount to propose H2 production catalysts that have well established recycle routes or planned sustainable ones.
The aim of this study is to test the autothermal reforming of acetic under thermodynamically determined optimum conditions to measure conversion, product gas composition and carbon deposition using cobalt-free catalysts with composition and structure with industrial reforming relevance in packed and structured bed. Particulates from broken pellets of Ni on Al2O3 and of CaO-Al2O3 quadralobe raschig rings, were already tested on HAc feedstock for SR [28,29,30], and chemical looping steam reforming [31], with sorption enhancement [32]. We compare these catalysts in packed bed in the same ATR conditions with made in house Rh-Al2O3 powder as well as structured catalysts made by TST Ltd., Hull, UK, i.e., Rh-Al2O3 and Rh/Ce-Al2O3 washcoated honeycomb cordierite monoliths. Both pelletised, particulate catalysts and washcoated honeycomb monolith supported catalysts are extensively produced and used in industry. Cost-effective recycling routes have been practiced on such spent catalysts for many years, and more efficient methods continue to be investigated [33,34]. Monolith catalyst structures have many advantages [35] and have shown good performance in POX’s short contact time applications to biofuels [36], and power to gas applications [37]. The experiments in the present study were designed to test the activity of the catalysts and their suitability for autothermal hydrogen and syngas production by evaluating feed conversion, product distribution, H2 yield, sensitivity to steam and feed flow, and resistance to coke formation.
2. Results and Discussion
2.1. Equilibrium Modelling of ATR of HAc
To determine the reaction conditions needed for optimum hydrogen production, and assess process efficiencies of the experimental runs, equilibrium calculations were performed using the Aspen PlusTM software. An adiabatic Gibbs reactor (RGibbs) was used with the amount of acetic acid, water and air flowing into the reactor adjusted depending on the experiment of interest. The Peng-Robinson equation of state with Boston-Mathias modification (PR-BM) was selected as thermodynamic property method due to its applicability to high temperature gas-processing, chemical and petrochemical processes [38,39]. Table 1 is an example of the flow values input to Aspen corresponding to a liquid acetic acid flow of 1.0 mL/h, S/C of 2 and equivalence ratio l of 1.
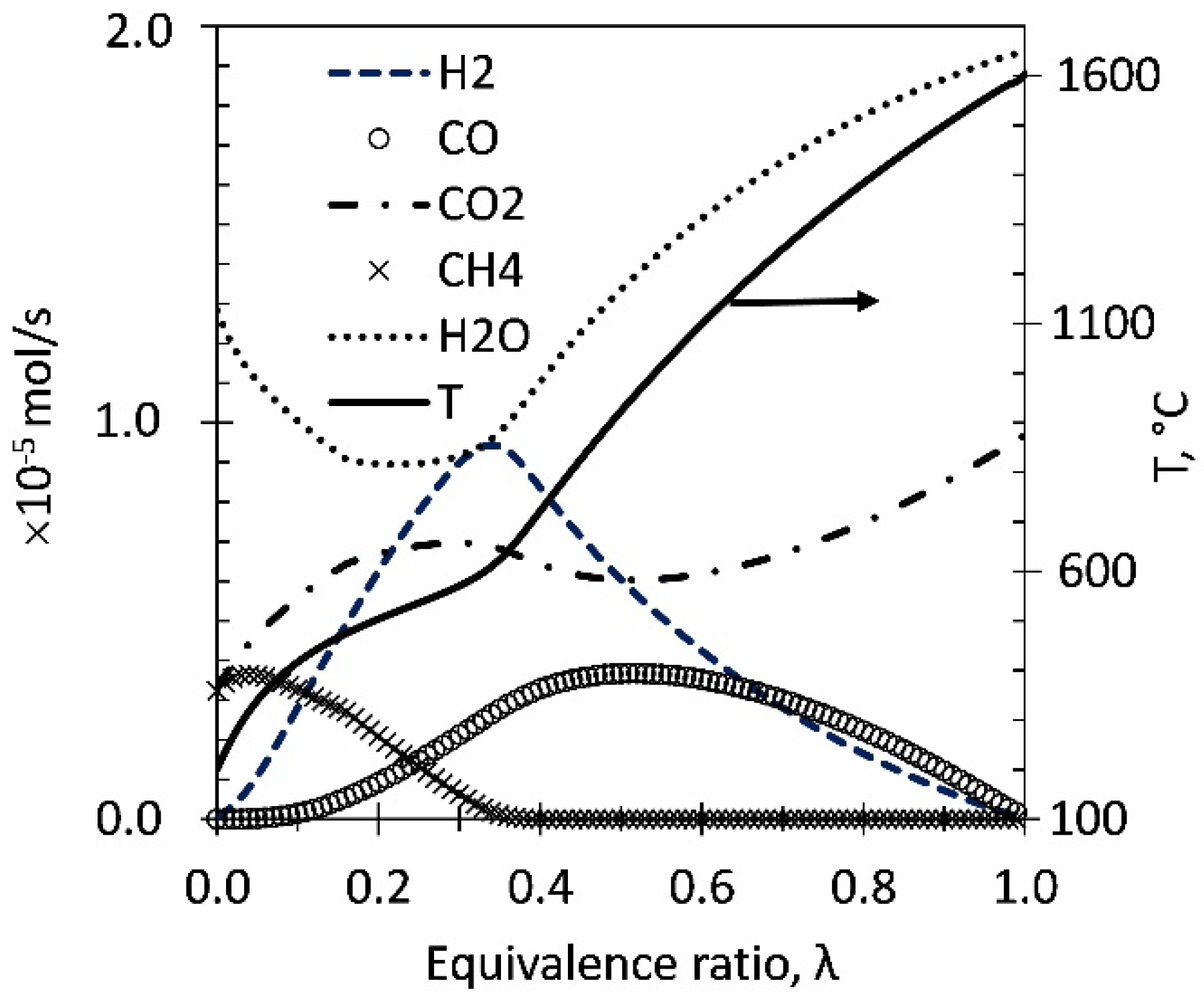
Figure 1 shows the equilibrium product gas distribution obtained for an HAc flow of 1 mL/h and feed steam to carbon molar ratio (S/C) of 2. The air flow was varied from 0 (λ = 0) to a maximum value of 4.62 × 10−5 mol/s corresponding to λ = 1. Maximum equilibrium hydrogen production occurs at λ = 0.35 at 570 °C, with 100% conversion of HAc and O2; this was used as the set point for experiments to determine the effect of catalyst on conversion, yield, selectivity and carbon formation. λ varies with S/C ratio (1 and 3).
2.2. Experiments with Ni- and Rh-Based Packed Bed Catalysts
Surface properties of the Ni- and Rh-based catalysts are listed in Table 2. The Ni-CaAl catalyst has a slightly larger (moderate) surface area compared to the Ni-Al catalyst whose surface area is very low due to its α-Al2O3 support. The higher surface area value for the Ni-CaAl catalyst can be attributed to its CaO content which has an initial high surface area of its own and could also be a result of the formation of mix phases during the support synthesis [40]. Upon reduction, the Ni-CaAl’s surface area is increased, unlike the Ni-Al, indicating the presence of CaO phase may have prevented sintering of the alumina.
The large specific surface area exhibited by the Rh-Al catalyst is due to its high γ-Al2O3 content. The 25 to 35% drop in surface area of the prepared catalysts compared to the alumina support can be attributed mostly to attrition occurring during catalyst preparation. This might not have been the case if a different method such as incipient wetness was used. Specchia et al. [41] prepared a 1 wt.% Rh/Al2O3 catalyst by incipient wetness using 1 mm γ-Al2O3 spheres with surface area of 157 m2/g. Their prepared catalyst showed minimal variation in properties with a surface area of 152 m2/g and pore volume of 0.45 cm3/g.
2.2.1. HAc Conversion on Ni and Rh Packed Bed Catalysts
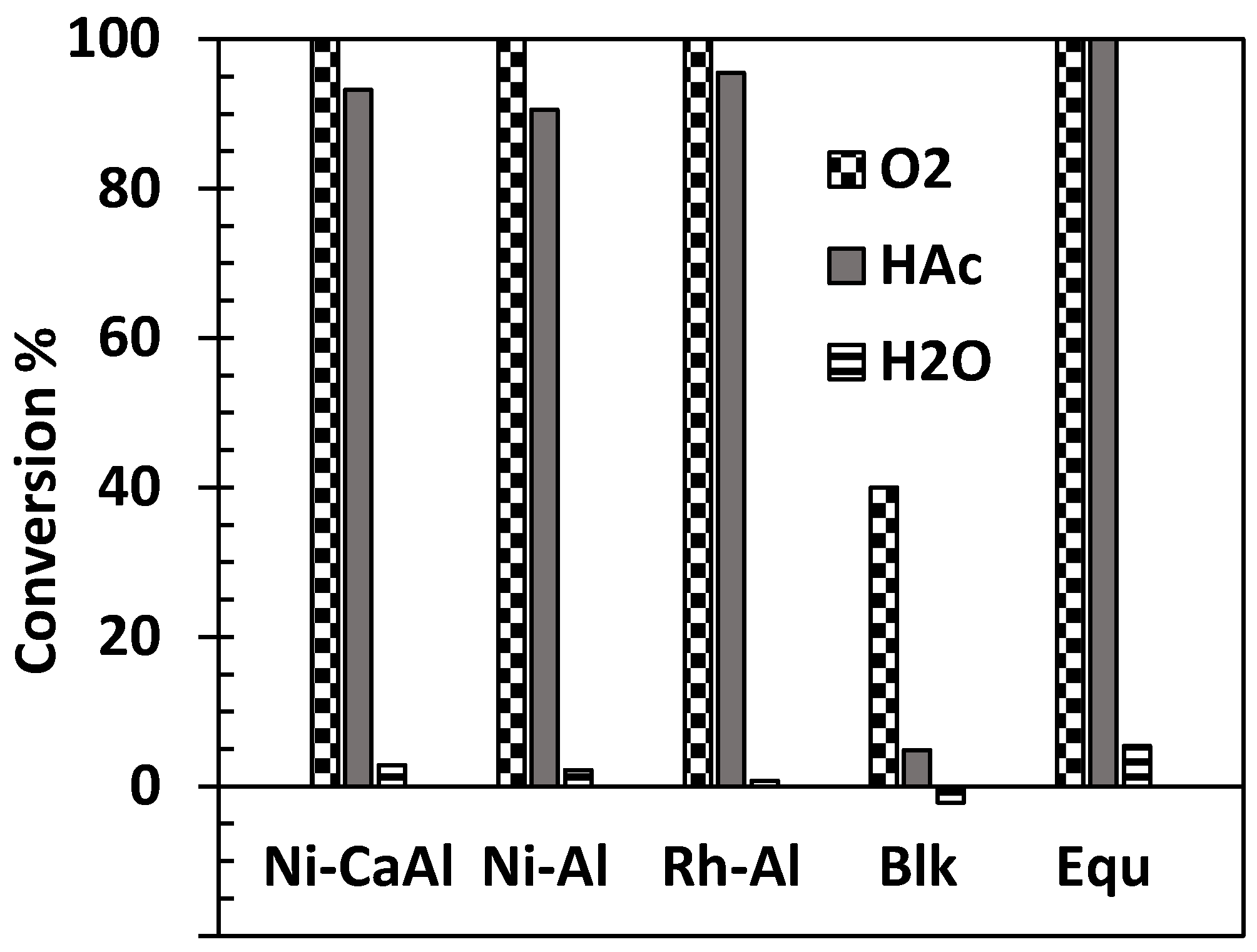
The conversion of acetic acid, water and oxygen obtained using the Ni-Al, the Ni-CaAl and Rh-Al catalysts in packed bed and a blank run performed with sand are compared to equilibrium in Figure 2.
The figure contains average values obtained over two runs using 1.0 g of either Ni catalysts or sand, or 0.2 g of Rh catalyst with liquid HAc feed rate set at 1 mL/h, liquid water flow at 1.257 mL/h (corresponding to a S/C of 2) and air flow set at 22 cm3/min STP (λ = 0.35). The product gas exits the catalyst bed at the pre-set ‘thermoneutral’ temperature of 570 °C measured by the reactor bed thermocouple.
The oxygen conversion on the three catalysts reached 100% showing their suitability in promoting oxidation reactions despite the relatively low reaction temperature used (compared to conventional SR, which is usually carried out above 650 °C). The Ni-Al catalyst however had a slightly lower HAc conversion of 91% compared to 93% obtained for the Ni-CaAl catalyst, with the Rh-Al exhibiting the highest conversion (95.5%) despite the much lower mass of catalyst and metal loading used. This incomplete acetic acid conversion is accompanied by low water conversion due to reduced SR or WGS catalytic activity. The blank experiment (SiO2 sand) resulted in less than 5% of acetic acid decomposed to C1 gases (CO, CO2 and CH4) at 570 °C. The water conversion for the blank experiment was negative indicating that water was possibly formed because of ketonization and combustion. It was not possible to detect acetone formation with the on-line micro-GC used for this study. However, total organic carbon analysis of condensates showed 78% liquid carbon recovery for the blank experiment. This indicates that more than 10% of the initial acetic acid is at least involved in either thermal decomposition or ketonization reactions. Basagiannis and Verykios [8] investigated the influence of temperature on the homogeneous reaction of acetic acid in the presence of steam. They reported the formation of acetone and CO2 at temperatures greater than 500 °C with H2, CO and CH4 formed because of thermal decomposition. The high acetic acid conversion obtained goes on to confirm selectivity of Ni-Al, Ni-CaAl and Rh-Al catalysts in promoting acetic acid ATR, with highest conversion obtained for Rh-Al. In terms of stability, the experiments involving Ni-Al catalyst were stopped after about 2 h due to carbon formation and resulting increased pressure drop in the reactor. Meanwhile, those performed with Ni-CaAl and Rh-Al catalysts lasted the full number of hours originally selected for the experiments (4 h and 3 h respectively). It is noteworthy that the Rh-Al catalyst had the lowest water conversion over two runs (0.7%) of the three catalysts (2.8% and 2.2% for Ni-CaAl and Ni-Al respectively), compared to the equilibrium value of 5.4%, indicating negligible activity in water gas shift in the Rh-Al catalyst.
