The Effects of the Metal Ion Substitution into the Active Site of Metalloenzymes: A Theoretical Insight on Some Selected Cases




Abstract
:1. Introduction
2. Computational Protocol
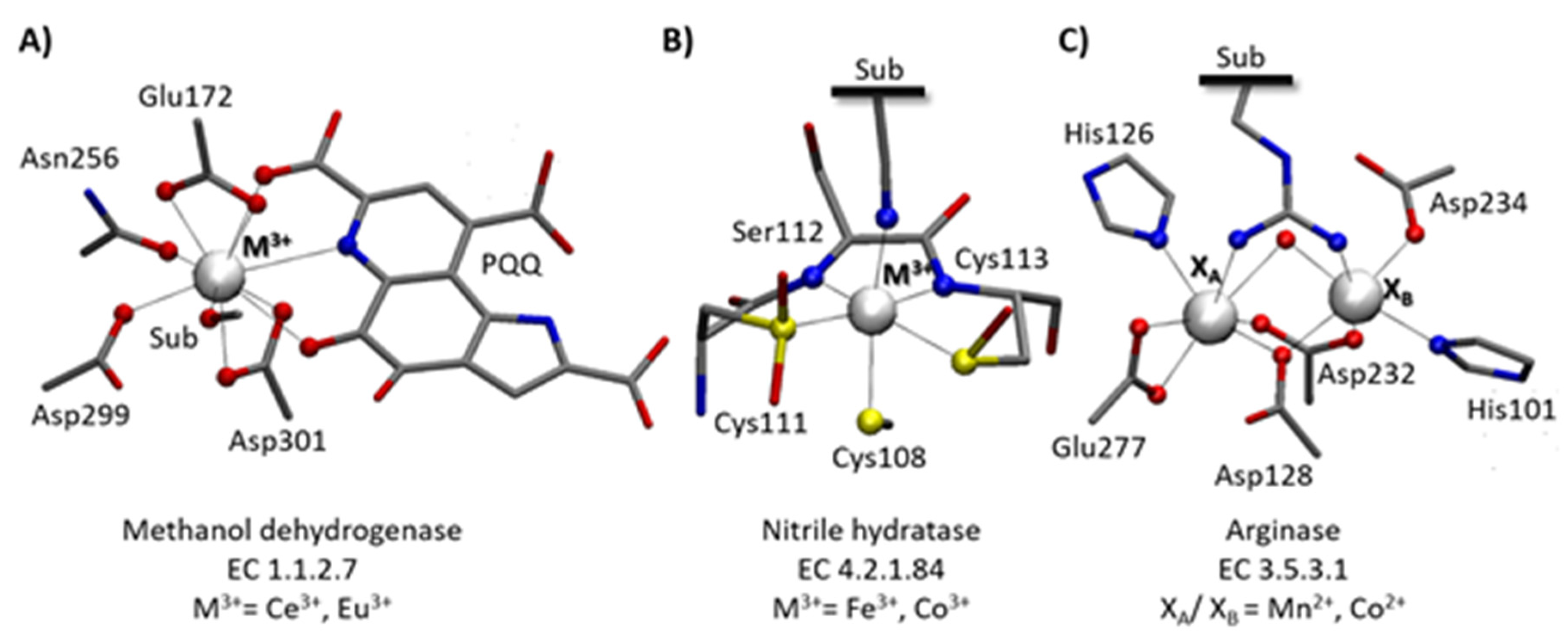
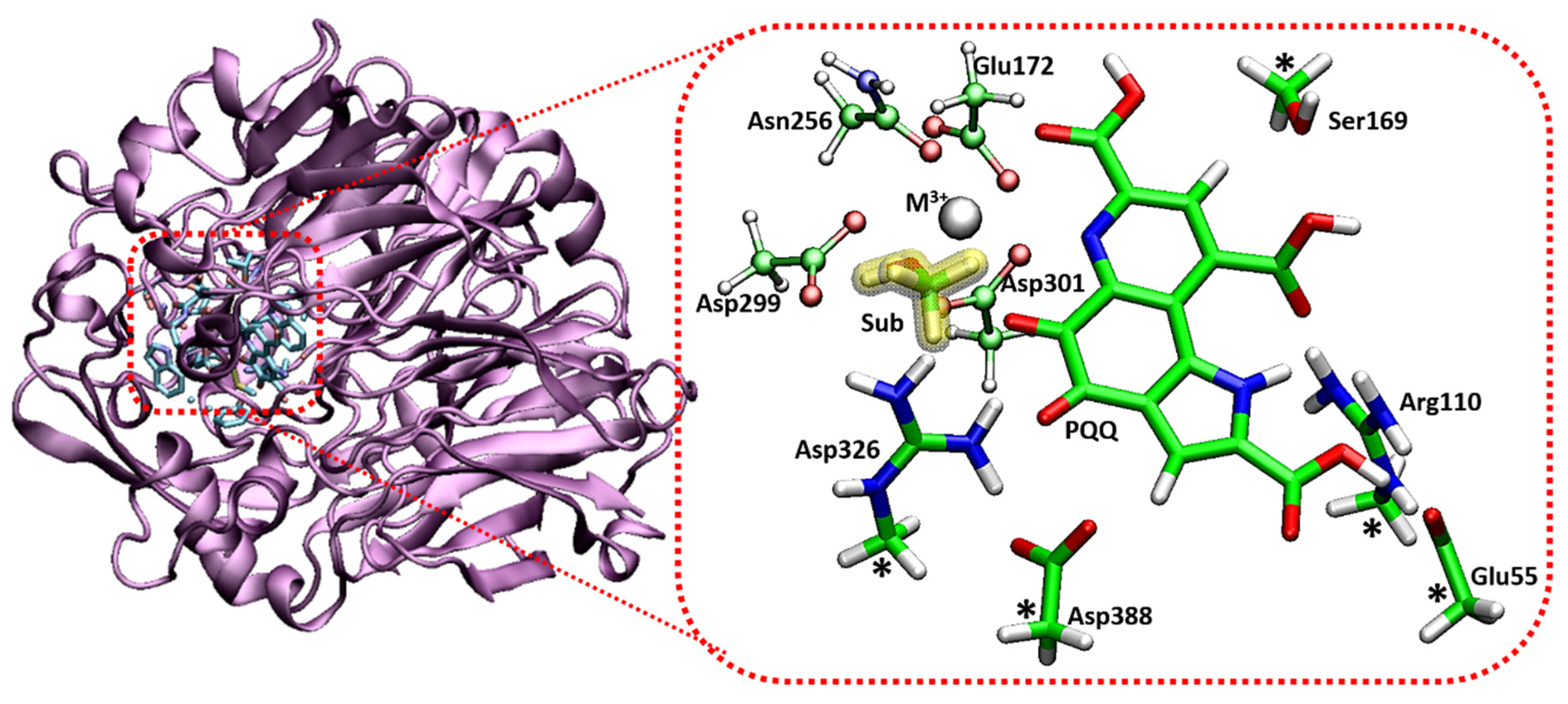
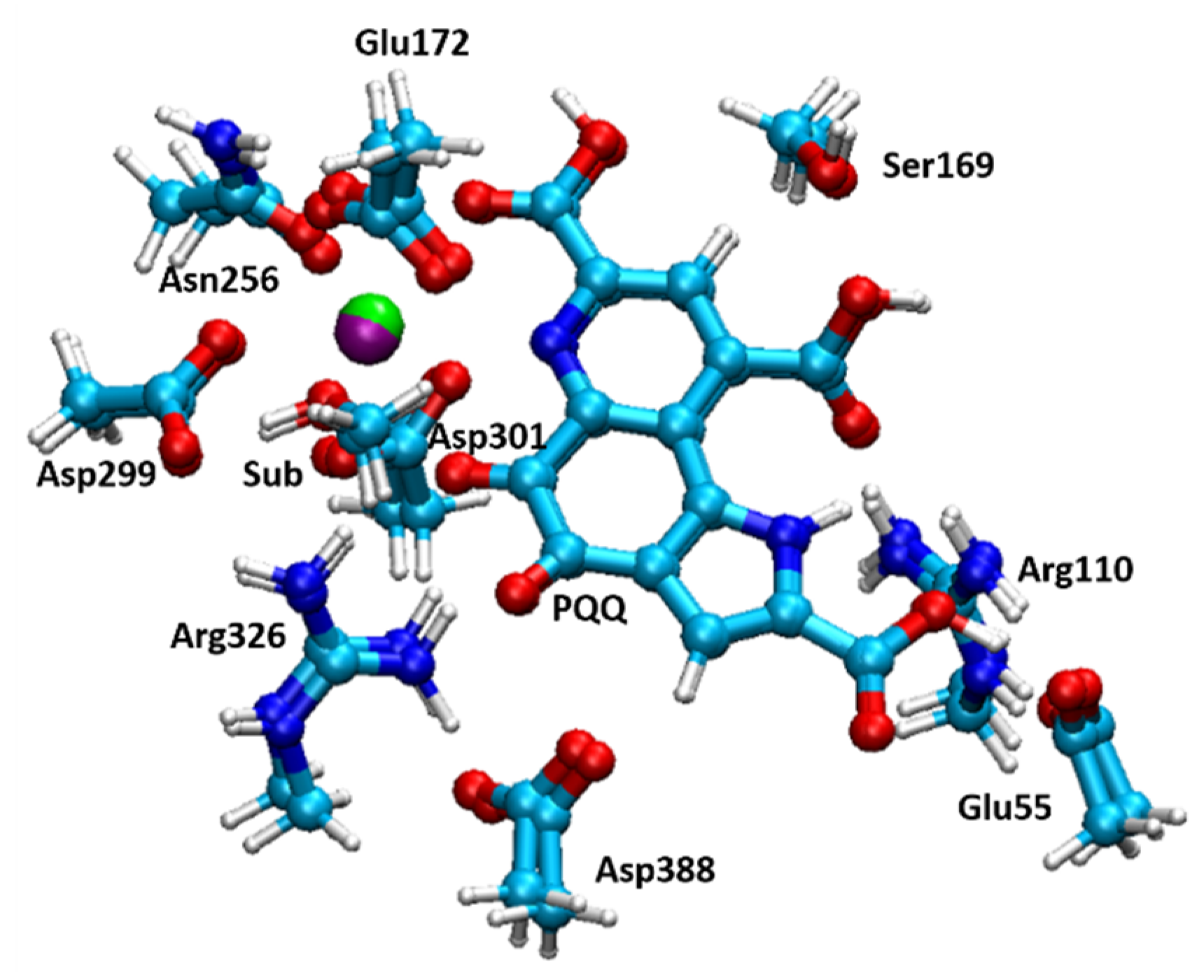
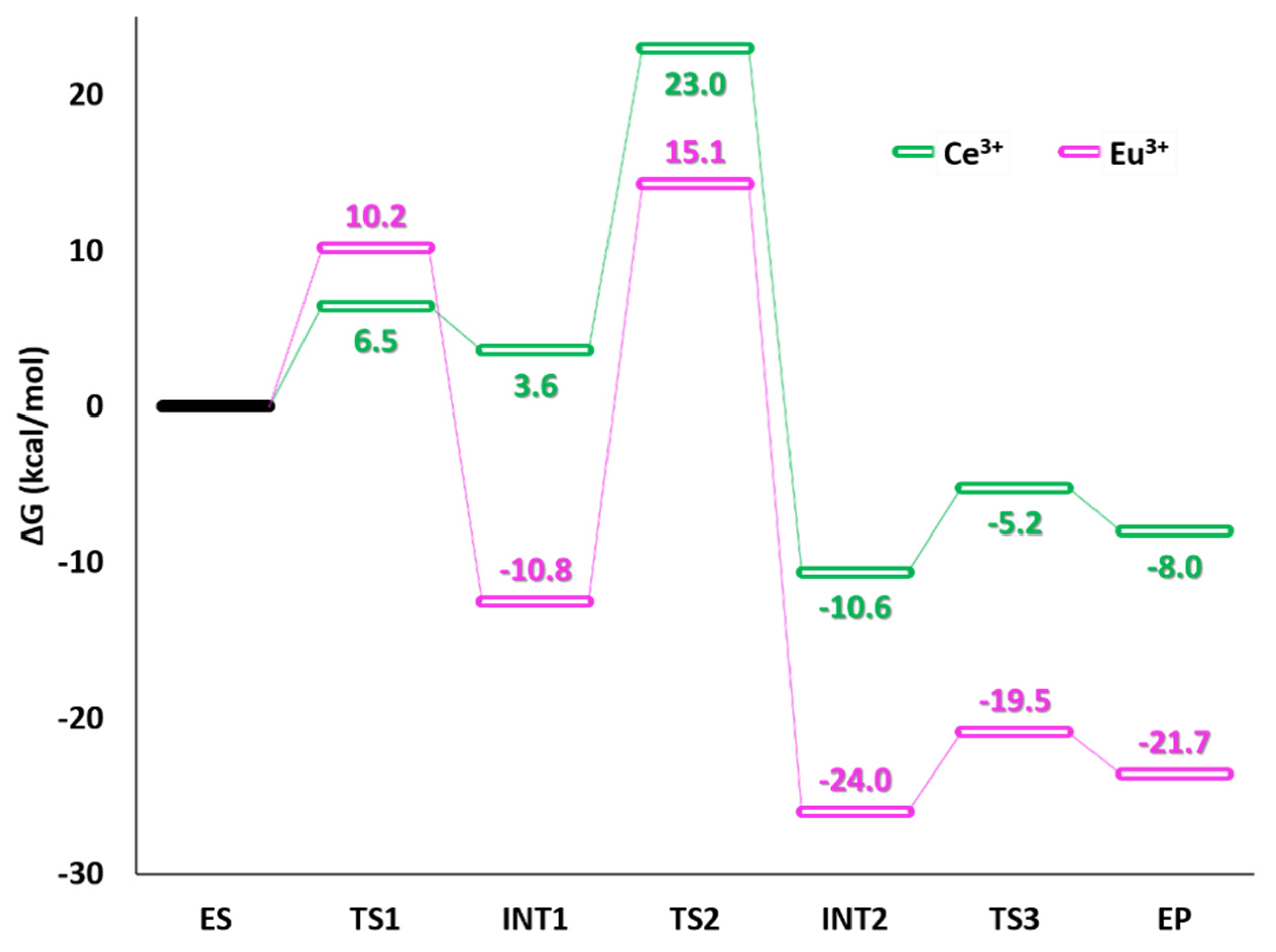
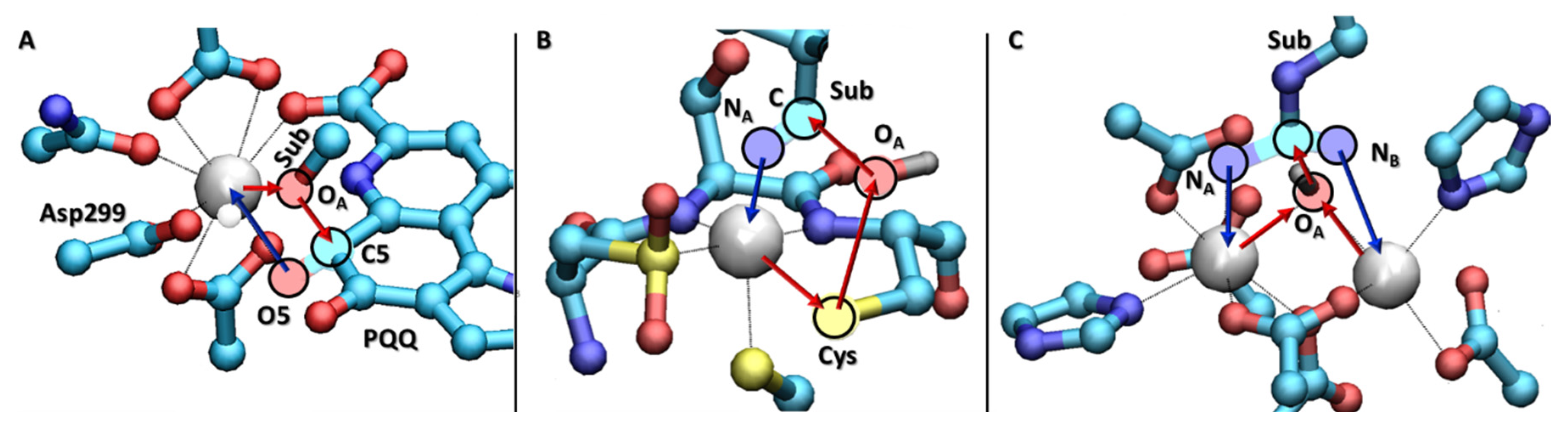
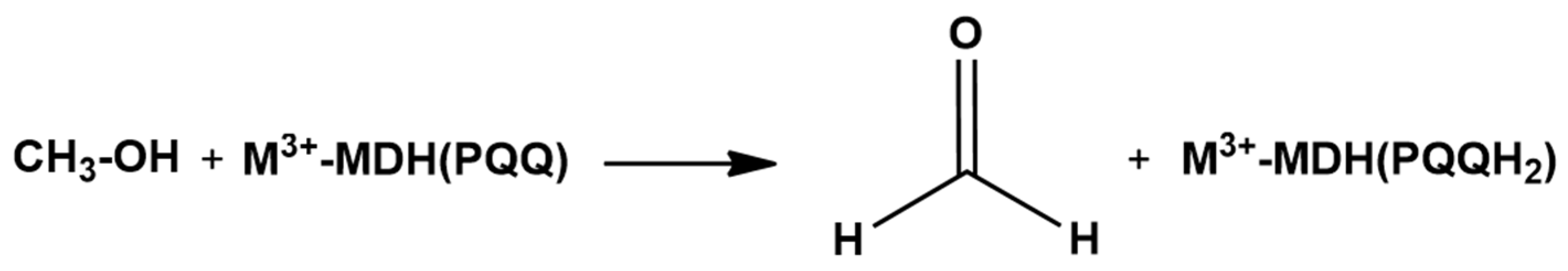
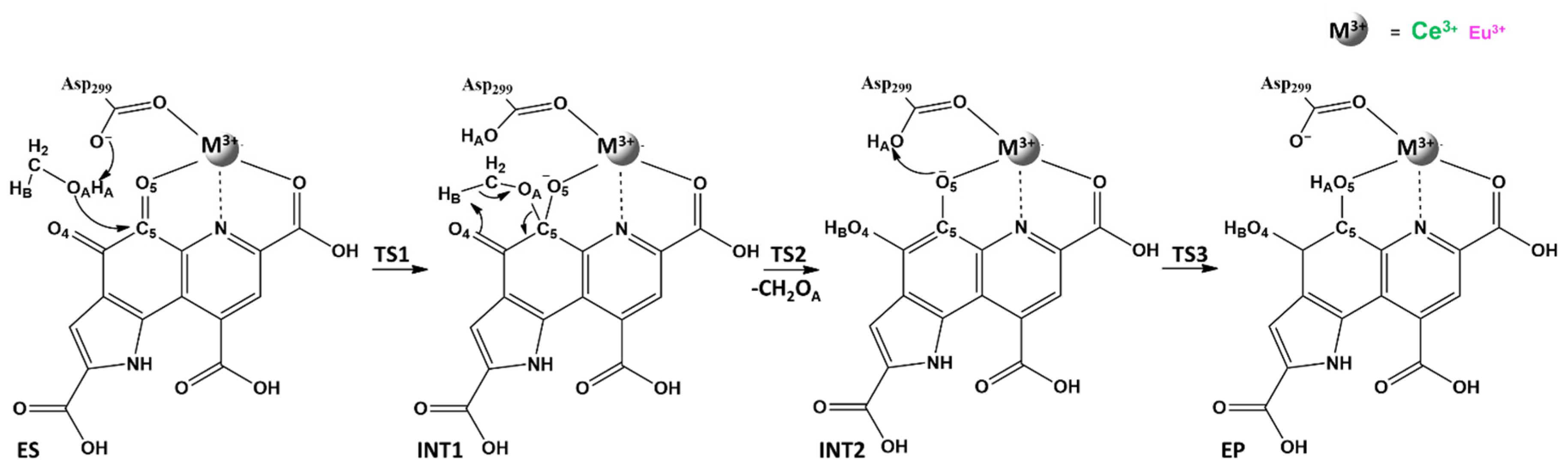
3. Ce3+ and Eu3+ Methanol Dehydrogenase
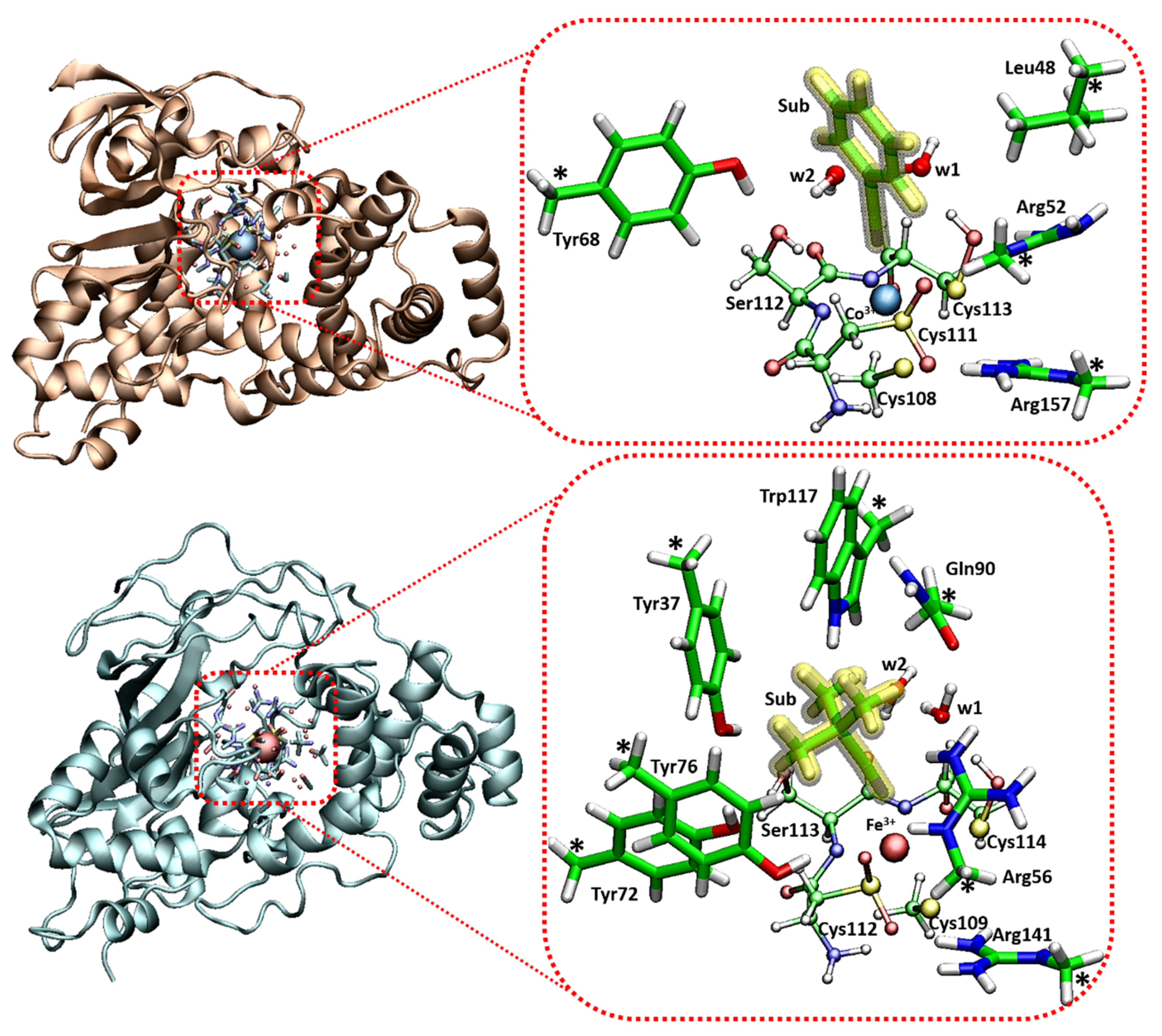
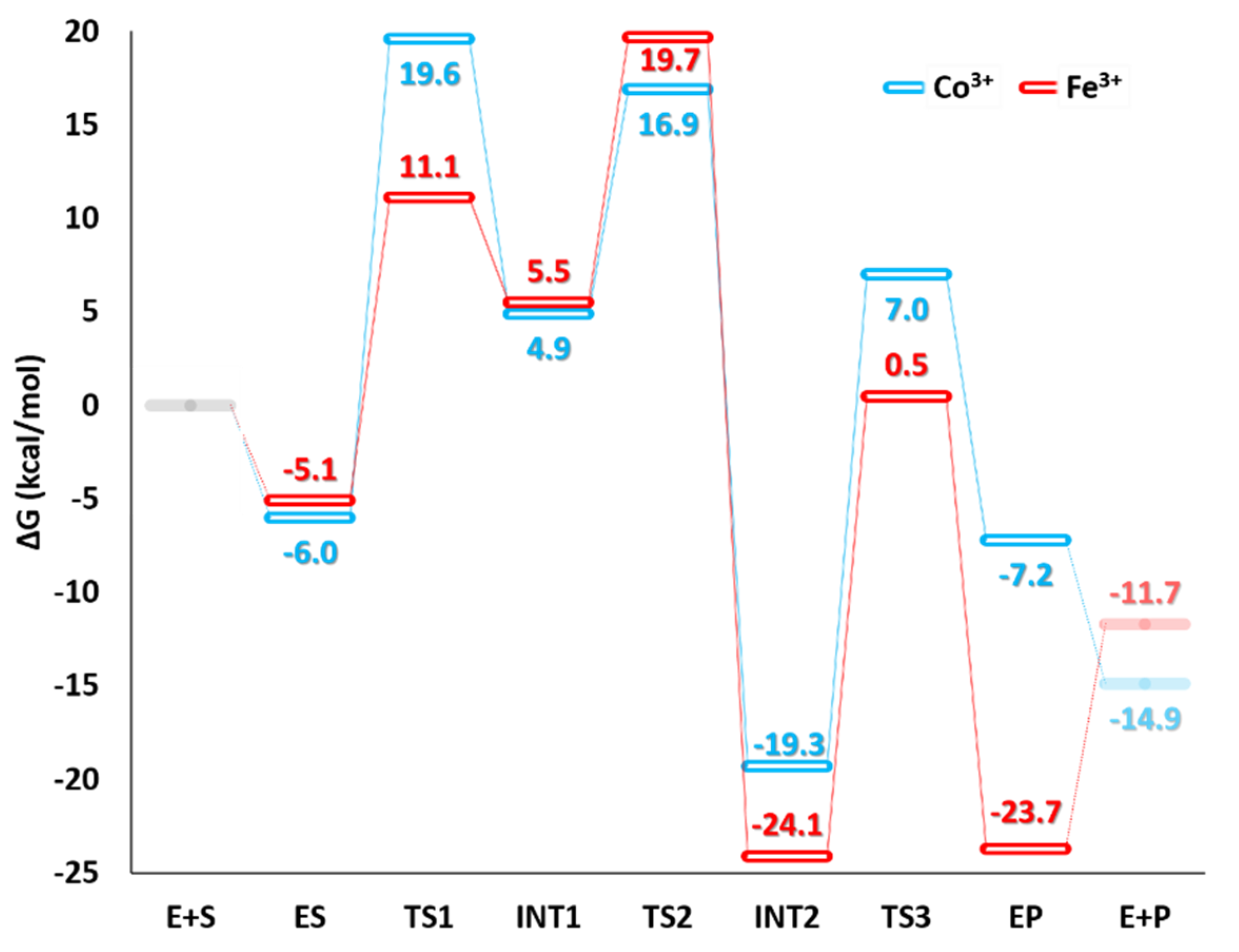
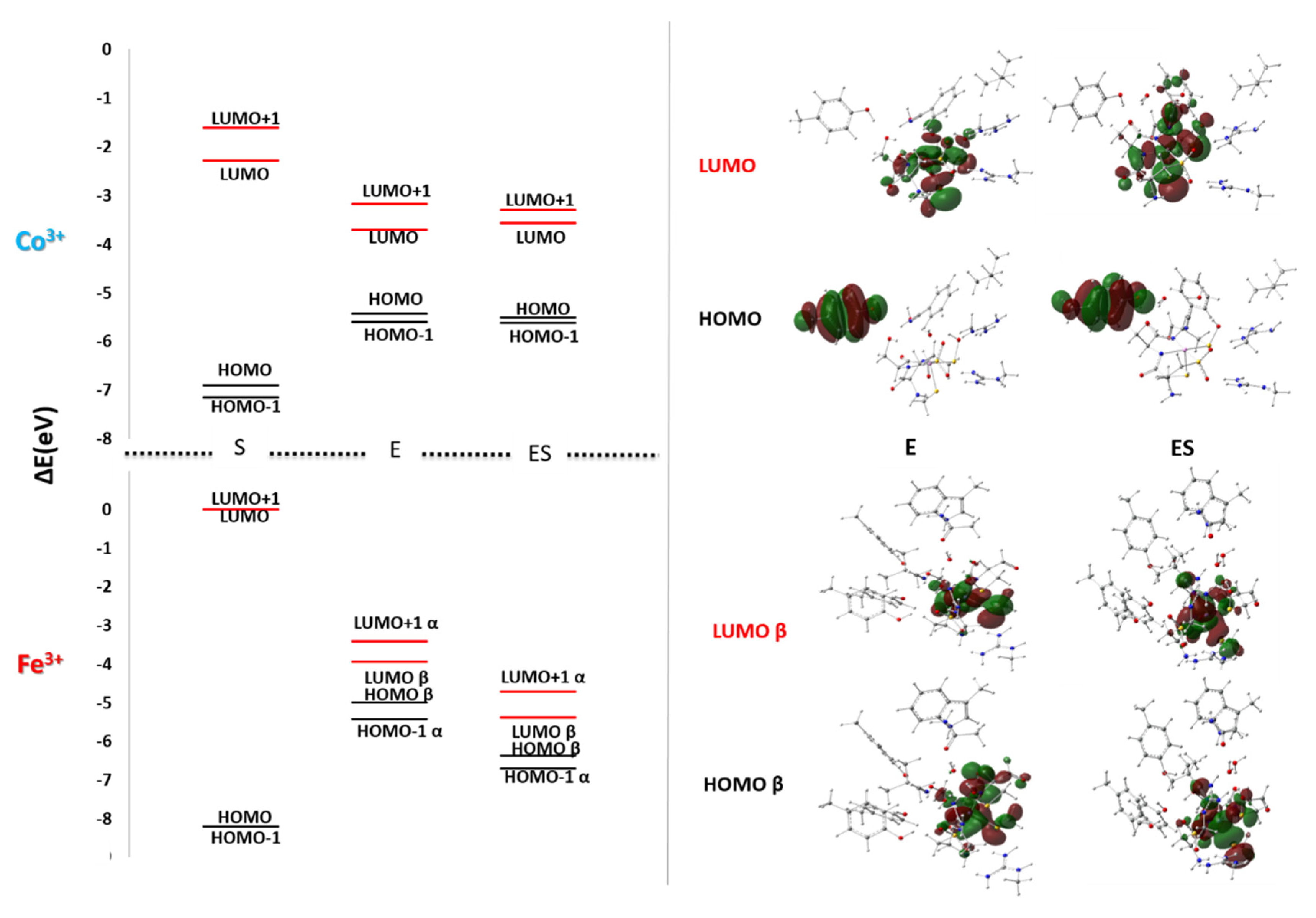
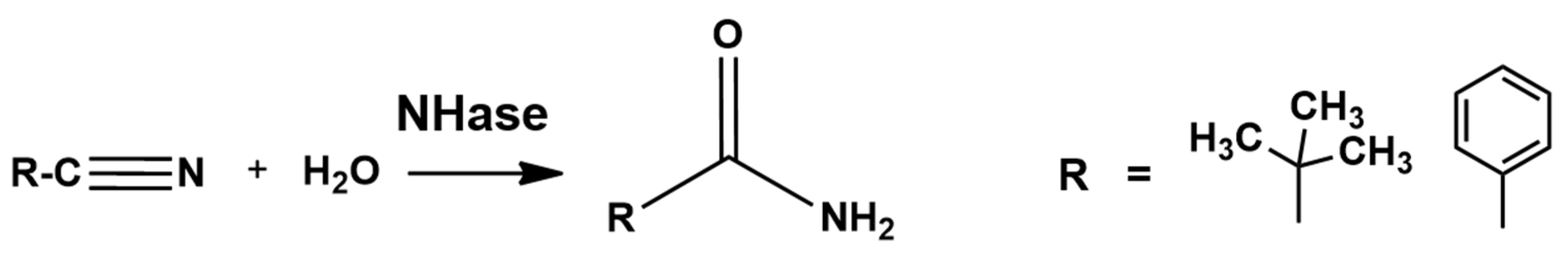
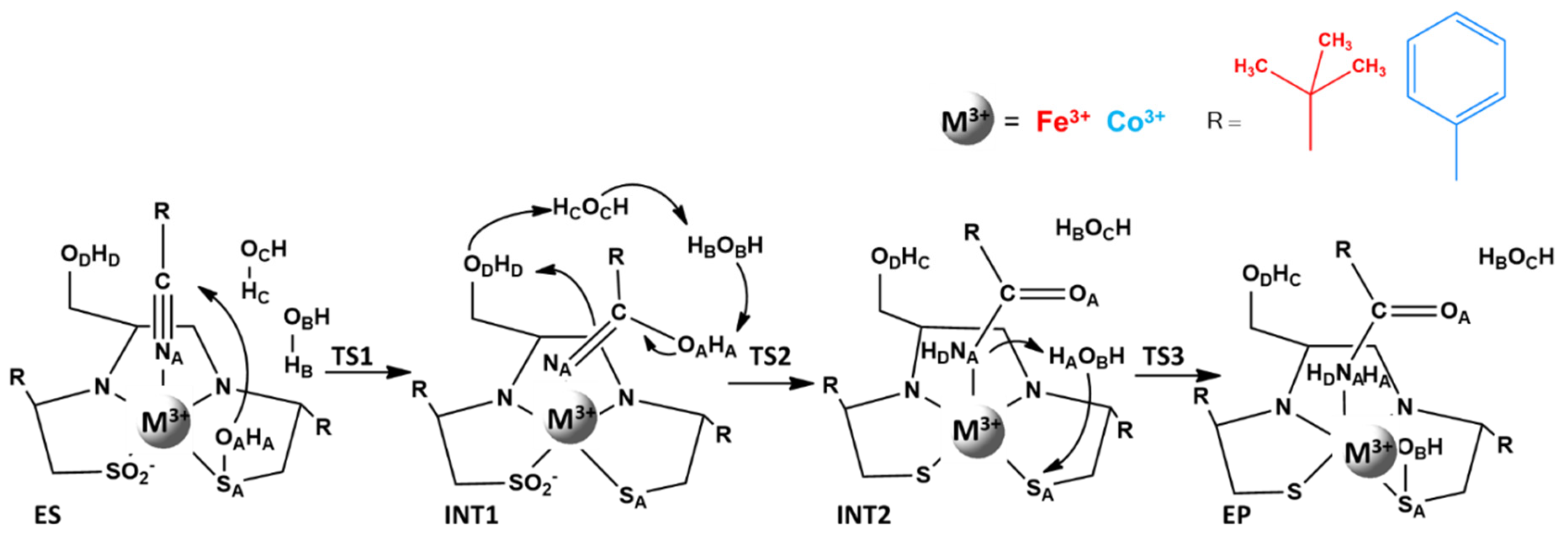
4. Low Spin Fe3+ and Co3+ Nitrile Hydratase
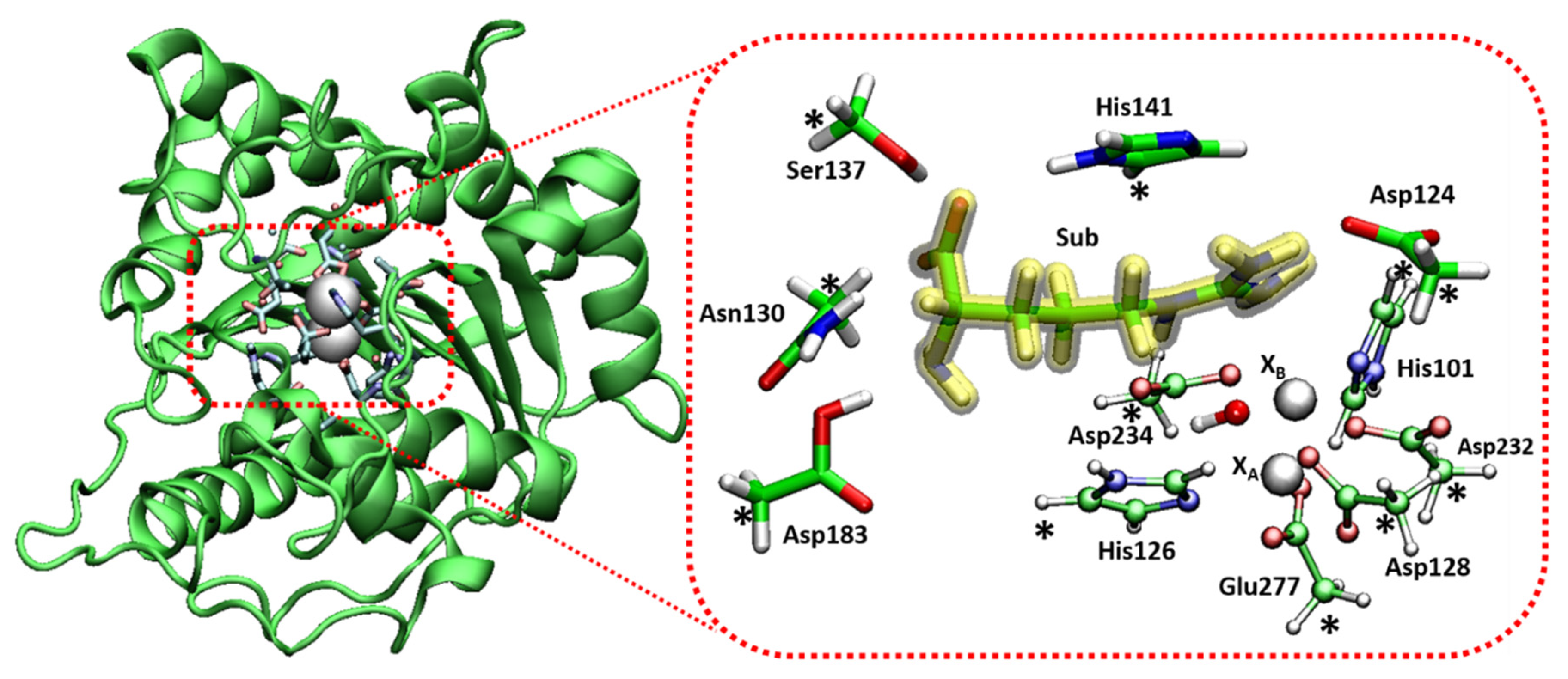
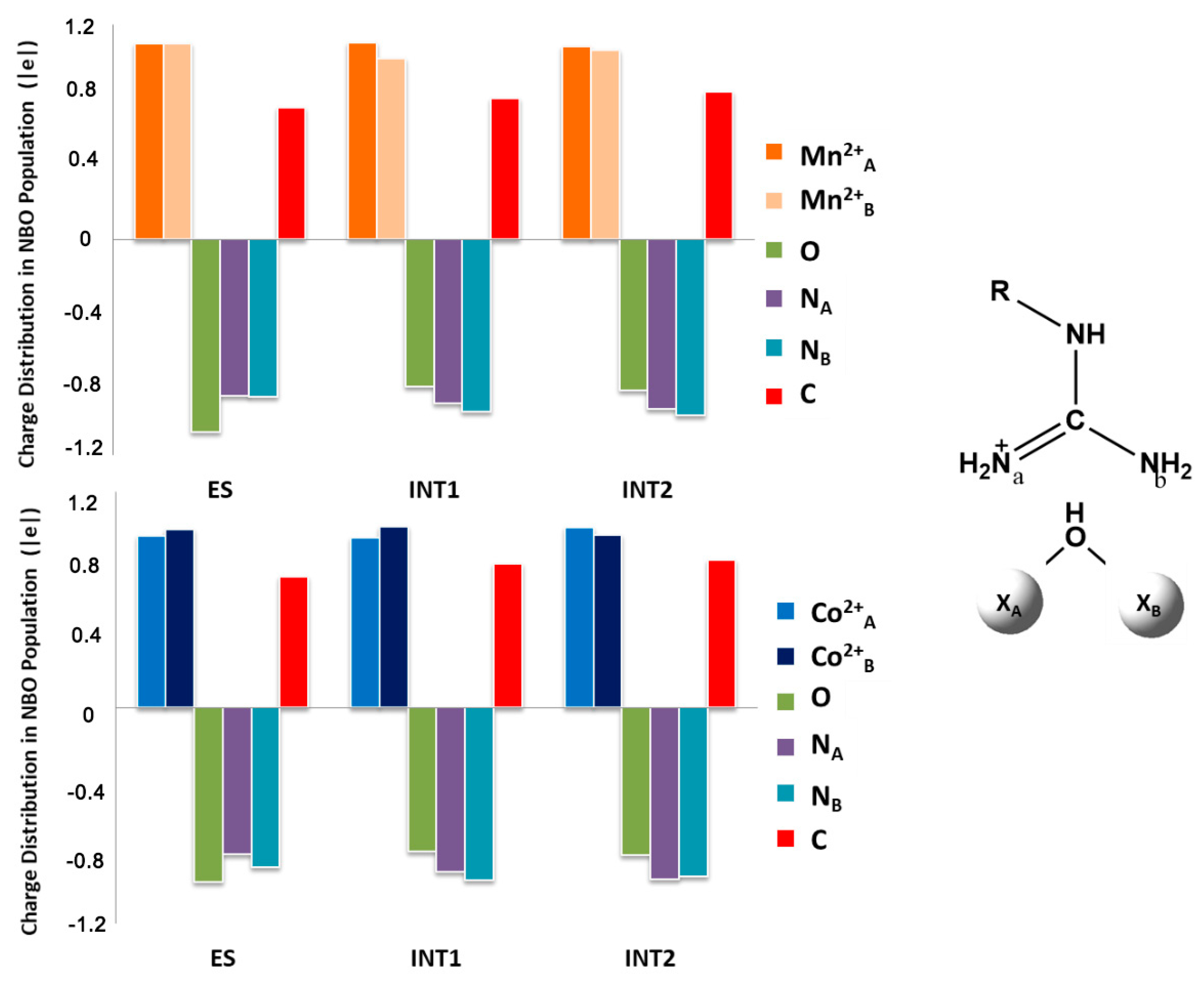
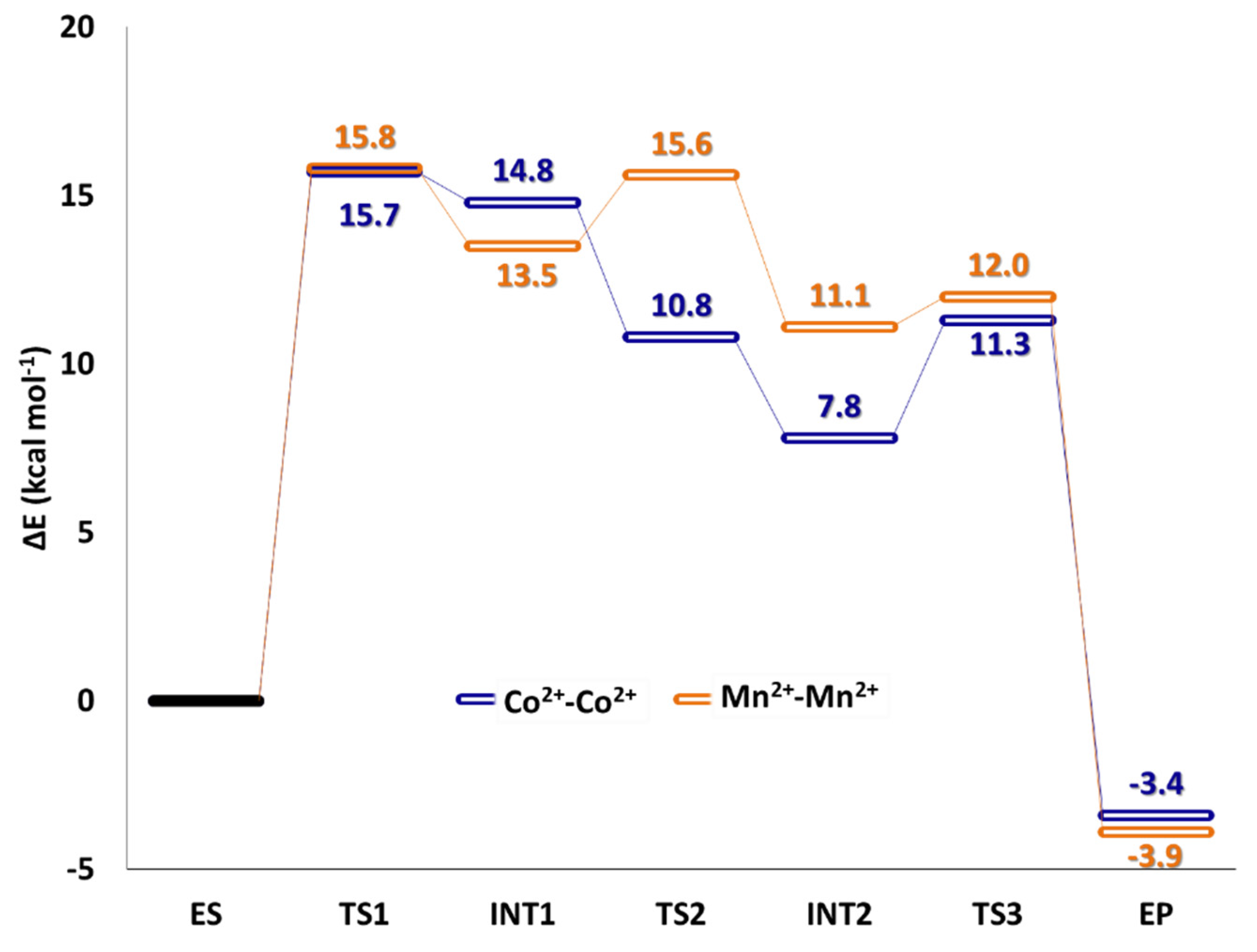
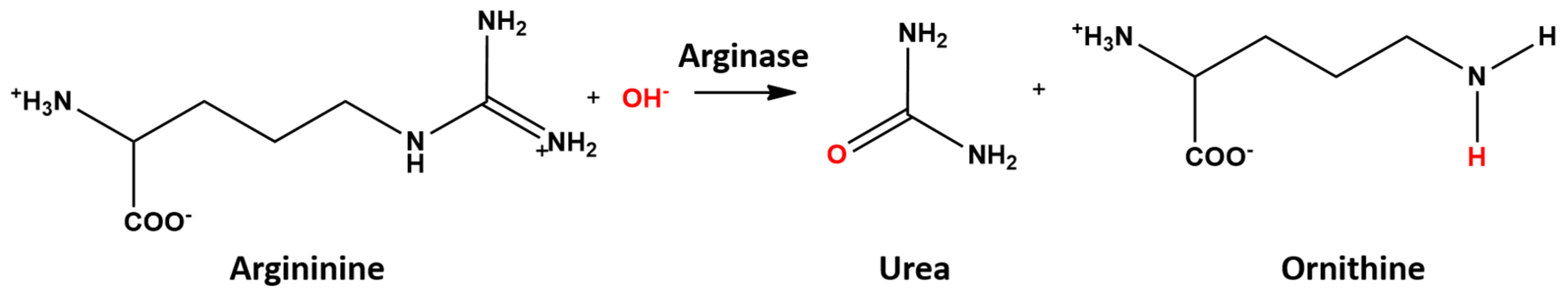
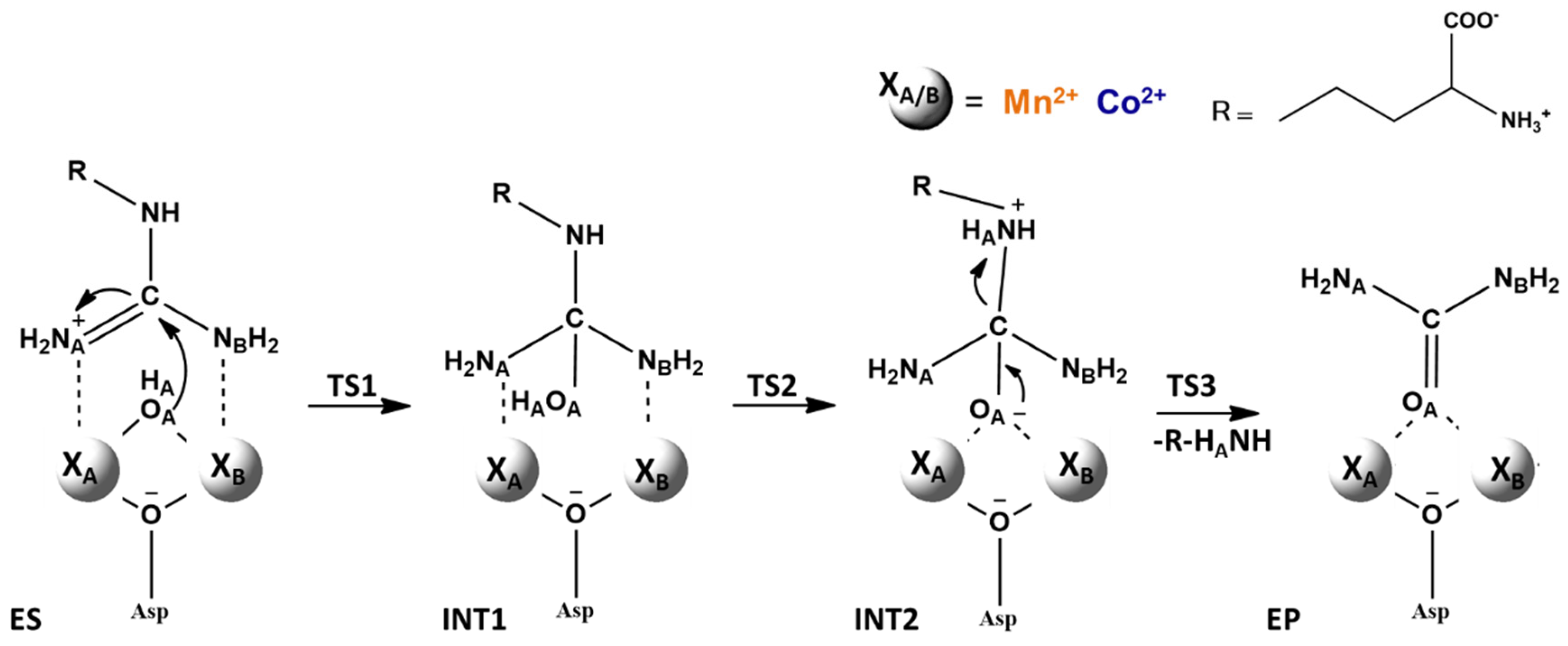
5. Co2+-Co2+ and Mn2+-Mn2+ Arginase
6. The Relevance of Metal Ions in the Three Selected Enzymes
7. Conclusions
- -
- The metals can play different roles that dictate the catalytic reaction mechanisms and the corresponding kinetic behaviors suggesting the principle “similar but not the same”;
- -
- The quantum mechanical-based theoretical methods can give reliable results not only in reproducing known data but mainly in elucidating the chemical processes in the fascination field of enzymology.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crabtree, R.H. The Organometallic Chemistry of Transition Metals, 7th ed.; Wiley: Hoboken, NJ, USA, 2014; pp. 436–468. [Google Scholar]
- Hilvert, D. Design of Protein Catalysts. Annu. Rev. Biochem. 2013, 82, 447–470. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gray, H.B.; Stiefel, E.I.; Valentine, J.S. Biological Inorganic Chemistry. Structure and Reactivity; University Science Books: Mill Valley, CA, USA, 2007; pp. 1–31. [Google Scholar]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Rauscher, F.J., III; Morris, J.F.; Tournay, O.E.; Cook, D.M.; Curran, T. Binding of the Wilms’ tumor locus zinc finger protein to the EGR-1 consensus sequence. Science 1990, 250, 1259–1262. [Google Scholar] [CrossRef]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.G. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Mikšovská, J.; Larsen, R.W. Structure-function relationships in metalloproteins. Methods Enzymol. 2003, 360, 302–329. [Google Scholar] [PubMed]
- Zheng, Y.J.; Xia, Z.X.; Chen, Z.W.; Mathews, F.S.; Bruice, T.C. Catalytic mechanism of quinoprotein methanol dehydrogenase: A theoretical and X-ray crystallographic investigation. Proc. Natl. Acad. Sci. USA 2001, 98, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.J.; Bruice, T.C. Conformation of coenzyme pyrroloquinoline quinone and role of Ca2+ in the catalytic mechanism of quinoprotein methanol dehydrogenase. Proc. Natl. Acad. Sci. USA 1997, 94, 11881–11886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Reddy, S.Y.; Bruice, T.C. Mechanism of methanol oxidation by quinoprotein methanol dehydrogenase. Proc. Natl. Acad. Sci. USA 2007, 104, 745–749. [Google Scholar] [CrossRef] [Green Version]
- Chistoserdova, L. Lanthanides: New life metals? World J. Microbiol. Biotechnol. 2016, 32, 138. [Google Scholar] [CrossRef]
- Skovran, E.; Martinez-Gomez, N.C. Just add lanthanides. Science 2015, 348, 862–863. [Google Scholar] [CrossRef]
- Barber-Zucker, S.; Shaanan, B.; Zarivach, R. Transition metal binding selectivity in proteins and its correlation with the phylogenomic classification of the cation diffusion facilitator protein family. Sci. Rep. 2017, 7, 16381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.M.; Wesson, L.; Eisenmant, G.; Eisenberg, D. Where metal ions bind in proteins (metafloprotein/protein structure/hydrophobicity contrast function). Proc. Natl. Acad. Sci. USA 1990, 87, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Dudev, T.; Lin, Y.; Dudev, M.; Lim, C. First−second shell interactions in metal binding sites in proteins: A PDB survey and DFT/CDM calculations. J. Am. Chem. Soc. 2003, 125, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Sobolev, V.; Edelman, M. First- and second-shell metal binding residues in human proteins are disproportionately associated with disease-related SNPs. Hum. Mutat. 2011, 32, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Dokmanić, I.; Sikić, M.; Tomić, S. Metals in proteins: Correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Rulísek, L.; Vondrásek, J. Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J. Inorg. Biochem. 1998, 71, 115–127. [Google Scholar] [CrossRef]