2.2.2. Product Distribution and H2 Yield, and Whether to Pre-Reduce or Not a Ni or Rh Catalyst for ATR of HAc
The products distributions with time on stream obtained using Ni-Al, Ni-CaAl and Rh-Al catalysts are shown in Supplementary data (Figure S1). The three catalysts were tested in reduced state and as-received (fresh). It is common practice when performing SR experiments to reduce Ni catalyst with H2 before their use. This is because it has been established that the Ni metal (not the oxide) provides the active site for reforming reactions [42,43]. Rh/Al2O3 readily oxidises and reduces according to the nature of gases present (H2, CO, O2) at the temperatures used in the present study [44,45]. In its Rh0 state, it promotes mainly the NOx reduction [46] and CO oxidation [47] in three-way catalysts, which represents 80% of the Rh global demand [48]. The presence of oxygen in the reactor feed could locally create a net oxidizing environment possibly leading to the oxidation of any previously reduced Ni or Rh on the catalyst surface. This raises the question of whether to reduce the catalyst before performing ATR experiments.
The only ATR product gases detected by the micro-GC were H2, CO, CH4 and CO2, thus confirming the high selectivity for equilibrium products of both catalysts by promoting reforming reactions (SR and POX) over thermal decomposition.
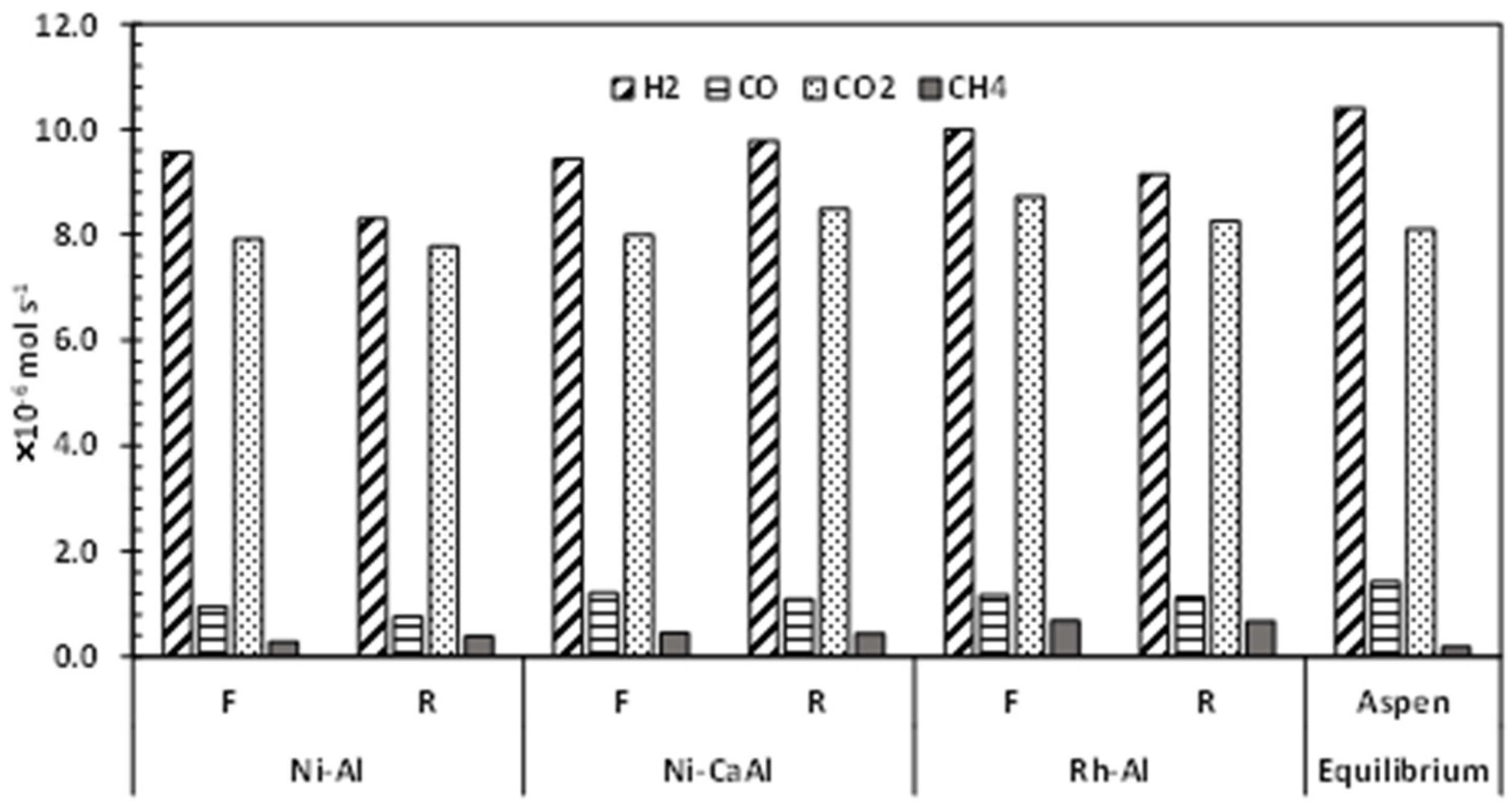
The average dry product gas composition and hydrogen yield obtained using Ni-Al, Ni-CaAl and Rh-Al in both fresh (F) and reduced (R) states are shown in Figure 3. The average is taken over several stable results for each gas. Ni-CaAl catalyst appears to be a more active ATR catalyst with average H2 production reaching 94% and 90% of the expected equilibrium value for the reduced (R) and fresh (F) catalyst respectively; compared to the 86% obtained for the fresh and reduced Ni-Al catalyst. The almost similar results obtained for the fresh and reduced forms of both catalysts confirms that the fresh catalyst is reduced by acetic acid, ‘auto-reduction’, at the start of the experiment to generate active Ni sites.
The ‘auto-reduction’ continues until an equilibrium Ni/NiO concentration is reached and steady production of synthesis gas is achieved. This equilibrium concentration of Ni and NiO is confirmed by the presence of both Ni and NiO peaks in the XRD patterns of a sample Ni-Al catalyst used in fresh, Ni-Al(F), and reduced, Ni-Al (R) states (see Figure S2a). The XRD results for Ni-CaAl catalyst are not included due to its large amorphous phase content. For the Ni catalysts, the presence of oxygen in the feed does not cause significant oxidation of the reduced catalyst as no NiO is detected in the used Ni-Al(R) catalyst. This suggests that the oxygen in the feed is completely consumed by reforming reactions and any NiO formed reacts almost immediately with H2 and CO to produce Ni(S), H2O, and CO2.
The maximum H2 yield obtained for the results shown in Figure 4 was 6.8 wt.% for the Ni-CaAl catalyst, which is well short of SR of HAc stoichiometric maximum, but close to that of HAc decomposition to H2 and CO. Reference [49] reported a H2 yield of 13.1 wt.% for the steam reforming of acetic acid using a co-precipitated Ni/Al/Ca catalyst in a fluidized bed reactor operated at 650 °C and a S/C molar ratio of 5.58.
The slightly better performance of the Ni-CaAl catalyst was expected, as the addition of CaO to the Al2O3 support hinders the formation of the spinel compound, NiAl2O4, which is known to impair the reducibility of NiO; while at the same time improving the same property by forming CaAl2O4 [50,51]. Another reason for the better performance of Ni-CaAl catalyst over that of Ni-Al’s was its superior resistance to coke formation; with only 13.9 and 11.6 mg/(g(cat).h) coke deposit detected compared to 25.7 and 54.0 mg/(g(cat).h) deposited for the Ni-Al catalyst in used fresh and reduced states respectively. It is noteworthy that the fresh, fully oxidised, as received Ni-Al catalyst experienced far less coking compared to the reduced form. This suggests that the ‘less’ active fresh (oxidised) versions of both Ni catalysts had a better resistance to coke formation than the more active (reduced) catalyst, and this can be attributed to the presence of less active NiO sites on the surface of the used fresh catalyst.
XRD spectra of the Rh-Al are shown in Figure S2b, where the small, well dispersed Rh loading on the support (shown in Figure S2c) makes the Rh and oxidised Rh peaks (RhO2, Rh2O3) undistinguishable from the overwhelming presence of the γ-Al2O3 peaks. The Rh-Al catalyst exhibited the least coke deposits (5.5 mg/(g(cat).h) fresh, 2.5 mg/(g(cat).h) reduced) despite using the lower mass of catalyst (0.2 g vs. 1 g) for same HAc feed flow. Closure of the carbon balance varied slightly for this set of experiments, within a 5.9% average discrepancy, with 2% minimum (Fresh Ni-Al) and 8% maximum (Fresh Rh-Al). Based on these observations, all further reforming experiments were performed using only unreduced ‘fresh’ catalysts.
2.2.3. Oxidation Test (POX)
The eventual success of an ideal ATR depends on the ability of the catalyst to promote the exothermic oxidation reaction(s) needed to support the endothermic steam reforming reaction. This ability was tested for the fresh Ni-Al, Ni-CaAl and Rh-Al catalysts by performing POX-only experiments (no steam) while maintaining the same reactor temperature, acetic acid flow and air flow as discussed above. The dry N2-free molar flow product gas composition obtained with time on stream for the three catalysts after 3 h is shown in Figure S3. A similar trend in product gas composition is seen for the three catalysts. Table 3 provides a summary of the POX experiments. The Ni-Al catalyst had a poor acetic acid conversion to reforming gases of just 78% compared to 91% obtained for the Ni-CaAl catalyst.
The low HAc conversion for the Ni-Al resulted in significant coking measured at 50.1 mg/g(cat).h compared to 34.9 for Ni-CaAl and 28.3 for Rh/Al, respectively. However, the Ni-Al catalyst had a better H2 selectivity with higher H2/CO ratio of 2.50. This implies that the Ni-Al catalyst is more active in promoting the WGS reaction.
This catalyst is also more selective for the complete oxidation reaction as confirmed by the slightly higher concentration in CO2. Meanwhile the Ni-CaAl catalyst is more active in promoting SR reaction and so performs better under ATR condition resulting in its higher hydrogen yield. All the catalysts perform better in ATR compared to POX as evidenced by the reduced coking for the former (see Table 2). The presence of steam during ATR helps to create a stronger oxidizing/reforming environment, which proves beneficial for the three catalysts.
2.2.4. Feed Molar Steam to Carbon Ratio (S/C)
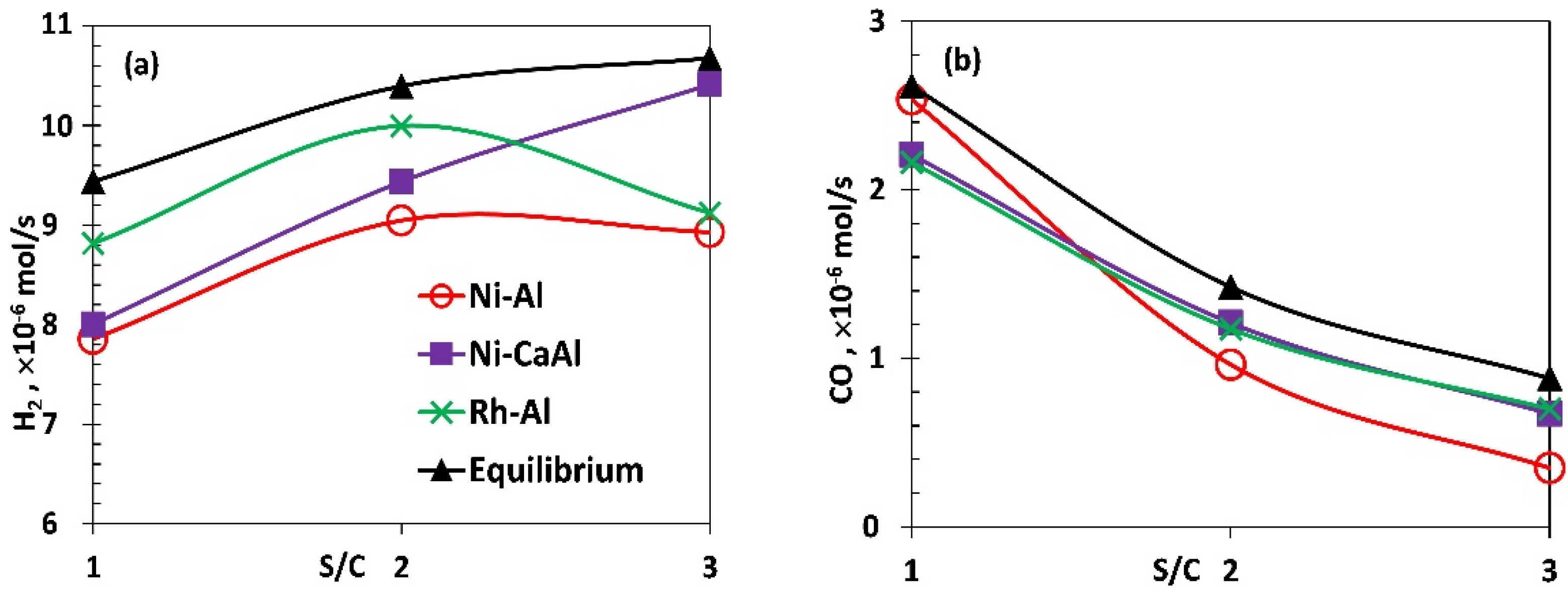
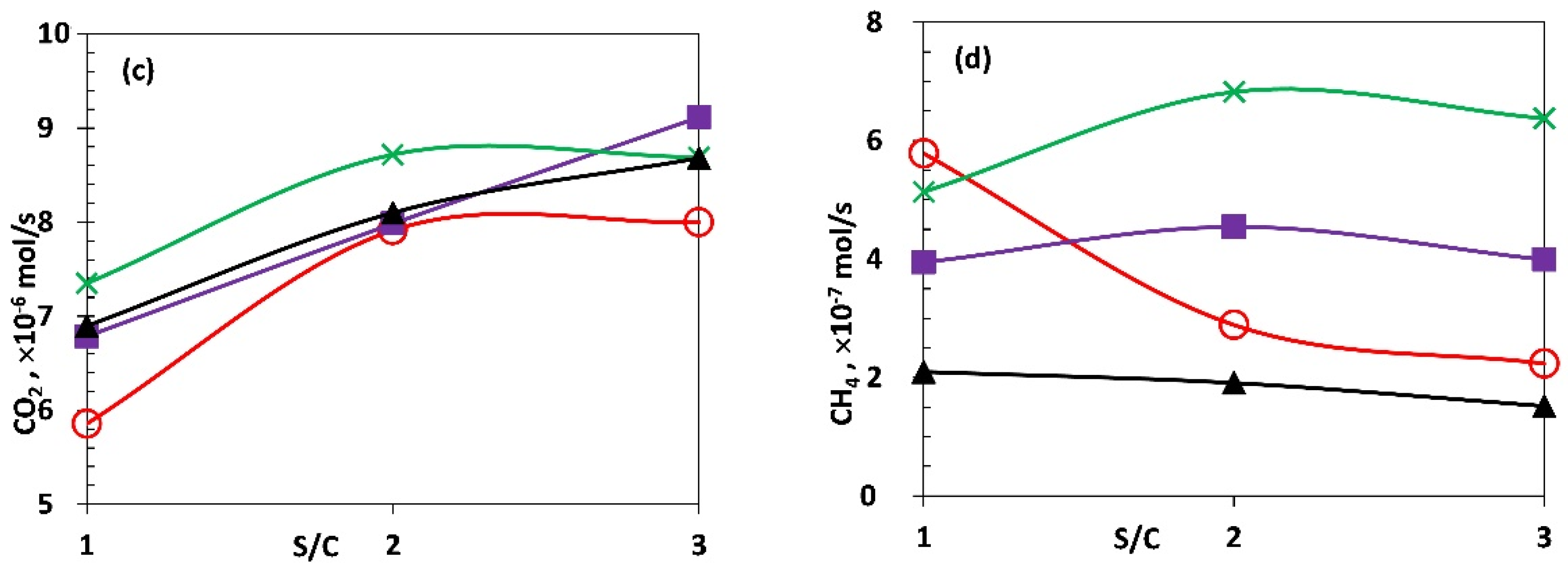
The influence of varying S/C on the ATR of acetic acid using fresh Ni-Al, Ni-CaAl and Rh-Al catalysts was investigated and the production rates of H2, CO, CO2 and CH4 shown in Figure 4a–d including equilibrium values. Based on equilibrium, it is expected that as S/C increases from the stoichiometric value of 1 to excess of steam (2 and 3), the amount of H2 and CO2 produced should increase towards the equilibrium limit while the amount of CO reduces, as Le Chatelier’s principle alters the balance of water gas shift towards more product. The trends obtained for H2, CO and CO2 were as expected between S/C of 1 and 2 for the three catalysts. Between S/C of 2 and 3, which represent between doubling and tripling of stoichiometric steam, results indicate non equilibrium effects (H2 and CO2 of Ni-Al plateauing), insensitivity of water gas shift activity beyond S/C of 2 (Rh-Al), and activity of methanation of CO and C in the Ni-CaAl and Rh-Al catalysts respectively as supported by the following section.
Increasing the S/C ratio from 1 to 3 apparently had no significant effect on the crystalline phases present on the used catalyst Ni-Al (Figure S4). Ni is the principal Ni-containing phase detected with only a minute NiO peak detected for the experiment carried out at S/C = 2. This confirms the earlier conclusion that most of the NiO in the fresh catalyst is reduced to Ni during ATR.
However, it should be expected that if for some reason higher air flows (higher λ) are used, then the concentration of surface NiO phase in the used catalyst might increase as seen in the work published by Medrano et al. [49].
2.2.5. Space Velocity Effect on Carbon Products
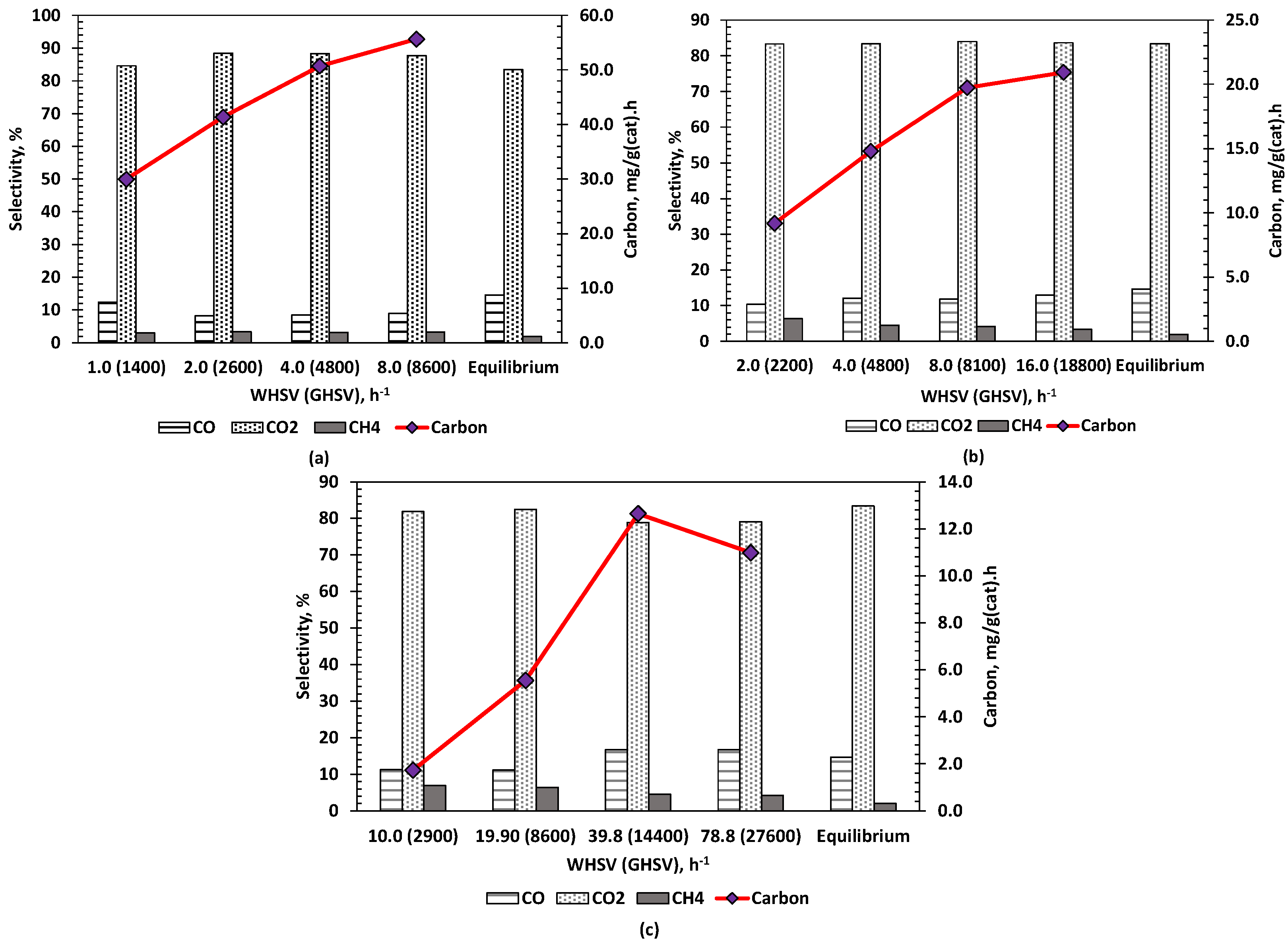
The effect of varying the space velocity on the performance of the ATR of HAc process and in particular carbon products using fresh Ni and Rh catalysts in packed bed was investigated. The space velocity (expressed as WHSV as well as GHSV) was investigated by varying HAc flow while keeping all other parameters constant at 1 g (Ni) and 0.2 g (Rh) of catalyst, S/C = 2, λ = 0.353, 570 °C and 1 bar. WHSV was defined as the total mass of feed flowing into the reactor at 20 °C divided by the mass of catalyst used (Equation (7)). The GHSV was calculated by dividing the total volumetric flow rate of all feed entering the reactor by the volume of catalyst (Equation (8)). Volumetric flows for HAc, water and air were determined by substituting the known molar flow rates into the ideal gas equation at NTP (20 °C and 1 atm). Figure S5 in Supplementary Data shows the influence of space velocity on H2 purity, H2 yield and conversion of acetic acid, water and oxygen. There are little differences between the three catalysts. The Ni-CaAl catalyst performed better than the Ni-Al for all space velocities examined with a hydrogen yield average of 6.6 wt.% compared to 5.7 wt.% for the latter. Both Ni catalysts have a similar and almost constant value for H2 purity, with Ni-CaAl having a higher acetic acid and water conversion. The lower water conversion for the Ni-Al catalyst is an indication of its affinity to promote acetic acid oxidation leading to lower H2 yield and slightly higher CO2. Counter-intuitively, HAc and water conversions were largest for the Ni-CaAl and Rh-Al catalyst at their highest GHSV (>14,000 h−1), achieving a H2 yield 97% and 91% of the equilibrium values, respectively.
Figure 5 shows the increase in catalyst coking, expressed in mg/g(cat).h, as the space velocity is increased, along with the selectivity to C1 gas products for the three catalysts. The latter exhibited very similar trends, with the Rh-Al catalyst displaying higher selectivity to CO, increasing with space velocity, eventually slightly exceeding the equilibrium value, due to possible kinetic limitations of water gas shift as there was a concurrent increase in HAc conversion. Methanation appeared also increasingly kinetically limited, as selectivity to methane decreased with space velocity for both Ni-CaAl and Rh-Al. No clear trend with GHSV was observed in the carbon in the condensates from the TOC analysis (not shown) which remained typically well below 1% of the carbon in the feed, with only outlier RhAl 14,400 GHSV at 4%, also corresponding to the highest solid Carbon yield of the Rh-Al runs. The coking rate increased in tandem with HAc feed rate for both Ni catalysts. Of the two Ni catalysts, the Ni-CaAl showed better resistance to coke formation with only about a third of that obtained using the Ni-Al catalyst for the same space velocity.
High flows caused rapid formation of coke on the Ni-Al catalyst and the experiment had to be stopped after 30 min due to pressure increase in the reactor. Gutierrez et al. [52] reported a similar problem when Ni/Al2O3 catalyst was used for the ATR of ethanol. Both Ni-Al and Ni-CaAl catalysts however showed a steady decrease in the fraction of fuel carbon deposit formed with increase in space velocity implying the possible existence of maximum or saturation value.
This could also imply that the Ni-CaAl catalyst performs better at higher feed flow within the range studied, as seen earlier. This can be due to the localised formation of hotspots in the catalyst bed which promote coke gasification. Formation of hotpots can however have a negative effect on the catalyst stability as it might lead to sintering and catalyst deactivation. Liguras et al. [53] obtained similar results for their work on the autothermal reforming (which they referred to as ‘catalytic partial oxidation’) of ethanol using Ni/La2O3 supported over cordierite monoliths as catalyst. By comparison to the Ni catalysts, the Rh-Al catalyst exhibited much lower carbon deposition, with the expected minimum found for the lowest WHSV (GHSV) values, roughly ¼ of that found for Ni-CaAl at similar GHSV (ca. 8000 h−1). The syngas yield (H2 + CO + CH4) of 1.19 × 10−5 mol s−1 was highest and closest to equilibrium (1.20 × 10−5 mol s−1) for the Rh-Al catalyst at the GHSV of 8600 h−1 and produced close to minimal carbon deposition rate (5.5 mg/(g(cat) h)) at the HAc feed rate 1 mL/h for a 0.2 g of fresh, unreduced, catalyst reactor load.