- Harding, M.M. The architecture of metal coordination groups in proteins. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, M.M. Geometry of metal-ligand interactions in proteins. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 401–411. [Google Scholar] [CrossRef]
- Harding, M.M. The geometry of metal-ligand interactions relevant to proteins. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1432–1443. [Google Scholar] [CrossRef]
- Harding, M.M. The geometry of metal-ligand interactions relevant to proteins. II. Angles at the metal atom, additional weak metal-donor interactions. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 857–867. [Google Scholar] [CrossRef]
- Zheng, H.; Chruszcz, M.; Lasota, P.; Lebioda, L.; Minor, W. Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 2008, 102, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A bioinformatic approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Good, N.M.; Vu, H.N.; Suriano, C.J.; Subuyuj, G.A.; Skovran, E.; Martinez-Gomez, N.C. Pyrroloquinoline quinone ethanol dehydrogenase in Methylobacterium extorquens AM1 extends lanthanide-dependent metabolism to multicarbon substrates. J. Bacteriol. 2016, 198, 3109–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panichev, A.M. Rare Earth Elements: Review of medical and biological properties and their abundance in the rock materials and mineralized spring waters in the context of animal and human geophagia reason evaluation. Achiev. Life Sci. 2015, 9, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Brittain, H.G.; Richardson, F.S.; Martin, R.B. Terbium(III) emission as a probe of calcium(II) binding sites in proteins. J. Am. Chem. Soc. 1976, 98, 8255–8260. [Google Scholar] [CrossRef]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Mechanisms of metalloenzymes studied by quantum chemical methods. Q. Rev. Biophys. 2003, 36, 91–145. [Google Scholar] [CrossRef]
- Chen, S.-L.; Fang, W.-H.; Himo, F. Technical aspects of quantum chemical modeling of enzymatic reactions: The case of phosphotriesterase. Theor. Chem. Account. 2008, 120, 515–522. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Modeling Aspects of Mechanisms for Reactions Catalyzed by Metalloenzymes. J. Comput. Chem. 2001, 22, 1634–1645. [Google Scholar] [CrossRef]
- Sheng, X.; Kazemi, M.; Planas, F.; Himo, F. Modeling enzymatic enantioselectivity using quantum chemical methodology. ACS Catal. 2020, 10, 6430–6449. [Google Scholar] [CrossRef]
- Himo, F. Recent trends in quantum chemical modeling of enzymatic reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazzetta, P.; Marino, T.; Russo, N.; Salahub, D. The role of metal substitution in the promiscuity of natural and artificial carbonic anhydrases. Coord. Chem. Rev. 2017, 345, 73–85. [Google Scholar] [CrossRef]
- Prejanò, M.; Marino, T.; Russo, N. QM cluster or QM/MM in computational enzymology: The test case of LigW-decarboxylase. Front. Chem. 2018, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.J.; Fernandes, P.A. Computational Enzymatic Catalysis. Acc. Chem. Res. 2008, 41, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Computational enzymatic catalysis—Clarifying enzymatic mechanisms with the help of computers. Phys. Chem. Chem. Phys. 2012, 14, 12431–12441. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. A 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lynch, B.J.; Truhlar, D.G. Development and assessment of a new hybrid density functional model for thermochemical kinetics. J. Phys. Chem. A 2004, 108, 2715–2719. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef] [PubMed]
- Shil, S.; Bhattacharya, D.; Sarkar, S.; Misra, A. Performance of the widely used minnesota density functionals for the prediction of heat of formations, ionization potentials of some benchmarked first row transition metal complexes. J. Phys. Chem. A 2013, 117, 4945–4955. [Google Scholar] [CrossRef] [PubMed]