Catalyst coking and deactivation pose a major problem during ATR of oxygenated bio-derived feedstocks in general. The overall low temperatures required for optimal H2 production (less than 600 °C) hinder coke gasification reactions thereby promoting carbon deposition on catalyst. The influence of process parameters on the rate of coke formation have been discussed individually in the preceding sections. Figure 6a–c shows respectively pictures of the used Ni-Al, Ni-CaAl and Rh-Al catalysts and their morphology as seen using SEM. The three catalysts were used in their fresh (unreduced) state for ATR experiments performed with a S/C molar ratio of 2 and λ = 0.353.
For all the catalysts, coking resulted in the formation of a carbonaceous mass surrounding individual (or group of) catalyst particles. These carbon deposits consisted of a dense filamentous network as revealed by the SEM images. Coking is a major problem when Ni based catalysts are used for reforming, especially at low temperatures. Marquevich et al. [54] reported rapid coking when commercial Ni catalysts were used in the SR of acetic acid at temperatures lower than 650 °C. One possible explanation is that the Boudouard reaction and the direct formation of carbon from hydrocarbon decomposition (cracking) are favoured below 700 °C [52]. The insert in Figure 6a is an image of used Ni-Al catalyst in which lumps of agglomerated coke particles measuring up to 5 mm could be measured. Coking was largely observed to have occurred only at the top of the catalyst bed exposed to the input feed. For this catalyst, this led, in most cases, to clogging and significant pressure drop in the reactor reducing the experimental duration in most cases to less than 2 h for the Ni-Al catalyst. In such cases, most of the Ni-Al catalyst particles found lower in the bed were almost without any coke, as seen on the SEM image of Figure 6a, which is void of any carbon filaments [55]. Cheng and Dupont [28] reported a similar result for the integrated catalyst reduction and acetic acid SR process for which the same 18 wt.% NiO/α-Al2O3 catalyst was used. The Ni-CaAl catalyst on the other hand, showed no agglomerated coking but an abundance of evenly distributed filamentous carbon deposits (multi wall carbon nanotubes) in the catalyst bed (Figure 6b). The carbon deposits formed in most cases were about the same size as the individual catalyst particles. The Rh-Al catalyst had no agglomerated coking and a much lower level of filamentous carbon (Figure 6c). No bed clogging was witnessed when using the Ni-CaAl or Rh-Al catalysts for all experiments performed up to three hours.
2.3. Experiments with Rh-Based Monoliths
2.3.1. HAc Conversion on Rh Monoliths
For experiments with the Rh monoliths, an acetic acid flow of 2 mL/h was selected, and all other flows determined based on the desired S/C molar ratio and λ. Three different ATR conditions were examined, and the flow settings are listed on Table 4.
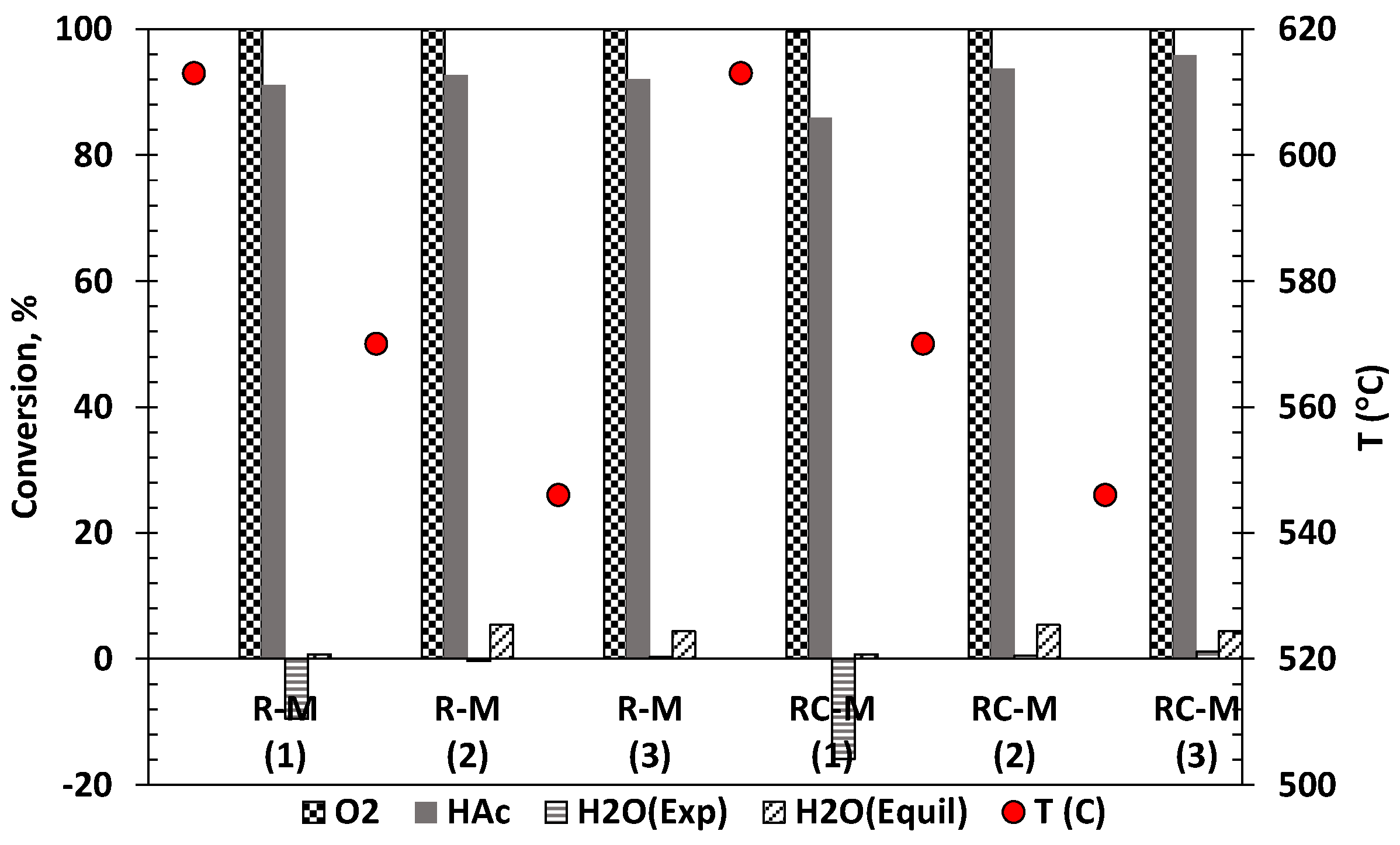
The conversion of acetic acid, water and oxygen and monolith temperature using both R-M and RC-M monoliths for the three conditions listed in Table 4 are shown in Figure 7. Acetic acid conversion varied only slightly from 91 to 92% as the S/C ratio was increased from 1 to 3 for the R-M monolith. The marginal increase in acetic acid conversion can be attributed to the slight increase in air flow as the ATR condition is varied from S/C of 1 to 3. The RC-M monolith showed improved acetic acid conversion as the ATR condition and corresponding S/C was varied from 1 to 3. This suggests that the ceria doped catalyst is sensitive to the amount of steam present in the reacting mixture. The ability of this catalyst to facilitate acetic acid conversion reactions is enhanced by the presence of water as a consequence of increased promotion of water consuming reactions (SR and WGS) and fuel decomposition.
This was confirmed by the slightly better water conversion obtained for the RC-M monolith compared to R-M. The oxygen conversion on both catalysts reached 100% confirming the ability of Rh based catalyst to promote oxidation reactions [55].
2.3.2. Product Distribution and H2 Yield
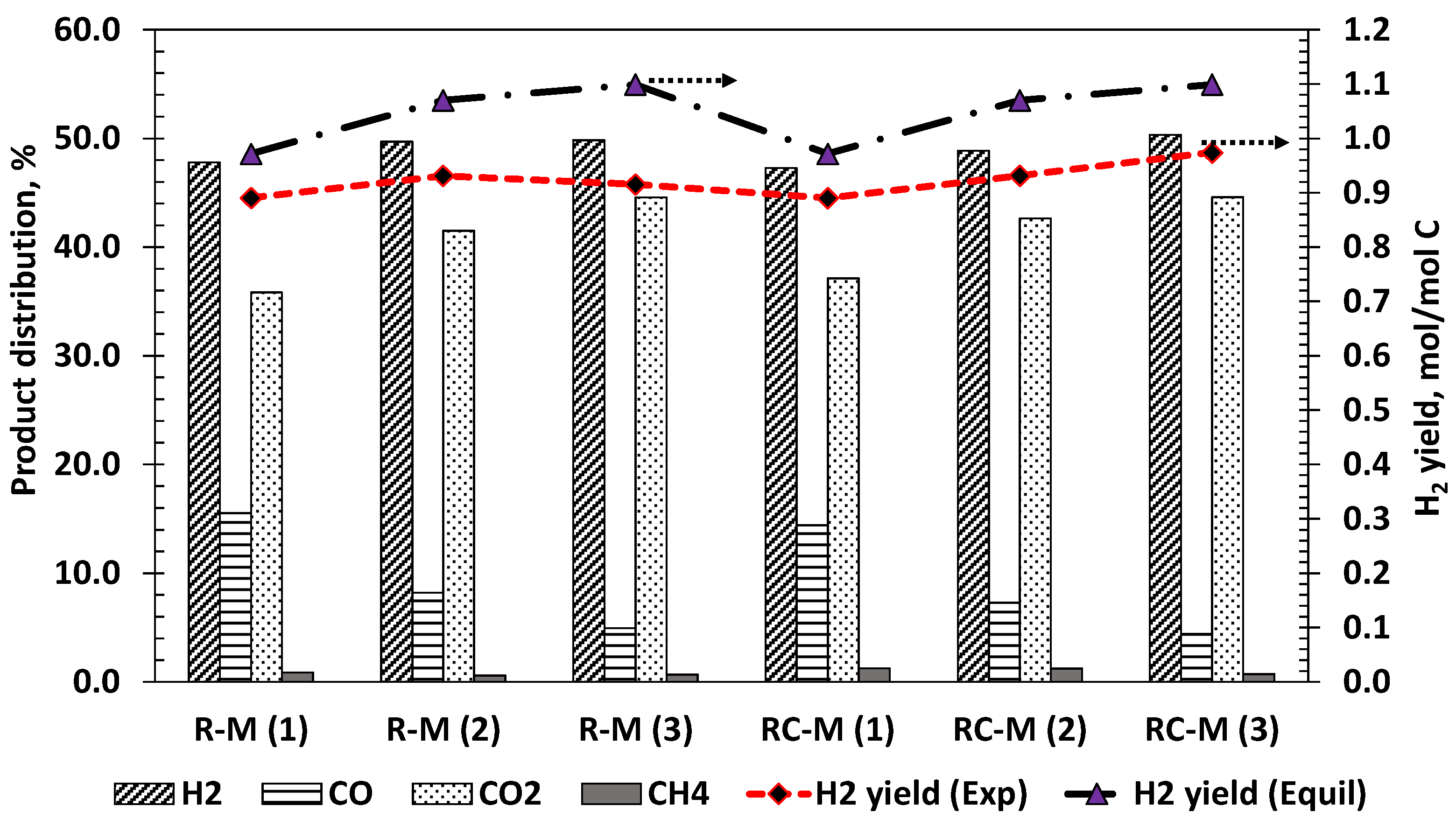
The product distribution obtained using the R-M and RC-M monoliths are shown in Figure S6. Both monoliths show a similar product distribution over time with the R-M monolith displaying more stable results than RC-M. An initial induction time was required to stabilise active sites on the catalyst before the detection of syngas by the micro-GC. This induction time was less than 5 min for the R-M monolith indicating that Rh2O3 was the active form of the metal during ATR. This result is in agreement with that obtained by Kaila et al. who showed that Rh2O3 was the active form of Rh in Zirconia supported RhPt bimetallic catalyst used for the ATR of simulated gasoline [55]. For both monoliths, H2 was the first gas component to spike followed almost immediately by a simultaneous increase in CO2 and CO concentration. This suggests that very little gaseous phase homogeneous decomposition occurs, rather, acetic acid, O2 and H2O all adsorb on active sites on the catalyst surface with the ensuing surface reactions leading ultimately to the desorption of H2, CO2, CO and CH4. Only small concentrations of H2 and CO2 were detected at the beginning of the experiment with the RC-M monolith (Figure S6b) with CO only appearing after the spike in H2 production. The delayed detection of CO2 suggests that the acetic acid decarboxylation is inhibited by adsorbed oxygen.