- Mata, R.A.; Suhm, M.A. Benchmarking quantum chemical methods: Are we heading in the right direction? Angew. Chem. In. Ed. 2017, 56, 11011–11018. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Truhlar, D.G. Status and challenges of density functional theory. Trends Chem. 2020, 2, 302–318. [Google Scholar] [CrossRef]
- Andrae, D.; Haußermann, H.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. Natural Bond Orbitals, version 3.1.; University of Wisconsin: Madison, WI, USA, 2001. [Google Scholar]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Amata, O.; Marino, T.; Russo, N. Catalytic activity of a æ-class zinc and cadmium containing carbonic anhydrase. Compared work mechanisms. Phys. Chem. Chem. Phys. 2011, 13, 3468–3477. [Google Scholar] [CrossRef]
- Chen, W.; Fang, W.H.; Himo, F. Reaction mechanism of the binuclear zinc enzyme glyoxalase II—A theoretical study. J. Inorg. Biochem. 2009, 103, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.Z.; Himo, F.; Yu, J.G.; Liu, R.Z. Dipeptide hydrolysis by the dinuclear zinc enzyme human renal dipeptidase: Mechanistic insights from DFT calculations. J. Inorg. Biochem. 2010, 104, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. The search for the mechanism of the reaction catalyzed by farnesyltransferase. Chem. Eur. J. 2009, 15, 4243–4247. [Google Scholar] [CrossRef] [PubMed]
- Alberto, M.E.; Marino, T.; Ramos, M.J.; Russo, N. Atomistic details of the catalytic mechanism of Fe(III)–Zn(II) purple acid phosphatase. J. Chem. Theory Comput. 2010, 6, 2424–2433. [Google Scholar] [CrossRef]
- Warshel, A. Computer Modeling of Chemical Reactions in Enzymes and Solutions; Wiley: New York, NY, USA, 1991. [Google Scholar]
- Piazzetta, P.; Marino, T.; Russo, N. Insight into the promiscuous activity of human carbonic anhydrase against the cyanic acid substrate from a combined QM and QM/MM investigation. Phys. Chem. Chem. Phys. 2014, 16, 16671–16676. [Google Scholar] [CrossRef]
- Ribeiro, A.J.M.; Alberto, M.E.; Ramos, M.J.; Fernandes, P.A.; Russo, N. The catalytic mechanism of protein phosphatase 5 established by DFT calculations. Chem. Eur. J. 2013, 114, 3601–3658. [Google Scholar] [CrossRef]
- Liao, R.Z.; Yu, G.; Himo, F. Mechanism of tungsten-dependent acetylene hydratase from quantum chemical calculations. Proc. Natl. Acad. Sci. USA 2010, 107, 22523–22527. [Google Scholar] [CrossRef] [Green Version]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modeling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Van der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Brás, N.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Swiderek, K.; Tunon, I.; Moliner, V. Predicting enzymatic reactivity: From theory to design. Comput. Mol. Sci. 2014, 4, 407–421. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Warshel, A. The empirical valence bond model: Theory and applications. Comput. Mol. Sci. 2011, 1, 30–45. [Google Scholar] [CrossRef]
- Brunk, E.; Rothlisberger, U. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem. Rev. 2015, 115, 6217–6263. [Google Scholar] [CrossRef]
- Anthony, C. Methanol Dehydrogenase, a PQQ-Containing Quinoprotein Dehydrogenase; Holzenburg, A., Scrutton, N.S., Eds.; Springer: New York, NY, USA, 2000; pp. 73–117. [Google Scholar]
- Kalyuzhnaya, M.G.; Hristova, K.R.; Lidstrom, M.E.; Chistoserdova, L. Characterization of a novel methanol dehydrogenase in representatives of burkholderiales: Implications for environmental detection of methylotrophy and evidence for convergent evolution. J. Bacteriol. 2008, 190, 3817–3823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duine, J.A.; Frank, J.; Westerling, J. Purification and properties of methanol dehydrogenase from Hyphomicrobium X. Biochim. Biophys. Acta 1978, 524, 277–287. [Google Scholar] [CrossRef]
- Ghosh, R.; Quayle, J.R. Purification and properties of the methanol dehydrogenase from Methylophilis methylotrophus. Biochem. J. 1981, 199, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Cornish, A.; Gossain, V.; Best, D.J. Purification, crystallization and preliminary X-ray diffraction characterization of methanol dehydrogenase from Methylosinus trichosporium OB3b. Eur. J. Biochem. 1987, 164, 223–227. [Google Scholar] [CrossRef]