The selectivity to H2 and C1 product gases obtained during acetic acid ATR at S/C of 2 is shown in Table 5, along with HAc and water conversions, syngas production rate and products ratios for the R-M and RC-M catalysts. Corresponding results for the packed bed catalysts experiments at 2 mL/h HAc feed, and same S/C and λ conditions are shown for comparison of outputs between catalysts structures. Of the two structured catalysts, the RC-M monolith yielded HAc and H2O conversions and products distribution closest to equilibrium. The RC-M monolith also had a slightly higher hydrogen yield, and this can be attributed to a better water conversion reflected in its higher H2/CO and CO2/CO ratios. This monolith is slightly more active in promoting the WGS and methanation reactions. For both monoliths, the H2 yield is short of the equilibrium value due to a HAc conversion in the 93–94% range compared to the 100% equilibrium value. Methanation activity can be attributed to the lower Rh loading of the powder catalyst compared to the monoliths as also found in [56,57]. The syngas production (represented by the sum of molar rates of H2, CO and CH4) shows that the Rh catalysts have very close outputs, of the order of 89% of the equilibrium, regardless of the structure, and despite the monoliths’s lower HAc conversions. The better acetic acid conversion obtained using the powder Rh catalyst (100%) however resulted in the highest hydrogen yield out of the three Rh catalysts. Comparing the monoliths’s outputs with the Ni packed bed catalysts, it can be seen that the Ni/CaAl and the RC-M offer very close HAc conversions with slightly different outcomes. The higher H2O conversion of the Ni/CaAl (2% vs. 0.5%) provides a small advantage on H2 yield (6.6 vs. 6.3 wt.%) despite a higher selectivity to CH4 (4.2 vs. 2.4%) over the RC-M’s counterparts. The results obtained using the Ni catalyst showed quite different selectivity to the Rh monoliths suggesting the involvement of different reaction mechanisms. The Ni based catalysts had lower CO and higher CH4 selectivity, indicating higher activities in both WGS and methanation.
It is worthy of note that despite the different nature and loading of active elements, support chemical composition, and physical structure of the catalysts, the range of outputs obtained in the experiments at S/C of 2, λ of 0.353 and HAc feed rate of 2 mL/h were remarkably similar and very promising. The results suggest that ultimately, rather than being based on superior catalytic activity, the selection of a suitable catalyst for ATR of HAc, will likely depend on cost versus longevity, mechanical integrity, ease of regeneration and recycling, sustainability of the materials and manufacture, scale of the process. For instance, the honeycomb monolith structure might offer advantages of durability (strength), lower health impacts (less fines) and lower operating costs (lower pressure drops) but would not be best suited for large throughput plants due to their much larger volume compared to packed beds. Research into more performant catalysts structures in the above selection criteria categories would be worthwhile.
2.3.3. Effect of the Feed Steam to Carbon Ratio
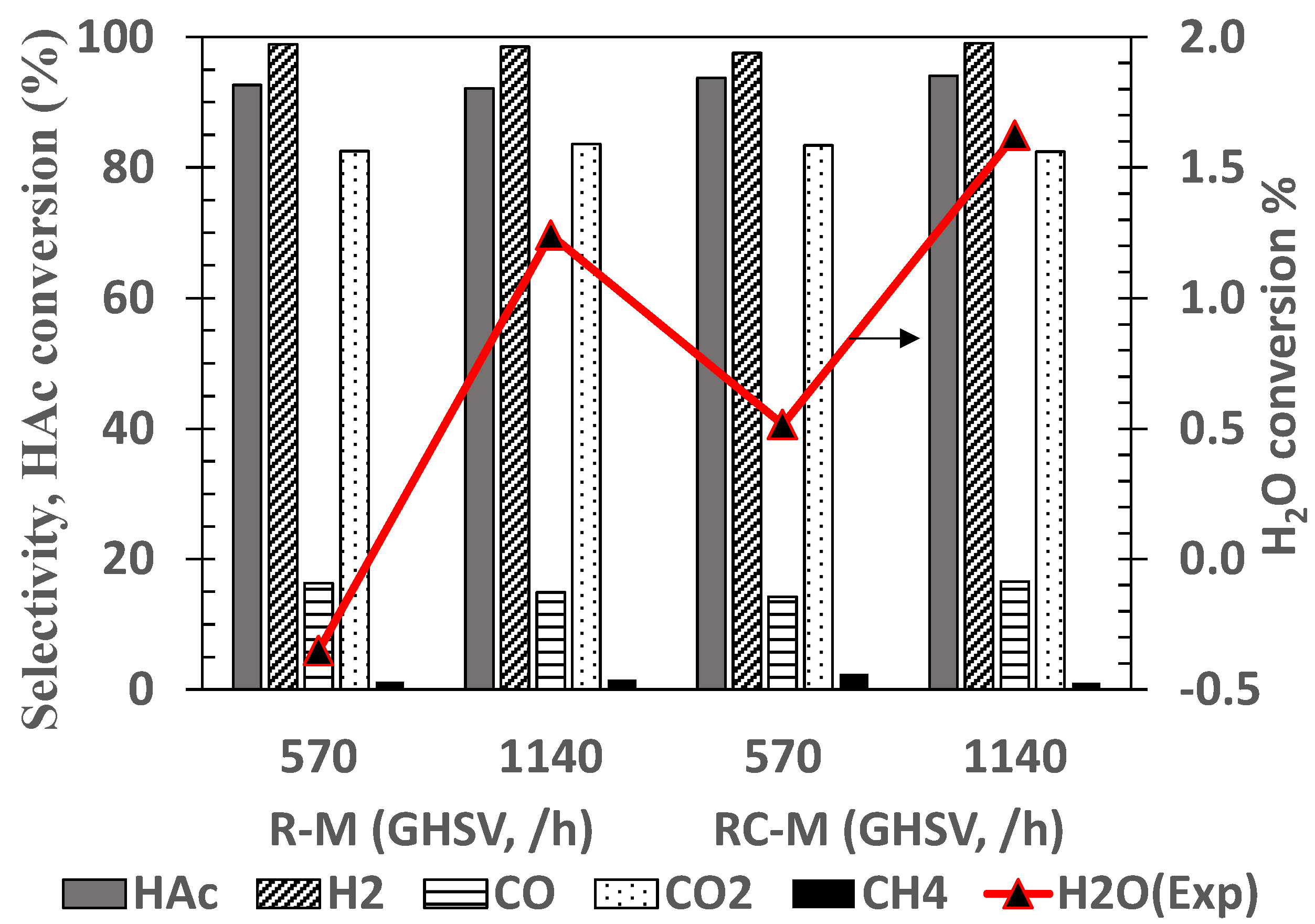
The influence of S/C molar ratio on the ATR of acetic acid using R-M and RC-M honeycomb monoliths was investigated and the results shown in Figure 8. At a given S/C, no significant differences of H2 yield were obtained between the two catalysts formulations. As S/C increases from 1 to 3, the increase in CO2 and corresponding decrease in CO production indicates an increase in WGS activity as with the case with all other catalysts presented so far in this work. Increasing the S/C molar ratio up to 3 did not have a ‘quenching’ effect mainly because the reacting system was supported by external heating.
This might not be the case in an actual ATR process, making it necessary to operate at the lower S/C ratio of 1 and 2. Rennard et al. [36] reported a decrease in WGS activity and hydrogen production during ATR of glycerol using Rh-Ce foam as the S/C ratio was increased above 1. From the balanced SR reaction, it is evident that only 2 moles of water are required per mole of acetic acid (S/C = 1) for a stoichiometric reaction with maximum H2 yield. For both monoliths, a choice of S/C = 2 might prove to be a good compromise leading to good process outputs and energy savings.
2.3.4. Effect of the Space Velocity
The influence of doubling the GHSV on feed HAc and water conversions and selectivity to gas products was investigated and the results for the R-M and RC-M monoliths are shown in Figure 9. There was no substantial change in the acetic acid conversion and dry gas composition as the GHSV was increased from 5.7 × 102 to 1.14 × 103 h−1. Selectivity to H2 and C1 gas products remained constant suggesting no significant increase in diffusion resistance for the GHSV examined. Monoliths structured catalyst have been demonstrated to be able to maintain good catalytic activity even at very high feed flows. Unfortunately, the rig setup used for these experiments did not permit higher space velocities to be tested. Hohn and Schmidt [58] studied the effect of high space velocities on product yield during the partial oxidation of methane using different Rh coated foam monoliths.
They found that the Rh monoliths were able to maintain high conversion and H2 yield at GHSV up to 105 h−1 with further increase leading eventually to a drop in the catalyst performance. The drop in activity was more pronounced for monoliths with lower Rh loadings. They suggested that the low front temperatures due to blow out resulting from convective heat transfer were responsible for the poor convention and product yield at very high GHSV. Higher flows would need to be tested to determine the value for which such conditions exist during acetic acid ATR.
For both monoliths the water conversion improved with the increase in GHSV, although remaining significantly lower than the equilibrium value (5.4%), confirming low activity in water gas shift and steam reforming reactions.
3. Experimental
3.1. Catalyst
Five catalysts in the form of either crushed pellets, powder and honeycomb monoliths were used in this investigation. Two pellet-type (broken up quadralobe Raschig rings) typical of commercial grade nickel catalysts, 18 wt.% NiO/Al2O3 and 15 wt.% NiO/CaO-Al2O3, henceforth termed Ni-Al and Ni-CaAl respectively were used in a first set of experiments, with original ring sizes of 11- and 16-mm diameter, respectively. The Ni catalysts formulations presented slightly different Ni loadings due to their respective different commercial uses. The Ni-based catalysts were crushed and sieved to obtain particle sizes in the range 0.355–1 mm before being used for reforming experiments. Precious metal rhodium is more catalytically active than nickel in catalytic partial oxidation of methane and less prone to deactivating carbon deposition, therefore the Rh loading in the catalysts was deliberately chosen to be one order of magnitude smaller than Ni’s, and closer to three way catalyst formulations. For the comparison with Rh catalysts, two compositions were used: 1 wt.% Rh on γ-Al2O3 (‘Rh-Al’), and 1 wt.%/3 wt.% Ce on γ-Al2O3 (‘RhCe-Al’), the former investigated in two forms of catalysts: powder in packed bed, followed by catalytic washcoat layer on cordierite honeycomb monolith, the latter only in the washcoated monolith form. The synthesis method for the Rh powder catalyst is described in [59]. For the structured monolith catalysts, two single blocks 19 mm outer diameter (o.d.) cordierite monolith washcoated with Rh-Al (hereafter termed R-M) and with RhCe-Al (henceforth termed RC-M), provided by Twigg Scientific & Technical Ltd. (Cambridge, UK), were used in the second set of experiments. The monoliths presented a density of 400 cpsi (cells per square inch), i.e., 62 cells/cm2 equivalent to 1.21 mm sides. The monoliths length was 42 mm with a 15 mm diameter, offering ca. 110 square channels to the flow. Typical wall and washcoat thicknesses are in the range 70–100 μm. The mass of Rh in both monoliths was estimated to be 4.3 mg, roughly twice that of the 2 mg used in the powder Rh catalyst.
3.2. Catalyst Characterisation
A Quantachrome Nova 2200e nitrogen gas adsorption/desorption isotherm was used to determine the surface area, the pore volume, and the pore size distribution of fresh and used catalysts. A Brucker D8 Advance diffractometer was used for the XRD analysis of the crystalline phase of prepared and used Nickel catalysts. The morphology of prepared and used Ni catalysts were examined using a Hitachi SU8230 high performance cold field emission scanning electron microscopy (CFE-SEM). The amount of carbon deposited on used Ni and Rh catalysts was determined using a Thermo Scientific Flash 2000 Elemental Analyzer.
3.3. Reactor Setup
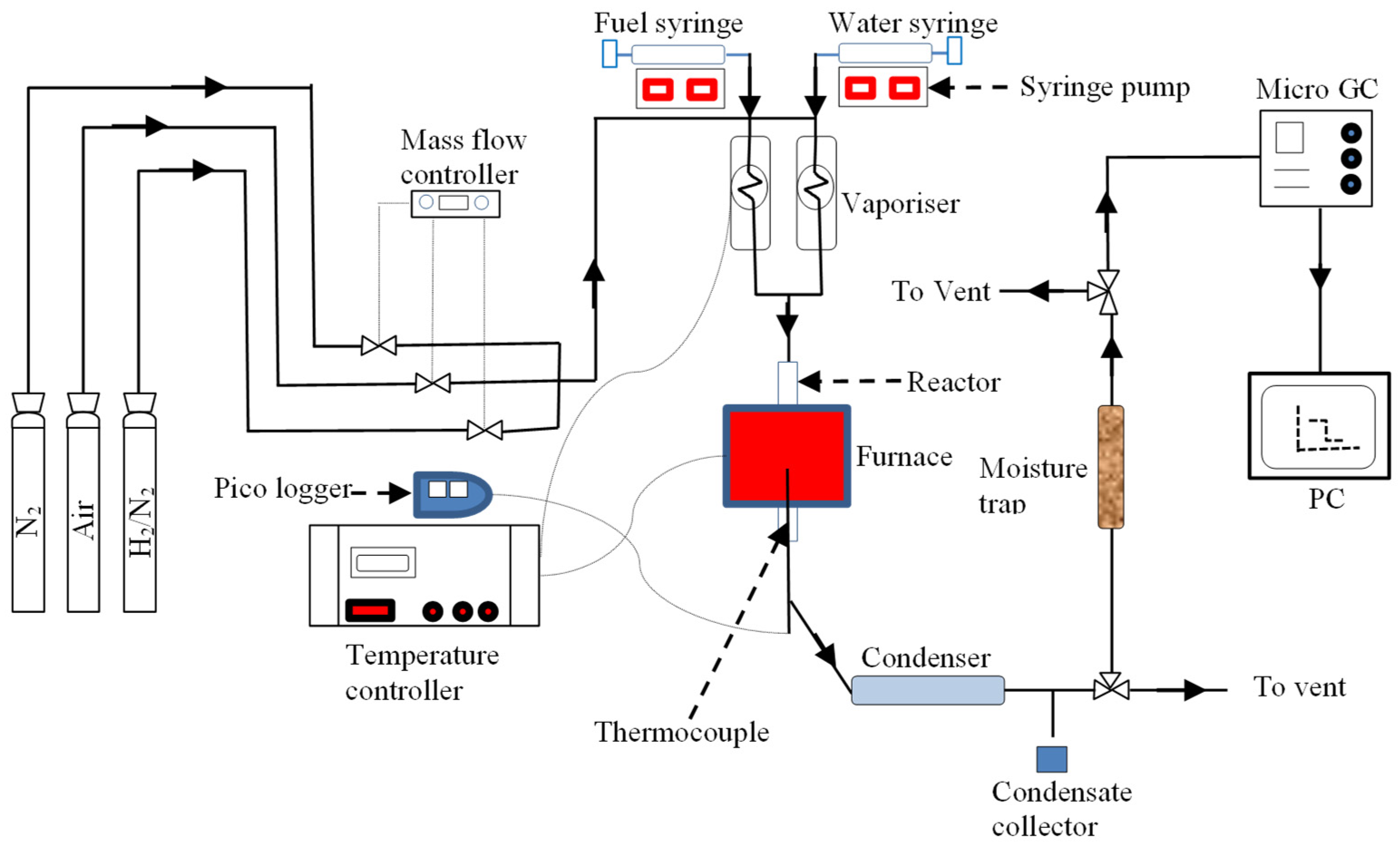
The schematic of the fixed bed reactor (experimental rig) used for ATR experiments (packed Ni catalyst pellets or Rh catalyst particles) is shown in Figure 10. The rig consists of a down flow 316 stainless steel tube reactor with 10 mm internal diameter placed inside an electric tube furnace (Elite Thermal Systems TSV10/20/85) to provide external heating.