- Williams, P.A.; Coates, L.; Mohammed, F.; Gill, R.; Erskine, P.T.; Coker, A.; Woods, S.P.; Anthony, C.; Cooper, J.B. The atomic resolution structure of methanol dehydrogenase from Methylobacterium extorquens. Acta Crystallogr. D 2005, 61, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Dai, W.; Zhang, Y.; White, S.A.; Boyd, G.D.; Mathews, F.S. Determination of the gene sequence and the three-dimensional structure at 2.4 angstroms resolution of methanol dehydrogenase from Methylophilus W3A1. J. Mol. Biol. 1996, 259, 480–501. [Google Scholar] [CrossRef]
- Chistoserdova, L. Metabolism of Formaldehyde in M. extorquens AM1. Molecular Genetic Analysis and Mutant Characterization; Lidstrom, M.E., Tabita, F.R., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996; pp. 16–24. [Google Scholar]
- Harms, N.; Ras, J.; Koning, S.; Reijinders, W.N.M.; Stouthamer, A.H.; van Spanning, R.J.M. Genetics of C1 Metabolism Regulation in Paracoccus Denitrificans; Lidstrom, M.E., Tabita, F.R., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996; pp. 126–132. [Google Scholar]
- Hibi, Y.; Asai, K.; Arafuka, H.; Hamajima, M.; Iwama, T.; Kawai, K. Molecular structure of La3+-induced methanol dehydrogenase-like protein in Methylobacterium radiotolerans. J. Biosci. Bioeng. 2011, 111, 547–549. [Google Scholar] [CrossRef]
- Anthony, C. The quinoprotein dehydrogenases for methanol and glucose. Arch. Biochem. Biophys. 2004, 428, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.M.; Anthony, C. The biochemistry, physiology and genetics of PQQ and PQQ-containing enzymes. Adv. Microb. Physiol. 1998, 40, 1–80. [Google Scholar] [PubMed]
- Pol, A.; Barends, T.R.M.; Dietl, A.; Khadem, A.F.; Eygensteyn, J.; Jetten, M.S.M.; Op den Camp, H.J.M. Rare earth metals are essential for methanotrophic life in volcanic mudpots. Enviromental Microbiol. 2014, 16, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Djinovic-Carugo, K.; Carugo, O. Structural biology of the lanthanides-mining rare earths in the Protein Data Bank. J. Inorg. Biochem. 2015, 143, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.B.; Richardson, F.S. Lanthanides as probes for calcium in biological systems. Q. Rev. Biophys. 1979, 12, 181–209. [Google Scholar] [CrossRef]
- Dos Remedios, C.G. Lanthanide ion probes of calcium-binding sites on cellular membranes. Cell Calcium 1981, 2, 29–51. [Google Scholar] [CrossRef]
- Mikkelson, R.B. Lanthanides as Calcium Probes; Chapman, D., Wallach, D.F., Eds.; Academic Press: New York, NY, USA, 1976; pp. 153–190. [Google Scholar]
- Anthony, C. Methanol dehydrogenase, a PQQ-containing quinoprotein dehydrogenase. Subcell. Biochem. 2000, 35, 73–117. [Google Scholar]
- Idupulapati, N.B.; Minardi, D.S. Coordination and binding of ions in Ca2+- and Ba2+-containing methanol dehydrogenase and interactions with methanol. Theochem 2009, 901, 72–80. [Google Scholar] [CrossRef]
- Good, N.M.; Fellner, M.; Demirer, K.; Hu, J.; Hausinger, R.P.; Martinez-Gomez, N.C. Lanthanide-dependent alcohol dehydrogenases require an essential aspartate residue for metal coordination and enzymatic function. J. Biol. Chem. 2020, 295, 8272–8284. [Google Scholar] [CrossRef]
- Reddy, S.Y.; Bruice, T.C. Determination of enzyme mechanisms by molecular dynamics: Studies on quinoproteins, methanol dehydrogenase, and soluble glucose dehydrogenase. Protein Sci. 2004, 13, 1965–1978. [Google Scholar] [CrossRef] [Green Version]
- Jahn, B.; Pol, A.; Lumpe, H.; Barends, T.R.M.; Dietl, A.; Hogendoorn, C.; Op den Camp, H.J.M.; Daumann, L.J. Similar but not the same: First kinetic and structural analyses of a methanol dehydrogenase containing a europium ion in the active site. ChemBioChem 2018, 19, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Idupulapati, N.B.; Mainardi, D. Quantum chemical modeling of methanol oxidation mechanisms by methanol dehydrogenase enzyme: Effect of substitution of calcium by barium in the active site. J. Phys. Chem. A 2010, 114, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Fitriyanto, N.A.; Fushimi, M.; Matsunaga, M.; Pertiwiningrum, A.; Iwama, T.; Kawai, K. Molecular structure and gene analysis of Ce3+—Induced methanol dehydrogenase of Bradyrhizobium sp. MAFF211645. J. Biosci. Bioeng. 2011, 111, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Mitsui, R.; Tani, A.; Sasa, K.; Tashiro, S.; Iwama, T.; Hayakawa, T.; Kawai, K. A Catalytic role of XoxF1 as La3+—Dependent methanol Dehydrogenase in Methylobacterium extorquens Strain AM1. PLoS ONE 2012, 7, e50480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daumann, L.J. Essential and Ubiquitous: The Emergence of Lanthanide Metallobiochemistry. Angew. Chem. Int. Ed. 2019, 58, 12795–12802. [Google Scholar] [CrossRef]