For the experimental runs with the monoliths, a 22 mm o.d. stainless-steel reactor was fitted in place, substituting the reactor described in the previous paragraph. The internal diameter of the reactor was increased from 18- to 19-mm to fit the monolith diameter by drilling over one end of the reactor tube, and was enclosed inside a furnace TSV12/38/120 supplied by Elite Thermal Systems Ltd. No other modifications were performed on the existing rig except using appropriate tube fittings where necessary to connect the new reactor.
3.4. Gas Analysis
Dry product gas leaving the rig by way of the moisture removal was analysed using a micro gas chromatograph (Micro GC) supplied by Varian Instruments, UK (model CP 4900). The moisture is removed using a condenser operating on a 30 vol.% ethylene glycol/water mixture flowing counter clockwise from a chiller (Fisher Scientific 3016S) set at −2 °C, followed by a silica gel moisture trap. The micro-GC is equipped with two thermal conductivity detectors (TCD) and two columns which are a Molecular Sieve 5A plot column, for the analysis of H2, O2, N2, CH4, and CO; and a Pora Plot Q column for the detection of CO2, CH4, C2H6, C2H4, C3H8 and C3H6. Both columns were tuned on argon as carrier gas.
The molar flow in mol/s of the dry gases produced during the ATR experiments and measured by the on-line micro-GC were determined by nitrogen elemental balance. The acetic acid conversion to C1, C2 and C3 gases was determined by carbon balance, and water conversion was determined by hydrogen balance. However C2 and C3 gases were never detected by the GC analysis and do not appear in the carbon balance. The total hydrogen produced as both gaseous H2 and bonded hydrogen contained in organic gases (C1 to C3) is attributed to the acetic acid and water feeds. The value of water conversion so calculated was compared and found to be on par with that obtained by closing the balance on elemental O as proposed by [36]. The total organic carbon (TOC) content of the condensate recovered after each experiment was determined by differential method using a Hach Large IL 550 TOC/TIC analyser. This was used to complete the carbon balance and typically accounted for less than 1% of the carbon feed.
Hydrogen yield (YH2) was expressed either as a mass fraction of the input fuel (wt.%), a mole fraction of input fuel (mol H2/mol HAc), or molar fraction per input carbon (mol/mol C) according to Equations (2)–(4).
where represents the molar flow rate of a species i, e.g., is the molar flow rate of acetic acid into the reactor and is the molar mass of acetic acid in g mol−1.
H2 selectivity in the absence of C2 and C3 species in the product gases is given by:
Determination of selectivity of the C1 gases CO, CO2 and CH4 is illustrated for CO using the following equation:
where yi is the mol fractions of relevant species i.
The hourly space velocity was calculated both as weight hourly space velocity (WHSV, h−1) and gas hourly space velocity (GHSV, h−1) using the following equations:
The volumetric flow rates used in calculating the GHSV was determined for all feed (acetic acid, water, and air) at normal conditions of 20 °C and 1 atm.
Over all the experiments performed on the packed bed catalysts, the average carbon balance closure was within 5.1%, which included carbon on catalyst (TGA runs), in condensates (total organic carbon content) and in gas products (GC).
3.5. Experimental Procedure
3.5.1. Ni- and Rh-Based Catalysts Packed Bed Experiments
About 1.0 g of crushed pellets (Ni) or 0.2 g powder (Rh) catalyst was loaded into the tubular reactor and held in place between two 4 μm fibre diameter quartz wool plugs (m.p. 1050 °C) obtained from Fisher Scientific UK Ltd., weighing 0.09 g each. Blank experiments were performed using sand (SiO2) obtained from Sigma-Aldrich, Germany, with a 50–70 mesh size. Depending on the experiment, the catalyst was either used in a fresh (oxidised) or pre-reduced state using hydrogen as the reducing agent. When performing pre-reduction of the catalysts, the reactor bed was heated to 650 °C under continuous N2 flow after which the N2 was turned off and a 5 vol% H2 (in N2) gas mixture was turned on with its flow set at 200 cm3/min. Catalyst pre-reduction was monitored by on-line micro GC and was deemed complete when the recorded H2 reading increased to 5% up from an initial value of about 3% at the start of the process. Once the catalyst reduction was complete, the H2 gas mixture was switched off and the system was flushed under high flow of N2 until no H2 was detected by the micro-GC. With no more H2 detected, and the water and fuel vaporisers heated to 50 and 120 °C respectively, the N2 flow was turned off and the ATR experiment was started first by switching on the water syringe pump, followed by the air mass flow controller and finally the acetic acid syringe pump. For most experiments the acetic acid flow was set to 1.000 mL/h on the syringe pump and each experiment was performed for at least 2 h. The HAc flow was only changed when the effect of flow rate (hourly space velocity) was investigated. The acetic acid flow rate was used to set the steam and air flow rate based on the desired S/C and air equivalence ratio (λ) respectively. λ was determined as a molar ratio given by:
The stoichiometric oxygen is calculated using the complete oxidation reaction (‘COX’) i.e., full combustion of HAc to CO2 and H2O products.
3.5.2. Rh-Based Honeycomb Monoliths Experiments
The experimental procedure used to investigate the Rh monolithic reactor was similar to that with the Ni and Rh packed beds. The monolith was lowered into the stainless-steel reactor and held in the middle by the protruding wall marking the end of the initial tube drilling carried out to increase the tube’s internal diameter (from 18 to 19 mm). A thermocouple was fitted at the bottom to measure the bulk gas temperature in the reactor. This temperature reading corresponded to that of the hot product gases flowing out of the monolith and was used to control the electric furnace. As with the packed bed experiments, the set-up was leak tested at several points under a N2 flow of 200 cm3/min using a portable flowmeter. The furnace was heated to the desired reaction temperature while the fuel and water vaporisers were heated to 50 and 120 °C, respectively. All this was done under continuous flow of N2 and the syringe pumps and flow meters turned on once the set temperatures were achieved as no catalyst reduction was required.
4. Conclusions
The autothermal reforming (ATR) of acetic acid (HAc) as a model bio-oil compound was examined in this study using unstructured (packed bed particles) and structured (honeycomb monolith) Ni and Rh catalysts. Ni was chosen despite the growing literature on Co for ATR of HAc due to its better sustainability credentials. Despite its high cost, Rh was chosen due to successful previous studies of partial oxidation of oxygenates with short contact time and because of established routes of recycling of spent catalysts. The Nickel catalysts performed better in the absence of pre-reduction treatment, with the Rh catalysts showing little difference with regards to pre-reduction, thus the experiments were run directly on the ‘fresh’ catalysts. Partial oxidation runs (in absence of steam) resulted in much higher carbon deposition rates than the ATR runs, revealing the benefits of steam feed. Increasing feed molar steam to carbon ratio (S/C) from 1 to 2 yielded significantly higher increases in conversion than from 2 to 3, thus S/C of 2 was deemed the most cost effective. In same conditions of HAc, air and steam feed, the fresh RC-M, and Ni-CaAl and Rh-Al catalysts, in order from less-to-more performant, exhibited the highest HAc conversions (≥94%), and hydrogen yields exceeding 88% of the equilibrium value, the shortfall attributed due to methanation activity. The syngas production of these three catalysts was over 89% of the equilibrium value. Discrepancies in main gas products among the Ni and Rh packed bed and monolith catalysts revealed a higher activity of steam consuming reactions (steam reforming, water gas shift) in the Ni catalysts, closest to the predicted equilibrium. In contrast, the Rh catalysts exhibited much less carbon deposition rates than the Ni catalysts, and in turn the Ni-CaAl much less than the Ni-Al, the latter forcing an early end of run due to coking induced blockage. The nature of the deposited carbon was also different between Ni-Al (lumpy, dense), from the Ni-CaAl and Rh-Al (filamentous, non-flow obstructing). The ATR of HAc is a first step into successfully conducting ATR of bio-oils and a biomass reliant route to green syngas and hydrogen, where biomasses and bio-oils offer ease of storage of energy dense material at scale, and an alternative to other forms of intermittent-reliant green hydrogen routes. It is felt that further research into catalysts structures suitable and compatible with local scales of sustainable biomass sources would be worthwhile.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11121504/s1, Figure S1: Dry N2-free product gas flow composition with time on stream obtained during ATR of acetic acid flowing at 1 mL/h, S/C = 2, λ = 0.353 (Air flow = 22 mL/min), hot product gas at 570 °C and 1 bar. (a) Fresh Ni-Al. (b) Reduced Ni-Al. (c) Fresh Ni-CaAl. (d) Reduced Ni-CaAl, (e) Fresh Rh-Al, (f) Reduced Rh-Al, Figure S2: XRD patterns of (a) unused fresh Ni-Al, used Ni-Al (used Ni-Al (F)) and used reduced Ni-Al (used Ni-Al (R)). Unlabelled peaks belong to the α-Al2O3 support, Ni-CaAl XRD are not shown due to large amorphous content (b) Rh-Al fresh (F) in orange and reduced Rh-Al (F) catalyst in red compared to the rhodium-free γ-Al2O3 support in yellow. (c) Elemental mapping of Rh-Al showing good dispersion of Rh, Figure S3: Dry N2-free molar flow product gas profiles with time on stream obtained for POX of acetic acid flowing at 1 mL/h, λ = 0.353 (Air flow = 22 mL/min), 570 °C and 1 bar. (a) Fresh Ni-Al (b) Fresh Ni-CaAl, (c) Fresh Rh-Al, Figure S4: XRD patterns of Ni-Al catalyst for experiments performed using fresh catalyst at S/C 1, 2 and 3 and 1 bar. Acetic acid flow, 1 mL/h, with corresponding λ and temperature values of (0.340, 610 °C), (0.353, 570 °C) and (0.374, 545 °C). All unlabelled peaks belong to the α-Al2O3 support, Figure S5: H2 purity, H2 yield, and conversion of acetic acid, water, and oxygen with WHSV (GHSV) for the (a) Ni-Al, (b) Ni-CaAl and (c) Rh-Al catalysts in packed bed. All flows at S/C = 2 and λ = 0.353 corresponding to a reactor exit temperature of 570 °C, Figure S6: Dry N2-free product gas with time on stream obtained during ATR of acetic acid flowing at 2 mL/h, S/C = 2, λ = 0.353 (Air flow = 43.3 mL/min), hot product gas at 570 °C and 1 bar. (a) R-M (b) RC-M.
Author Contributions
Conceptualisation, L.N.T. and V.D.; methodology, L.N.T.; software, L.N.T.; formal analysis, L.N.T.; investigation, L.N.T.; writing—original draft preparation, L.N.T. and E.R.-M.; writing—review and editing, M.V.T. methodology, editing, E.R.-M. and V.D.; supervision, V.D.; funding acquisition, V.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Engineering and Physical Sciences Research Council (EPSRC), grant number EP/R030243/1 and Consejo Nacional de Ciencia y Tecnología (CONACYT, PhD scholarship of E.R.-M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Associated data is available at https://doi.org/10.5518/1075, accessed on 30 November 2021.