- Lumpe, H.; Pol, A.; Op den Camp, H.J.M.; Daumann, L.J. Impact of the lanthanide contraction on the activity of a lanthanide-dependent methanol dehydrogenase—A kinetic and DFT study. Dalton Trans. 2018, 47, 10463–10472. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Yu, Z.; Groom, J.; Cheng, J.F.; Tarver, A.; Yoshikuni, Y.; Chistoserdova, L. Rare earth element alcohol dehydrogenases widely occur among globally distributed, numerically abundant and environmentally important microbes. ISME J. 2019, 13, 2005–2017. [Google Scholar] [CrossRef]
- Lumpe, H.; Daumann, L.J. Studies of redox cofactor pyrroloquinoline quinone and its interaction with Lanthanides(III) and Calcium(II). Inorg. Chem. 2019, 58, 8432–8441. [Google Scholar] [CrossRef]
- Bogart, J.A.; Lewis, A.J.; Schelter, E.J. DFT Study of the Active Site of the XoxF-Type Natural, Cerium-dependent methanol dehydrogenase enzyme. Chem. Eur. J. 2015, 21, 1743–1748. [Google Scholar] [CrossRef]
- McSkimming, A.; Cheisson, T.; Carroll, P.J.; Schelter, E.J. Functional synthetic model for the lanthanide-dependent quinoid alcohol dehydrogenase active site. J. Am. Chem. Soc. 2018, 140, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Marino, T.; Prejanò, M.; Russo, N. How Metal Coordination in the Ca-, Ce-, and Eu-Containing Methanol Dehydrogenase Enzymes Can Influence the Catalysis: A Theoretical Point of View; Broclawick, E., Borowski, T., Radoń, M., Eds.; Springer: Cham, Switzerland, 2019; pp. 487–501. [Google Scholar]
- Cotton, S.A.; Raithby, P.R. Systematics and surprises in lanthanide coordination chemistry. Coord. Chem. Rev. 2017, 340, 220–231. [Google Scholar] [CrossRef]
- Seitz, M.; Oliver, A.G.; Raymond, K.N. The lanthanide contraction revisited. J. Am. Chem. Soc. 2007, 129, 11153–11160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyykkö, P. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Hothi, P.; Sutcliffe, M.J.; Scrutton, N.S. Kinetic isotope effects and ligand binding in PQQ-dependent methanol dehydrogenase. Biochem. J. 2005, 388, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Prejanò, M.; Marino, T.; Russo, N. How can methanol dehydrogenase from Methylacidiphilum fumariolicum work with the alien CeIII ion in the active center? A theoretical study. Chem. Eur. J. 2017, 23, 8652–8657. [Google Scholar] [CrossRef]
- Prejanò, M.; Russo, N.; Marino, T. How the lanthanide ions affect the addition-elimination step of methanol dehydrogenases. Chem. Eur. J. 2020. [Google Scholar] [CrossRef]
- Leopoldini, M.; Russo, N.; Toscano, M. The preferred reaction path for the oxidation of methanol by PQQ-containing methanol dehydrogenase: Addition-elimination versus hydride transfer mechanism. Chem. Eur. J. 2007, 13, 2109–2117. [Google Scholar] [CrossRef]
- Hazardous Substances Databank; U.S. National Library of Medicine: Bethesda, MD, USA, 1995.
- Woutersen, R.A. Psychosocial factors at work and subsequent depressive symptoms in the Gazel cohort. Scand. J. Work. Environ. Health 1998, 24, 197–205. [Google Scholar]
- Wyatt, J.M.; Knowles, C.J. Microbial degradation of acrylonitrile waste effluents: The degradation of effluents and condensates from the manufacture of acrylonitrile. Int. Biodeterior. Biodegrad. 1995, 35, 227–248. [Google Scholar] [CrossRef]
- Martinez, S.; Wu, R.; Sanishvili, R.; Liu, D.; Holz, R. The Active site sulfenic acid ligand in nitrile Hydratases can function as a nucleophile. J. Am. Chem. Soc. 2014, 136, 1186–1189. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimizu, S. Metalloenzyme nhase: Structure, regulation and application to biotechnology. Nat. Biotechnol. 1998, 16, 733–736. [Google Scholar] [CrossRef]