Acknowledgments
Our thanks to Sergio Ramírez Solís, for training support of catalyst synthesis and XRD methodology, whose PhD was also funded by CONACYT.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kang, I.; Bae, J. Autothermal reforming study of diesel for fuel cell application. J. Power Sources 2006, 159, 1283–1290. [Google Scholar] [CrossRef]
- Czernik, S.; French, R. Distributed production of hydrogen by auto-thermal reforming of fast pyrolysis bio-oil. Int. J. Hydrogen Energy 2014, 39, 744–750. [Google Scholar] [CrossRef]
- Bridgwater, A.V.; Meier, D.; Radlein, D. An overview of fast pyrolysis of biomass. Org. Geochem. 1999, 30, 1479–1493. [Google Scholar] [CrossRef]
- Xiu, S.; Shahbazi, A. Bio-oil production and upgrading research: A review. Renew. Sustain. Energy Rev. 2012, 16, 4406–4414. [Google Scholar] [CrossRef]
- Hu, X.; Gholizadeh, M. Biomass pyrolysis: A review of the process development and challenges from initial researches up to the commercialisation stage. J. Energy Chem. 2019, 39, 109–143. [Google Scholar] [CrossRef] [Green Version]
- Pimenidou, P.; Dupont, V. Characterisation of palm empty fruit bunch (PEFB) and pinewood bio-oils and kinetics of their thermal degradation. Bioresour. Technol. 2012, 109, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Zin, R.M.; Lea-Langton, A.; Dupont, V.; Twigg, M.V. High hydrogen yield and purity from palm empty fruit bunch and pine pyrolysis oils. Int. J. Hydrogen Energy 2012, 37, 10627–10638. [Google Scholar] [CrossRef] [Green Version]
- Basagiannis, A.C.; Verykios, X.E. Reforming reactions of acetic acid on nickel catalysts over a wide temperature range. Appl. Catal. A Gen. 2006, 308, 182–193. [Google Scholar] [CrossRef]
- Trane, R.; Dahl, S.; Skjøth-Rasmussen, M.S.; Jensen, A.D. Catalytic steam reforming of bio-oil. Int. J. Hydrogen Energy 2012, 37, 6447–6472. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, W.; Jia, X.Y.; Chen, B.Q.; An, S.; Xie, X.Y.; Huang, L.H. Ca-Al layered double hydroxides-derived Ni-based catalysts for hydrogen production via auto-thermal reforming of acetic acid. Int. J. Hydrogen Energy 2019, 44, 20007–20016. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Q.; An, S.; Yang, H.; Huang, L.H. Effect of Fe on CoxAl3FeyOm +/-delta Catalysts for Hydrogen Production by Auto-thermal Reforming of Acetic Acid. J. Inorg. Mater. 2019, 34, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.M.; Chen, H.; Jia, X.Y.; Wang, Q.; Huang, L.H. Y-Mn-O Supported Ni-Based Catalyst for Hydrogen Production via Auto-thermal Reforming of Acetic Acid. Chin. J. Inorg. Chem. 2021, 37, 555–560. [Google Scholar]
- Hu, X.M.; Yang, J.L.; Sun, W.J.; Wang, N.; An, S.; Wang, Q.; Zhang, Y.; Xie, X.Y.; Huang, L.H. Y-Zr-O solid solution supported Ni-based catalysts for hydrogen production via auto-thermal reforming of acetic acid. Appl. Catal. B-Environ. 2020, 278, 119264. [Google Scholar] [CrossRef]
- Song, Y.X.; Chen, B.Q.; Hu, X.M.; Wang, Q.; Xie, X.Y.; Dai, H.; Huang, L.H. Highly Efficient Al-Doped Ni-Mn-O Catalysts for Auto-Thermal Reforming of Acetic Acid: Role of MnAl2O4 for Stability of Ni Species. Energy Fuels 2020, 34, 14647–14655. [Google Scholar] [CrossRef]
- An, S.; Zhang, Y.; Hu, X.M.; Xie, X.Y.; Wang, Q.; Chen, H.; Huang, L.H. Durable Mn(II)Cr(III)O-x composites-supported Ni-based catalysts with wide dynamic range for hydrogen production via auto-thermal reforming of acetic acid. Fuel 2020, 278, 118227. [Google Scholar] [CrossRef]
- Li, H.G.; Jia, X.Y.; Wang, N.; Chen, B.Q.; Xie, X.Y.; Wang, Q.; Huang, L.H. Auto-thermal reforming of acetic acid over hydrotalcites-derived co-based catalyst: A stable and anti-coking Co/Sr-Alx-O catalyst. Appl. Catal. B-Environ. 2020, 267, 118370. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhong, X.Y.; Xie, X.Y.; Jia, X.Y.; Chen, B.Q.; Wang, N.; Huang, L.H. Auto-thermal reforming of acetic acid for hydrogen production by ordered mesoporous Ni-xSm-Al-O catalysts: Effect of samarium promotion. Renew. Energy 2020, 145, 2316–2326. [Google Scholar] [CrossRef]
- Banza Lubaba Nkulu, C.; Casas, L.; Haufroid, V.; de Putter, T.; Saenen, N.D.; Kayembe-Kitenge, T.; Obadia, P.M.; Mukoma, D.K.W.; Ilunga, J.-M.L.; Nawrot, T.S.; et al. Sustainability of artisanal mining of cobalt in DR Congo. Nat. Sustain. 2018, 1, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Shedd, K.B. Cobalt, U.S. Geological Survey Minerals Yearbook—2016; U.S. Geological Survey, Department of the Interior: Reston, VA, USA, 2016. [Google Scholar]
- Tkaczyk, A.H.; Bartl, A.; Amato, A.; Lapkovskis, V.; Petranikova, M. Sustainability evaluation of essential critical raw materials: Cobalt, niobium, tungsten and rare earth elements. J. Phys. D Appl. Phys. 2018, 51, 203001. [Google Scholar] [CrossRef] [Green Version]
- European Comission. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the regions on the 2017 List of Critical Raw Matrials for the EU; COM(2017) 490 final 2017; European Comission: Brussel, Belgium, 2017. [Google Scholar]
- Zeng, X.; Li, J. On the sustainability of cobalt utilization in China. Resour. Conserv. Recycl. 2015, 104, 12–18. [Google Scholar] [CrossRef]
- Miceli, M.; Frontera, P.; Macario, A.; Malara, A. Recovery/Reuse of Heterogeneous Supported Spent Catalysts. Catalysts 2021, 11, 591. [Google Scholar] [CrossRef]
- Saguru, C.; Ndlovu, S.; Moropeng, D. A review of recent studies into hydrometallurgical methods for recovering PGMs from used catalytic converters. Hydrometallurgy 2018, 182, 44–56. [Google Scholar] [CrossRef]
- Jia, L.-P.; Huang, J.-J.; Ma, Z.-L.; Liu, X.-H.; Chen, X.-Y.; Li, J.-T.; He, L.-H.; Zhao, Z.-W. Research and development trends of hydrometallurgy: An overview based on Hydrometallurgy literature from 1975 to 2019. Trans. Nonferrous Met. Soc. China 2020, 30, 3147–3160. [Google Scholar] [CrossRef]
- Viera, M.; Pogliani, C.; Donati, E. Recovery Of Zinc, Nickel, Cobalt And Other Metals By Bioleaching. In Microbial Processing of Metal Sulfides; Donati, E., Sand, W., Eds.; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Sverdrup, H.U.; Olafsdottir, A.H. Assessing the Long-Term Global Sustainability of the Production and Supply for Stainless Steel. BioPhys. Econ. Resour. Qual. 2019, 4, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Dupont, V. Nickel catalyst auto-reduction during steam reforming of bio-oil model compound acetic acid. Int. J. Hydrogen Energy 2013, 38, 15160–15172. [Google Scholar] [CrossRef]
- Cheng, F.; Dupont, V. Steam Reforming of Bio-Compounds with Auto-Reduced Nickel Catalyst. Catalysts 2017, 7, 114. [Google Scholar] [CrossRef]
- Cheng, F.; Dupont, V.; Twigg, M.V. Direct reduction of nickel catalyst with model bio-compounds. Appl. Catal. B Environ. 2017, 200, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Omoniyi, O.A.; Dupont, V. Chemical looping steam reforming of acetic acid in a packed bed reactor. Appl. Catal. B Environ. 2018, 226, 258–268. [Google Scholar] [CrossRef]
- Omoniyi, O.A.; Dupont, V. Optimised cycling stability of sorption enhanced chemical looping steam reforming of acetic acid in a packed bed reactor. Appl. Catal. B Environ. 2019, 242, 397–409. [Google Scholar] [CrossRef]
- Zhao, Z.; Qiu, Z.; Yang, J.; Ma, B.; Li, Z.; Lu, S.; Xu, Y.; Cao, L.; Zhang, W. Recovery of Rare Earth Element Cerium from Spent Automotive Exhaust Catalysts Using a Novel Method. Waste Biomass Valorization 2019, 11, 4967–4976. [Google Scholar] [CrossRef]
- Prasetyo, E.; Anderson, C. Platinum Group Elements Recovery from Used Catalytic Converters by Acidic Fusion and Leaching. Metals 2020, 10, 485. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, A.; Moulijn, J.A. Structured Catalysts and Reactors, 2nd ed.; Chemical Industries; CRC press, Taylor & Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar]
- Rennard, D.C.; Kruger, J.S.; Schmidt, L.D. Autothermal catalytic partial oxidation of glycerol to syngas and to non-equilibrium products. ChemSusChem 2009, 2, 89–98. [Google Scholar] [CrossRef]
- Biegger, P.; Kirchbacher, F.; Medved, A.; Miltner, M.; Lehner, M.; Harasek, M. Development of Honeycomb Methanation Catalyst and Its Application in Power to Gas Systems. Energies 2018, 11, 1679. [Google Scholar] [CrossRef] [Green Version]
- Doherty, W.; Reynolds, A.; Kennedy, D. Aspen Plus Simulation of Biomass Gasification in a Steam Blown Dual Fluidised Bed. In Materials and Processes for Energy: Communicating Current Research and Technological Developments; Mendez-Vilas, A., Ed.; Formatex Research Centre: Montreal, Canada, 2013; ISBN 978-84-939843-7-3. [Google Scholar]
- Halvorsen, B.M.; Adhikari, U.; Eikeland, M.S. Gasification of Biomass for Production of Syngas for Biofuel. In Proceedings of the 56th Conference on Simulation and Modelling, Linköping, Sweden, 7–9 October 2015. [Google Scholar]
- Vagia, E.; Lemonidou, A. Thermodynamic analysis of hydrogen production via autothermal steam reforming of selected components of aqueous bio-oil fraction. Int. J. Hydrogen Energy 2008, 33, 2489–2500. [Google Scholar] [CrossRef]
- Specchia, S.; Vella, L.D.; de Rogatis, L.; Montini, T.; Specchia, V.; Fornasiero, P. Rh-based catalysts for syngas production via SCT-CPO reactors. Catal. Today 2010, 155, 101–107. [Google Scholar] [CrossRef]
- Bengaard, H.S.; Nørskov, J.K.; Sehested, J.; Clausen, B.S.; Nielsen, L.P.; Molenbroek, A.M.; Rostrup-Nielsen, J.R. Steam Reforming and Graphite Formation on Ni Catalysts. J. Catal. 2002, 209, 365–384. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Activity of Nickel Catalysts for Steam Reforming of Hydrocarbons. J. Catal. 1973, 31, 173–199. [Google Scholar] [CrossRef]
- Vis, J.C.; Buk, H.F.J.V.; Huizinga, T.; van Grondelle, J.; Prins, R. Reduction and oxidation of Rh/Al2O3 and Rh/TiO2 catalysts as studied by temperature-programmed reduction and oxidation. J. Mol. Catal. 1984, 25, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Carpenter, J.E. The oxidation state and catalytic activity of supported rhodium. J. Catal. 1983, 80, 472–478. [Google Scholar] [CrossRef]
- Zheng, Q.; Farrauto, R.; Deeba, M.; Valsamakis, I. Part I: A Comparative Thermal Aging Study on the Regenerability of Rh/Al2O3 and Rh/CexOy-ZrO2 as Model Catalysts for Automotive Three Way Catalysts. Catalysts 2015, 5, 1770–1796. [Google Scholar] [CrossRef]
- Gustafson, J.; Balmes, O.; Zhang, C.; Shipilin, M.; Schaefer, A.; Hagman, B.; Merte, L.R.; Martin, N.M.; Carlsson, P.-A.; Jankowski, M.; et al. The Role of Oxides in Catalytic CO Oxidation over Rhodium and Palladium. ACS Catal. 2018, 8, 4438–4445. [Google Scholar] [CrossRef]
- Rood, S.; Eslava, S.; Manigrasso, A.; Bannister, C. Recent advances in gasoline three-way catalyst formulation: A review. Proc. Inst. Mech. Eng. Part D J. Automob. Eng. 2019, 234, 936–949. [Google Scholar] [CrossRef]
- Medrano, J.; Oliva, M.; Ruiz, J.; Garcia, L.; Arauzo, J. Catalytic steam reforming of acetic acid in a fluidized bed reactor with oxygen addition. Int. J. Hydrogen Energy 2008, 33, 4387–4396. [Google Scholar] [CrossRef]
- Cabello, A.; Gayán, P.; García-Labiano, F.; de Diego, L.F.; Abad, A.; Izquierdo, M.T.; Adánez, J. Relevance of the catalytic activity on the performance of a NiO/CaAl2O4 oxygen carrier in a CLC process. Appl. Catal. B Environ. 2014, 147, 980–987. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.Z.; Yu, C.C.; Liu, Y.; Shen, S.K. Mechanistic investigations on the partial oxidation of methane to synthesis gas over a nickel-on-alumina catalyst. Appl. Catal. Gen. 1998, 174, 121–128. [Google Scholar] [CrossRef]
- Gutierrez, A.; Karinen, R.; Airaksinen, S.; Kaila, R.; Krause, A.O.I. Autothermal reforming of ethanol on noble metal catalysts. Int. J. Hydrogen Energy 2011, 36, 8967–8977. [Google Scholar] [CrossRef]
- Liguras, D.K.; Goundani, K.; Verykios, X.E. Production of hydrogen for fuel cells by catalytic partial oxidation of ethanol over structured Ni catalysts. J. Power Sources 2004, 130, 30–37. [Google Scholar] [CrossRef]
- Marquevich, M.; Czernik, S.; Chornet, E.; Montane, D. Hydrogen from biomass: Steam reforming of model compounds of fast-pyrolysis oil. Energy Fuels 1999, 13, 1160–1166. [Google Scholar] [CrossRef]
- Kaila, R.K.; Gutiérrez, A.; Slioor, R.; Kemell, M.; Leskelä, M.; Krause, A.O.I. Zirconia-supported bimetallic RhPt catalysts: Characterization and testing in autothermal reforming of simulated gasoline. Appl. Catal. B Environ. 2008, 84, 223–232. [Google Scholar] [CrossRef]
- Cavallaro, S. Ethanol Steam Reforming on Rh/Al2O3Catalysts. Energy Fuels 2000, 14, 1195–1199. [Google Scholar] [CrossRef]
- Aupretre, F.; Descorme, C.; Duprez, D. Hydrogen production for fuel cells from the catalytic ethanol steam reforming. Top. Catal. 2004, 30–31, 487–491. [Google Scholar] [CrossRef]
- Hohn, K.L.; Schmidt, L.D. Partial oxidation of methane to syngas at high space velocities over Rh-coated spheres. Appl. Catal. Gen. 2001, 211, 53–68. [Google Scholar] [CrossRef]
- Tande, L.N.; Resendiz-Mora, E.; Dupont, V. Bioh2, Heat and Power from Palm Empty Fruit Bunch via Pyrolysis-Autothermal Reforming: Plant Simulation, Experiments, and CO2 Mitigation. Energies 2021, 14, 4767. [Google Scholar] [CrossRef]
Figure 1.