- Schmid, A.; Dordick, J.S.; Hauer, B.; Kiener, A.N.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [Google Scholar] [CrossRef]
- Cheng, Z.; Xia, Y.; Zhou, Z. Recent advances and promises in nitrile hydratase: From mechanism to industrial applications. Front. Bioeng. Biotechnol. 2020, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.; Cummings, S. The current and future applications of microorganism in the bioremediation of cyanide contamination. Antonie Leeuwenhoek 2006, 90, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ashima, Y.; Suto, M. Development of an enzymatic process for manufacturing acrylamide and recent progress. Bioprocess. Technol. 1993, 16, 91–107. [Google Scholar]
- Kobayashi, M.; Nagasawa, T.; Yamada, H. Enzymatic synthesis of acrylamide: A success story not yet over. Trends Biotechnol. 1992, 10, 402–408. [Google Scholar] [CrossRef]
- Yamada, H.; Kobayashi, M. Nitrile hydratase and its application to industrial production of acrylamide. Biosci. Biotechnol. Biochem. 1996, 60, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Payne, M.S.; Wu, S.; Fallon, R.D.; Tudor, G.; Stieglitz, B.; Turner, I.M., Jr.; Nelson, M.J. A stereoselective cobalt-containing nitrile hydratase. Biochemistry 1997, 36, 5447–5454. [Google Scholar] [CrossRef]
- Mascharak, P.K. Structural and functional models of nitrile hydratase. Coord. Chem. Rev. 2002, 225, 201–214. [Google Scholar] [CrossRef]
- Hopmann, K.H.; Himo, F. Theoretical Investigation of the Second-Shell Mechanism of Nitrile Hydratase. Eur. J. Inorg. Chem. 2008, 20, 1406–1412. [Google Scholar] [CrossRef]
- Light, K.M.; Yamanaka, Y.; Odaka, M.; Solomon, E.I. Spectroscopic and computational studies of nitrile hydratase: Insights into geometric and electronic structure and the mechanism of amide synthesis. Chem. Sci. 2015, 6, 6280–6294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, J.A. Synthetic analogues of cysteinate-ligated non-heme iron and non-corrinoid cobalt enzymes. Chem. Rev. 2004, 104, 825–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, N.; Gumataotao, N.; Hajnas, N.; Wu, R.; Lankathilaka, K.P.W.; Bornscheuer, U.T.; Liu, D.; Fiedler, A.T.; Holz, R.C.; Bennett, B. Multiple states of nitrile hydratase from rhodococcus equi TG328−2: Structural and mechanistic insights from electron paramagnetic resonance and density functional theory studies. Biochemistry 2017, 56, 3068–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Sharma, R.; Banerjee, U. The nitrile-degrading enzymes: Current status and future prospects. Appl. Microbiol. Biotechnol. 2002, 60, 33–44. [Google Scholar] [CrossRef]
- Mylerova, V.; Martinkova, L. Synthetic applications of nitrileconverting enzymes. Curr. Org. Chem. 2003, 7, 1279–1295. [Google Scholar]
- Miyanaga, A.; Fushinobu, S.; Ito, K.; Wakagi, T. Crystal structure of cobalt-containing nitrile hydratase. Biochem. Biophys. Res. Commun. 2001, 288, 1169–1174. [Google Scholar] [CrossRef]
- Hashimoto, K.; Suzuki, H.; Taniguchi, K.; Noguchi, T.; Yohda, M.; Odaka, M. Catalytic mechanism of nitrile hydratase proposed by time-resolved X-ray crystallography using a novel substrate, tertButylisonitrile. J. Biol. Chem. 2008, 283, 36617–36623. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Chow, M.; Taniguchi, K.; Lugo-Mas, P.; Davin, S.; Maeda, M.; Kovacs, J.A.; Odaka, M.; Hodgson, K.O.; Hedman, B.; et al. Sulfur K-Edge XAS and DFT calculations on nitrile hydratase: Geometric and electronic structure of the non-heme iron active site. J. Am. Chem. Soc. 2006, 128, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, Y.; Kato, Y.; Hashimoto, K.; Iida, K.; Nagasawa, K.; Nakayama, H.; Dohmae, N.; Noguchi, K.; Noguchi, T.; Yohda, M.; et al. Time-resolved crystallography of the reaction intermediate of nitrile hydratase: Revealing a role for the cysteine sulfenic acid ligand as a catalytic nucleophile. Angew. Chem. Int. Ed. 2015, 54, 10763–10767. [Google Scholar] [CrossRef]
- Hopmann, K.H.; Guo, J.-D.; Himo, F. Theoretical investigation of the first-shell mechanism of nitrile hydratase. Inorg. Chem. 2007, 46, 4850–4856. [Google Scholar] [CrossRef]
- Prejanò, M.; Marino, T.; Rizzuto, C.; Madrid Madrid, J.C.; Russo, N.; Toscano, M. Reaction mechanism of low-spin Iron(III)- and Cobalt(III)-containing nitrile hydratases: A quantum mechanics investigation. Inorg. Chem. 2017, 58, 13390–13400. [Google Scholar] [CrossRef] [PubMed]
- Hopmann, K.H.; Himo, F. On the role of tyrosine as catalytic base in nitrile hydratase. Eur. J. Inorg. Chem. 2008, 22, 3452–3459. [Google Scholar] [CrossRef]
- Hopmann, K.H. Full reaction mechanism of nitrile hydratase: A cyclic intermediate and an unexpected disulfide switch. Inorg. Chem. 2014, 53, 2760–2762. [Google Scholar] [CrossRef] [PubMed]
- Kayanuma, M.; Hanaoka, K.; Shoji, M.; Shigeta, Y. A QM/MM study of the initial steps of catalytic mechanism of nitrile hydratase. Chem. Phys. Lett. 2015, 623, 8–13. [Google Scholar] [CrossRef]
- Kayanuma, M.; Shoji, M.; Yohda, M.; Odaka, M.; Shigeta, Y. Catalytic mechanism of nitrile hydratase subsequent to cyclic intermediate formation: A QM/MM study. J. Phys. Chem. B 2016, 120, 3259–3266. [Google Scholar] [CrossRef]
- Takarada, H.; Kawano, Y.; Hashimoto, K.; Nakayama, H.; Ueda, S.; Yohda, M.; Kamiya, N.; Dohmae, N.; Maeda, M.; Odaka, M. Mutational study on αGln90 of Fe-type nitrile hydratase from Rhodococcus sp. N771. Biosci. Biotechnol. Biochem. 2006, 70, 881–889. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Sabio, G.; Mora, A.; Rodriguez, P.C.; Ochoa, A.C.; Centeno, F.; Christianson, D.W. Crystal structure of human arginase I at 1.29-A resolution and exploration of inhibition in the immune response. Proc. Natl. Acad. Sci. USA 2005, 102, 13058–13063. [Google Scholar] [CrossRef] [Green Version]
- Di Costanzo, L.; Moulin, M.; Haertlein, M.; Meilleur, F.; Christianson, D.W. Expression, purification, assay, and crystal structure of perdeuterated human arginase I. Arch. Biochem. Biophys. 2007, 465, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Hellerman, L.A.; Perkins, M.E. Activation of enzymes III. The role of metal Ions in the activation od arginase. The hydrolysis of arginine induced by certain metal ions with urease. J. Biol. Chem. 1935, 112, 175–194. [Google Scholar]
- .Hellerman, L.A. Reversible inactivations of certain hydrolytic enzymes. Physiol. Rev. 1937, 17, 454–484. [Google Scholar] [CrossRef]
- Stone, E.M.; Glazer, E.S.; Chantranupong, L.; Cherukuri, P.; Breece, R.M.; Thierney, D.L.; Curley, S.A.; Iverson, B.L.; Georgiou, G. Replacing Mn2+ with Co2+ in human arginase i enhances cytotoxicity toward l-arginine auxotrophic cancer cell lines. ACS Chem. Biol. 2010, 5, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Antonio, E.L.; Chirstianson, D.W. Crystal structures of complexes with cobalt-reconstituted human arginase I. Biochemistry 2011, 50, 8018–8027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penninckx, M.; Simon, J.P.; Wiame, J.M. Interaction between arginase and L-ornithine carbamoyltransferase in Saccharomyces cerevisiae. Purification of S. cerevisiae enzymes and evidence that these enzymes as well as rat-liver arginase are trimers. Eur. J. Biochem. 1974, 49, 429–442. [Google Scholar] [CrossRef]
- Kanyo, Z.F.; Scolnick, L.R.; Ash, D.E.; Christianson, D.W. Structure of a unique binuclear manganese cluster in arginase. Nature 1996, 383, 554–557. [Google Scholar] [CrossRef]
- Cox, J.D.; Kim, N.N.; Traish, A.M.; Christianson, D.W. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat. Struct. Biol. 1999, 6, 1043–1048. [Google Scholar]
- Khangulov, S.V.; Sossong, T.M., Jr.; Ash, D.E.; Dismukes, G.C. l-Arginine binding to liver arginase Requires proton transfer to gateway residue His141 and coordination of the guanidinium group to the Dimanganese(II,II) center. Biochemistry 1998, 37, 8539–8550. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.C.; Jeffrey, P.D.; Patchett, M.L.; Kanyo, Z.F.; Aker, E.N. Crystal structures of Bacillus caldovelox arginase in complex with substrate and inhibitors reveal new insights into activation, inhibition and catalysis in the arginase superfamily. Structure 1999, 7, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Cama, E.; Emig, F.A.; Ash, D.E.; Christianson, D.W. Structural and functional importance of first-shell metal ligands in the binuclear manganese cluster of arginase I. Biochemistry 2003, 42, 7748–7758. [Google Scholar] [CrossRef] [PubMed]
- Leopoldini, M.; Russo, N.; Toscano, M. Determination of the catalytic pathway of a manganese arginase enzyme through density functional investigation. Chem. Eur. J. 2009, 15, 8026–8036. [Google Scholar] [CrossRef]