Syngas equilibrium flow composition (mol s−1) and temperature (in °C) obtained at S/C = 2 while varying λ from 0 to 1 for ATR of HAc (feed rate 1 mL/h). HAc is fully converted, hence not shown.
Figure 1.
Syngas equilibrium flow composition (mol s−1) and temperature (in °C) obtained at S/C = 2 while varying λ from 0 to 1 for ATR of HAc (feed rate 1 mL/h). HAc is fully converted, hence not shown.

Figure 2.
Conversion in % of HAc (1 mL/h), water and oxygen using 1 g Ni-Al, 1 g Ni-CaAl, 0.2 g Rh-Al catalysts, 1 g sand (Blank experiment ‘Blk’), and equilibrium (Equ) at S/C 2, reactor exit gas temperature of 570 °C, λ = 0.353 and pressure of 1 bar.
Figure 2.
Conversion in % of HAc (1 mL/h), water and oxygen using 1 g Ni-Al, 1 g Ni-CaAl, 0.2 g Rh-Al catalysts, 1 g sand (Blank experiment ‘Blk’), and equilibrium (Equ) at S/C 2, reactor exit gas temperature of 570 °C, λ = 0.353 and pressure of 1 bar.

Figure 3.
Average nitrogen-free dry product gas composition obtained during ATR of acetic acid flowing at 1 mL/h using Ni-Al, Ni-CaAl and Rh-Al catalyst in fresh (F) and reduced (R) state. S/C = 2, λ = 0.353 (Air flow = 22 mL/min), 570 °C and 1 bar.
Figure 3.
Average nitrogen-free dry product gas composition obtained during ATR of acetic acid flowing at 1 mL/h using Ni-Al, Ni-CaAl and Rh-Al catalyst in fresh (F) and reduced (R) state. S/C = 2, λ = 0.353 (Air flow = 22 mL/min), 570 °C and 1 bar.

Figure 4.
Average molar production rates (a) H2, (b) CO, (c) CO2 and (d) CH4 for the packed bed Ni and Rh catalysts compared to equilibrium. HAc flow, 1 mL/h with corresponding λ and temperature values of (0.340, 610 °C), (0.353, 570 °C) and (0.374, 545 °C) for S/C of 1, 2 and 3 respectively.
Figure 4.
Average molar production rates (a) H2, (b) CO, (c) CO2 and (d) CH4 for the packed bed Ni and Rh catalysts compared to equilibrium. HAc flow, 1 mL/h with corresponding λ and temperature values of (0.340, 610 °C), (0.353, 570 °C) and (0.374, 545 °C) for S/C of 1, 2 and 3 respectively.


Figure 5.
Effect of increasing WHSV (and GHSV), on CO, CO2 and CH4 selectivity or dry gas composition (%) and carbon formation (mg g(cat)−1h−1). All flows at S/C = 2 and λ = 0.353 corresponding to a reactor exit temperature of 570 °C. (a) Ni-Al (b) Ni-CaAl (c) Rh-Al.
Figure 5.
Effect of increasing WHSV (and GHSV), on CO, CO2 and CH4 selectivity or dry gas composition (%) and carbon formation (mg g(cat)−1h−1). All flows at S/C = 2 and λ = 0.353 corresponding to a reactor exit temperature of 570 °C. (a) Ni-Al (b) Ni-CaAl (c) Rh-Al.

Figure 6.
Naked eye (inserts) and SEM images of the used (a) Ni-Al, (b) Ni-CaAl, and (c) Rh-Al catalysts in packed bed. Acetic acid flow, 1 mL/h, 1 g of catalyst, S/C = 2, λ = 0.353 (Air flow = 22 mL/min), 570 °C and 1 bar.
Figure 6.
Naked eye (inserts) and SEM images of the used (a) Ni-Al, (b) Ni-CaAl, and (c) Rh-Al catalysts in packed bed. Acetic acid flow, 1 mL/h, 1 g of catalyst, S/C = 2, λ = 0.353 (Air flow = 22 mL/min), 570 °C and 1 bar.

Figure 7.
Temperature (in °C, right ordinate) and conversions of HAc, O2, and H2O (exp. and equil., left ordinate) using R-M and RC-M monoliths for ATR with water flow set at S/C molar ratio of 1, 2, 3 and pressure of 1 bar. S/C is given in parentheses below the catalyst name.
Figure 7.
Temperature (in °C, right ordinate) and conversions of HAc, O2, and H2O (exp. and equil., left ordinate) using R-M and RC-M monoliths for ATR with water flow set at S/C molar ratio of 1, 2, 3 and pressure of 1 bar. S/C is given in parentheses below the catalyst name.

Figure 8.
Product distribution and H2 yield obtained during ATR of HAc flowing at 2 mL/h, S/C shown in brackets = 1,2,3, and equivalence ratio λ = 0.353 (Air flow = 43.3 mL/min), hot product gas at 570 °C and 1 bar.
Figure 8.
Product distribution and H2 yield obtained during ATR of HAc flowing at 2 mL/h, S/C shown in brackets = 1,2,3, and equivalence ratio λ = 0.353 (Air flow = 43.3 mL/min), hot product gas at 570 °C and 1 bar.

Figure 9.
Influence of GHSV on feed conversion and dry product gas selectivity. S/C = 2, λ = 0.353 and hot product gas at 570 °C.
Figure 9.
Influence of GHSV on feed conversion and dry product gas selectivity. S/C = 2, λ = 0.353 and hot product gas at 570 °C.

Figure 10.
Schematic diagram of experimental rig.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Input values used for equilibrium analysis in Aspen flows used for all reactants.
| Component | Flow, mol/s | Flow, mL/h (NTP) |
|---|---|---|
| Acetic acid, liq. | 4.85 × 10−6 | 1.000 |
| Water (S/C = 2), liq. | 1.941 × 10−5 | 1.261 |
| O2 needed for COX *, STP | 9.71 × 10−6 | 840 |
| Air needed for COX *, STP | 4.62 × 10−5 | 3999.0 |
* COX is complete oxidation to CO2 and H2O, air is 79% N2/21%O2 (mol).
Table 2.
Morphological properties of Ni and Rh packed bed catalysts.
| Catalyst | BET (m2/g) | BJH Surface Area (m2/g) | Pore Size (nm) | Pore Volume (cm3/g) |
|---|---|---|---|---|
| Ni-Al (fresh) | 4.251 | 4.485 | 1.372 | 0.012 |
| Ni-Al (reduced) | 3.756 | 4.125 | 1.255 | 0.011 |
| Ni-CaAl (fresh) | 24.239 | 19.39 | 0.738 | 0.052 |
| Ni-CaAl (reduced) | 31.652 | 32.145 | 1.215 | 0.061 |
| Rh-Al (fresh) | 228.8 | 245.4 | 6.001 | 0.4386 |
| Rh-Al (reduced) | 229.3 | 244.4 | 6.056 | 0.7930 |
| γ-Al2O3 | 303.9 | 322.6 | 6.175 | 1.051 |
Table 3.
HAc conversion, Products distribution, H2/CO and carbon deposition (CD) of the POX of HAc on fresh packed bed Ni and Rh catalysts. Acetic acid flow 1 mL/h, λ = 0.353 (Air flow = 22 mL/min), hot product gas at 570 °C. CD from ATR at S/C 2 shown for comparison.
Table 3.
HAc conversion, Products distribution, H2/CO and carbon deposition (CD) of the POX of HAc on fresh packed bed Ni and Rh catalysts. Acetic acid flow 1 mL/h, λ = 0.353 (Air flow = 22 mL/min), hot product gas at 570 °C. CD from ATR at S/C 2 shown for comparison.
| Catalyst | Conv, % HAc | Product Distribution, mol% | CD, mg/(g(cat).h) ATR | |||||
|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | CO2 | H2/CO | POX | |||
| Ni-Al | 83.6 | 36.1 | 14.4 | 6.6 | 43.0 | 2.5 | 50.1 | 25.7 |
| Ni-CaAl | 91.1 | 34.7 | 16.7 | 8.5 | 40.0 | 2.1 | 34.9 | 13.9 |
| Rh-Al | 88.5 | 32.0 | 15.9 | 8.8 | 43.3 | 2.0 | 28.3 | 5.5 |
| Equilibrium | 86.3 | 38.0 | 18.5 | 4.2 | 39.3 | 2.1 | 0.0 | 0.0 |
Table 4.
Flows for ATR using R-M and RC-M monoliths (HAc 2 mL/h).
| ATR Condition | Water, mL/h | Air, mL/min | S/C | λ | Temperature, °C |
|---|---|---|---|---|---|
| 1 | 1.257 | 42.3 | 1 | 0.341 | 613 |
| 2 | 2.514 | 43.3 | 2 | 0.353 | 570 |
| 3 | 3.771 | 46.4 | 3 | 0.375 | 546 |
Table 5.
Products selectivity and hydrogen yield obtained for ATR of acetic acid flowing at 2 mL/h, S/C = 2, λ = 0.353 (Air flow = 43.3 mL/min), hot product gas at 570 °C and 1 bar.
Table 5.
Products selectivity and hydrogen yield obtained for ATR of acetic acid flowing at 2 mL/h, S/C = 2, λ = 0.353 (Air flow = 43.3 mL/min), hot product gas at 570 °C and 1 bar.
| Conversion % | Selectivity % | Syngas mol/s H2 + CO + CH4 | H2 Yield wt.% | Products Ratio | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HAc | H2O | H2 | CO | CO2 | CH4 | H2/CO | CO2/CO | |||
| Equil | 100.0 | 5.4 | 98.2 | 14.6 | 83.4 | 2.0 | 2.40 × 10−5 | 7.2 | 7.3 | 5.7 |
| R-M | 92.6 | −0.4 | 98.8 | 16.3 | 82.5 | 1.2 | 2.13 × 10−5 | 6.2 | 6.1 | 5.1 |
| RC-M | 93.7 | 0.5 | 97.5 | 14.2 | 83.4 | 2.4 | 2.12 × 10−5 | 6.3 | 6.7 | 5.9 |
| Rh/Al | 100.0 | 3.4 | 95.2 | 16.7 | 78.8 | 4.5 | 2.14 × 10−5 | 6.5 | 5.3 | 4.7 |
| Ni/Al | 90.5 | 2.2 | 96.3 | 9.1 | 87.2 | 3.7 | 2.01 × 10−5 | 6.0 | 10.7 | 9.6 |
| Ni/CaAl | 94.0 | 2.0 | 95.9 | 11.9 | 84.0 | 4.2 | 2.16 × 10−5 | 6.6 | 8.1 | 7.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tande, L.N.; Resendiz-Mora, E.; Dupont, V.; Twigg, M.V. Autothermal Reforming of Acetic Acid to Hydrogen and Syngas on Ni and Rh Catalysts. Catalysts 2021, 11, 1504. https://doi.org/10.3390/catal11121504
AMA Style
Tande LN, Resendiz-Mora E, Dupont V, Twigg MV. Autothermal Reforming of Acetic Acid to Hydrogen and Syngas on Ni and Rh Catalysts. Catalysts. 2021; 11(12):1504. https://doi.org/10.3390/catal11121504
Chicago/Turabian StyleTande, Lifita N., Erik Resendiz-Mora, Valerie Dupont, and Martyn V. Twigg. 2021. "Autothermal Reforming of Acetic Acid to Hydrogen and Syngas on Ni and Rh Catalysts" Catalysts 11, no. 12: 1504. https://doi.org/10.3390/catal11121504
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.