- Marino, T.; Russo, N.; Toscano, M. What occurs by replacing Mn2+ with Co2+ in human arginase I: First-principles computational analysis. Inorg. Chem. 2013, 52, 655–659. [Google Scholar] [CrossRef]
- Srivastava, A.; Dwivedi, N.; Samanta, U.; Sau, A.K. Insight into the role of a unique SSEHA motif in the activity and stability of Helicobacter pylori arginase. IUBMB Life 2011, 63, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Libera, J.L.; Caballero, J.; Tuñón, I.; Hernández-Rodríguez, E.W.; Ruiz-Pernía, J. On the nature of the enzyme–substrate complex and the reaction mechanism in human arginase I. A combined molecular dynamics and QM/MM study. ACS Catal. 2020, 10, 8321–8333. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
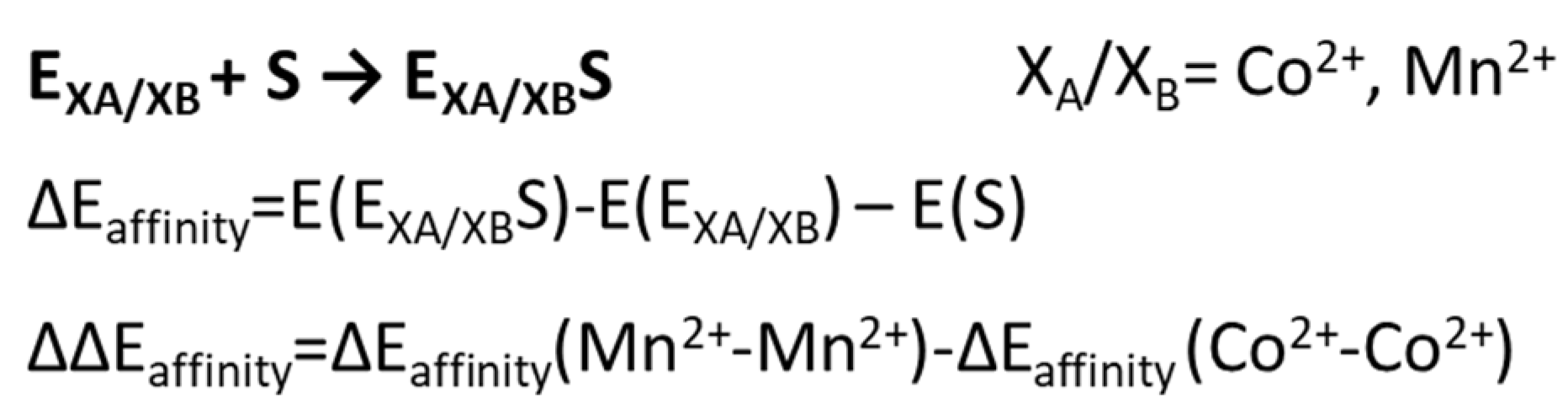{kind=link}
| Distances | ES | TS1 | INT1 | TS2 | INT2 | TS3 | EP |
|---|---|---|---|---|---|---|---|
| M3+-O | 2.57 (2.45) | 2.85 (2.83) | 2.84 (2.83) | 4.83 (2.89) | 5.73 (4.82) | n.a. | n.a. |
| C5PQQ-OA | 4.00 (3.72) | 1.89 (1.63) | 1.48 (1.48) | 2.37 (1.74) | 4.15 (3.56) | n.a. | n.a. |
| OAsp299-HA | 1.52 (1.60) | 1.04 (1.00) | 1.00 (0.99) | 1.00 (1.00) | 1.02 (1.04) | 1.12 (1.11) | 1.51 (1.37) |
| O4PQQ-HB | 2.84 (2.84) | 3.16 (3.21) | 2.93 (3.03) | 1.35 (1.24) | 0.97 (0.97) | 0.99 (0.97) | 0.98 (0.97) |
| O5PQQ-HA | 3.40 (3.39) | 3.82 (2.83) | 2.76 (2.75) | 1.65 (2.34) | 1.54 (1.47) | 1.22 (1.25) | 1.02 (1.05) |
| Distances | Ce3+-MDH | Eu3+-MDH |  |
| M3+-O1Glu172 | 2.55 | 2.47 | |
| M3+-O2Glu172 | 2.58 | 2.48 | |
| M3+-OAsn256 | 2.49 | 2.45 | |
| M3+-O5PQQ | 2.83 | 2.83 | |
| M3+-N6PQQ | 3.00 | 3.00 | |
| M3+-O7PQQ | 2.79 | 2.79 | |
| M3+-O1Asp301 | 2.55 | 2.52 | |
| M3+-O2Asp301 | 2.55 | 2.55 | |
| M3+-O1Asp299 | 2.42 | 2.53 |
| Distances | ES | TS1 | INT1 | TS2 | INT2 | TS3 | EP |
|---|---|---|---|---|---|---|---|
| M3+-NA | 2.00 (2.04) | 1.89 (1.88) | 1.87 (1.88) | 1.87 (2.12) | 1.89 (1.91) | 2.07 (2.10) | 3.87 (3.99) |
| C-OA | 3.53 (3.94) | 2.01 (1.79) | 1.41 (1.36) | 1.36 (1.37) | 1.28 (1.27) | 1.23 (1.22) | 1.26 (1.26) |
| NA-HD | 2.37 (3.43) | 2.22 (2.10) | 1.83 (3.20) | 1.71 (1.29) | 1.02 (1.02) | 1.02 (1.02) | 1.01 (1.01) |
| OB-HA | 1.70 (1.69) | 2.05 (1.73) | 1.63 (1.51) | 1.14 (1.57) | 0.98 (0.98) | 1.29 (1.35) | 1.01 (1.03) |
| OC-HB | 1.69 (1.83) | 1.83 (4.23) | 1.63 (1.82) | 1.25 (1.49) | 0.98 (0.98) | 1.05 (1.01) | 0.96 (0.97) |
| OD-HC | 1.96 (4.00) | 2.26 (1.81) | 2.00 (1.93) | 1.75 (2.03) | 0.97 (0.99) | 0.96 (0.97) | 0.98 (0.98) |
| NA-HA | 2.66 (2.92) | 3.11 (3.12) | 2.94 (3.08) | 2.98 (3.06) | 4.02 (2.87) | 1.24 (1.19) | 1.02 (1.02) |
| OB-SA | 3.85 (3.80) | 4.68 (4.29) | 5.11 (5.19) | 5.03 (5.42) | 5.06 (3.44) | 2.69 (2.47) | 1.70 (1.66) |
| Distances | ES | TS1 | INT1 | TS2 | INT2 | TS3 | EP |
|---|---|---|---|---|---|---|---|
| XA-OA | 2.02 (2.07) | 2.17 (2.28) | 2.89 (2.69) | 2.41 (2.26) | 2.19 (2.29) | 2.54 (2.35) | 3.20 (2.54) |
| XB-OA | 1.98 (2.07) | 2.12 (2.21) | 2.25 (2.16) | 2.21 (2.32) | 2.13 (2.16) | 2.05 (2.18) | 1.98 (2.14) |
| OA-C | 2.79 (2.83) | 1.80 (1.75) | 1.44 (1.48) | 1.41 (1.43) | 1.34 (1.34) | 1.30 (1.30) | 1.27 (1.28) |
| N-HA | 2.83 (2.81) | 2.47 (2.43) | 2.53 (2.64) | 2.60 (2.47) | 1.06 (1.06) | 1.03 (1.04) | 1.02 (1.02) |
| N-C | 1.36 (1.34) | 1.40 (1.40) | 1.45 (1.45) | 1.45 (1.45) | 1.59 (1.61) | 1.96 (1.96) | 3.42 (3.31) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prejanò, M.; Alberto, M.E.; Russo, N.; Toscano, M.; Marino, T. The Effects of the Metal Ion Substitution into the Active Site of Metalloenzymes: A Theoretical Insight on Some Selected Cases. Catalysts 2020, 10, 1038. https://doi.org/10.3390/catal10091038
Prejanò M, Alberto ME, Russo N, Toscano M, Marino T. The Effects of the Metal Ion Substitution into the Active Site of Metalloenzymes: A Theoretical Insight on Some Selected Cases. Catalysts. 2020; 10(9):1038. https://doi.org/10.3390/catal10091038
Chicago/Turabian StylePrejanò, Mario, Marta Erminia Alberto, Nino Russo, Marirosa Toscano, and Tiziana Marino. 2020. "The Effects of the Metal Ion Substitution into the Active Site of Metalloenzymes: A Theoretical Insight on Some Selected Cases" Catalysts 10, no. 9: 1038. https://doi.org/10.3390/catal10091038