
Sudden Cardiac Death in Biventricular Arrhythmogenic Cardiomyopathy: A New Undescribed Variant of the MYH6 Gene
 , , and
, , and 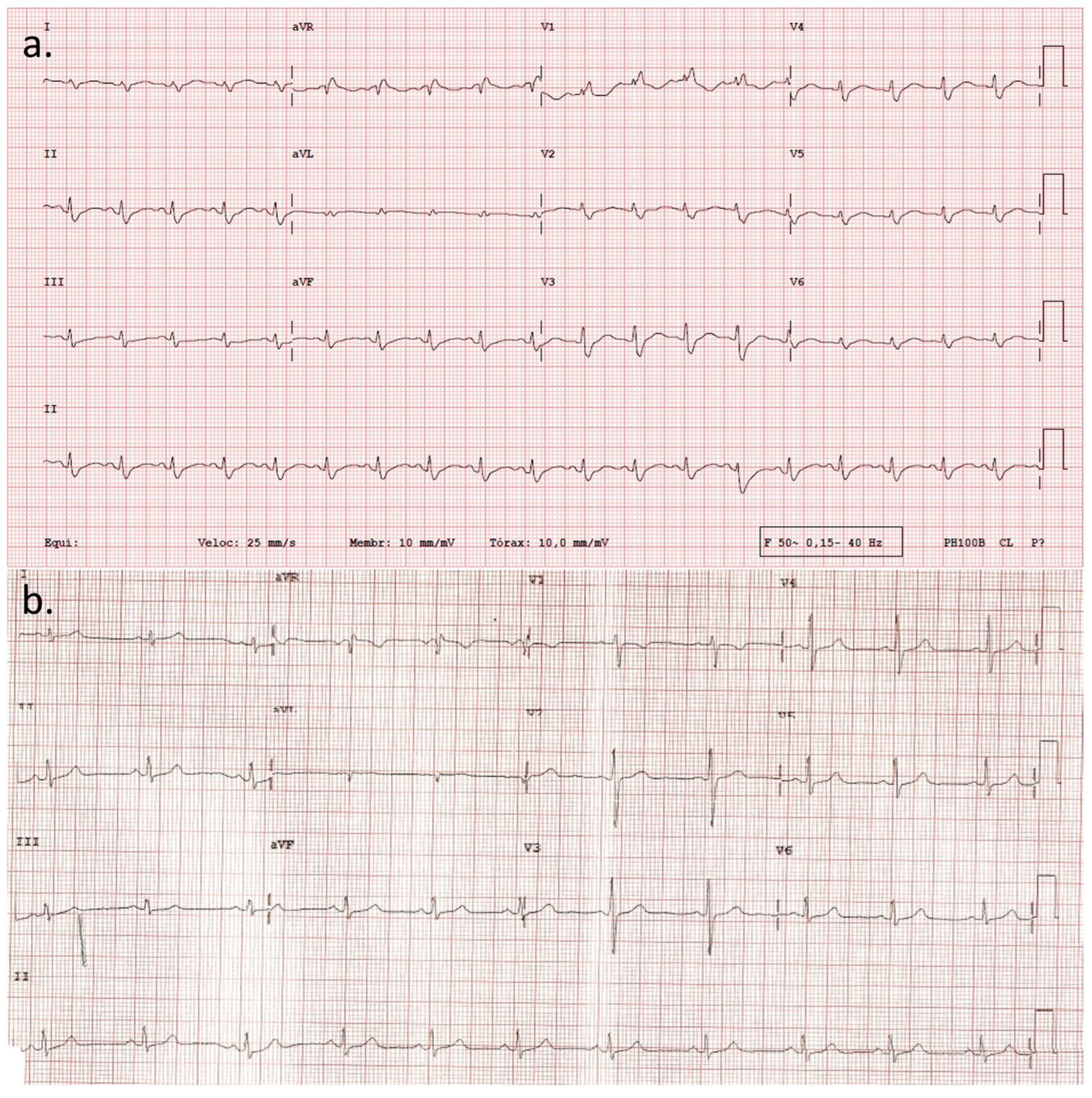{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
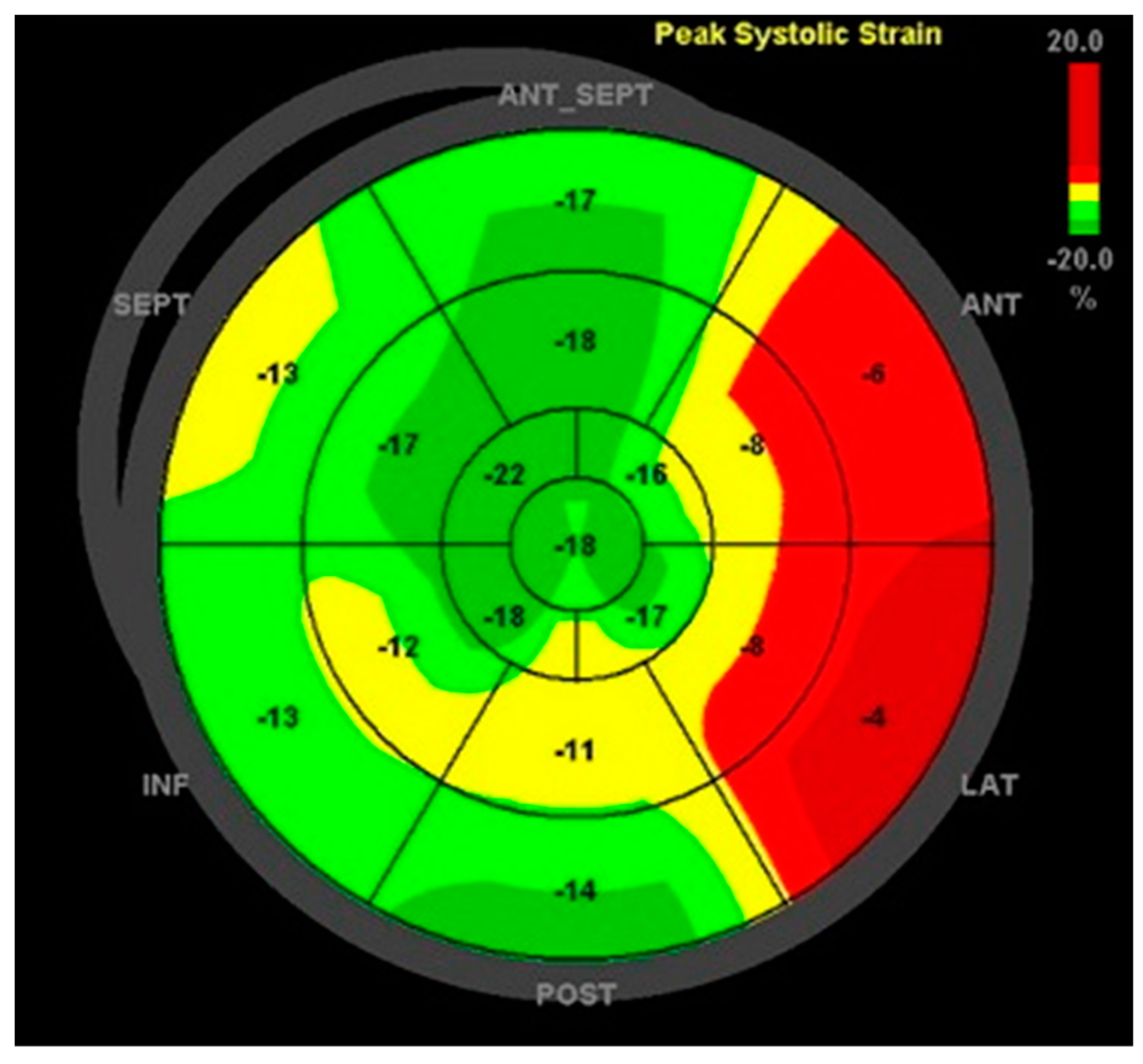
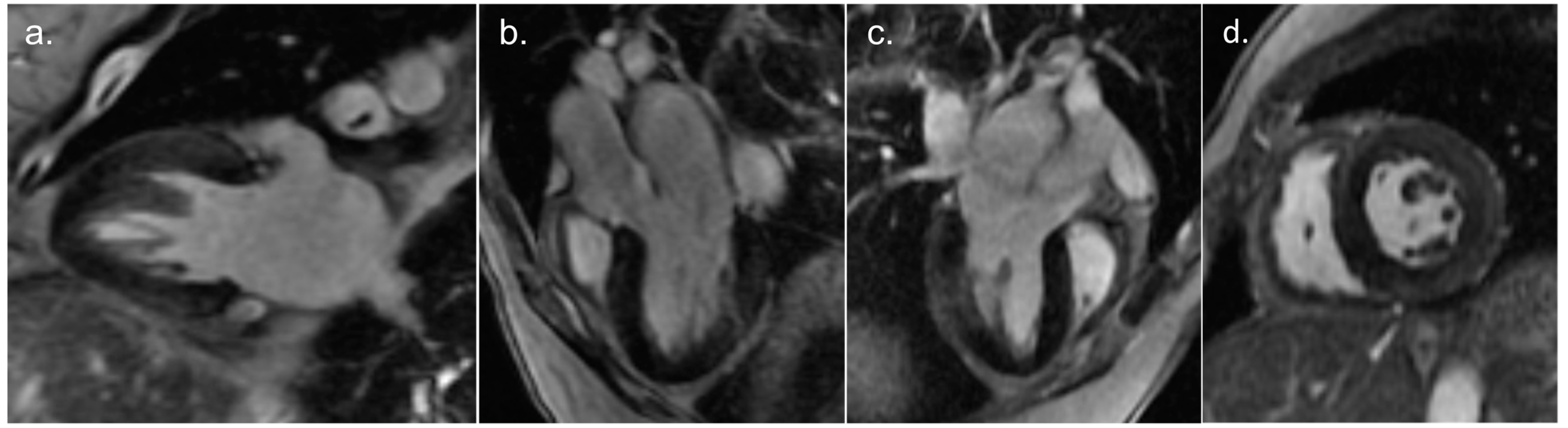
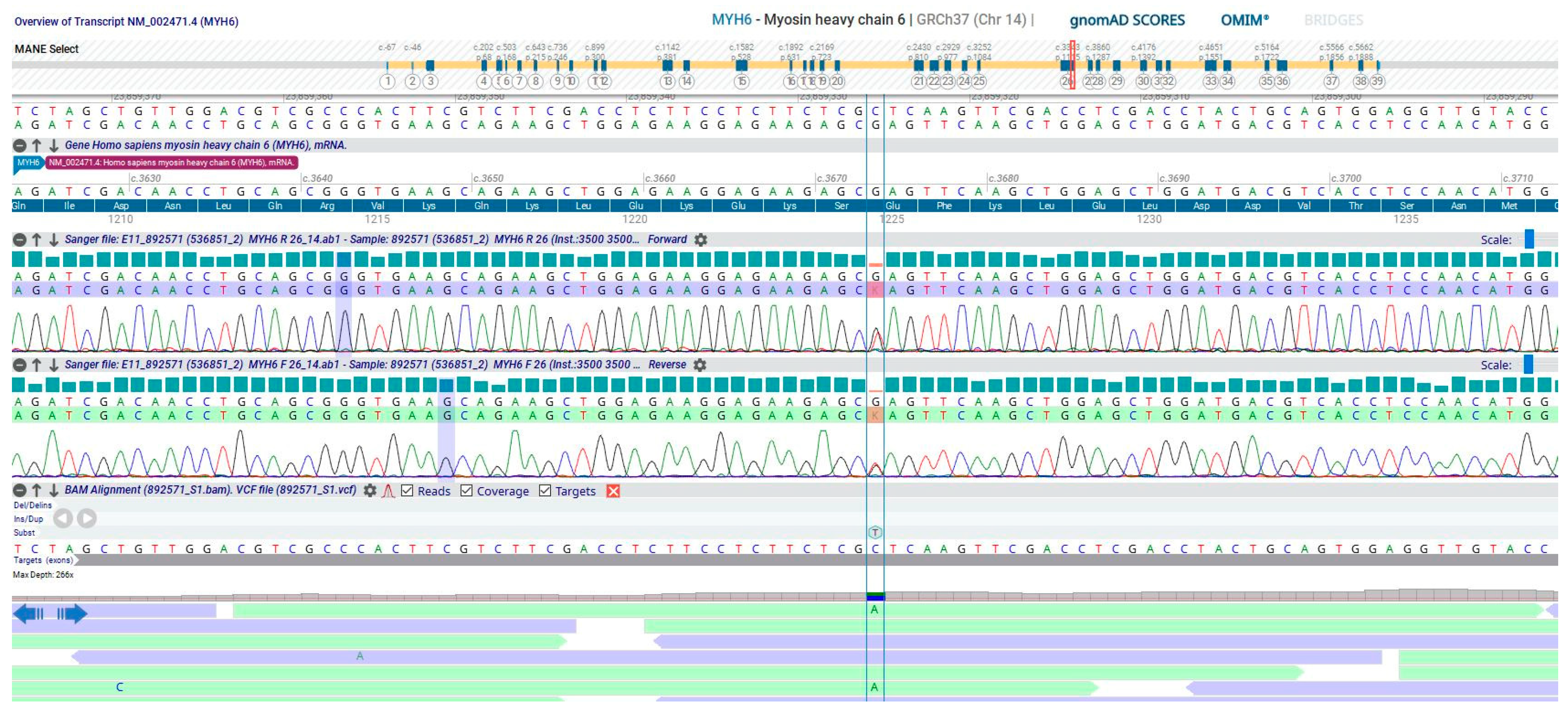
2. Detailed Case Description
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C. Arrhythmogenic left ventricular cardiomyopathy. Heart 2022, 108, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Patel, D.M.; Green, K.J. Desmosomes in the Heart: A Review of Clinical and Mechanistic Analyses. Cell Commun. Adhes. 2014, 21, 109–128. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Šarić, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual Color Photoactivation Localization Microscopy of Cardiomyopathy-associated Desmin Mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Ortiz-Genga, M.; Gomez-Milanes, I.; Lopez-Cuenca, D.; Ruiz-Espejo, F.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Monserrat, L.; Gimeno, J.R. A mutation in the Z-line Cypher/ZASP protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2015, 88, 172–176. [Google Scholar] [CrossRef]
- Good, J.-M.; Fellmann, F.; Bhuiyan, Z.A.; Rotman, S.; Pruvot, E.; Schläpfer, J. ACTN2 variant associated with a cardiac phenotype suggestive of left-dominant arrhythmogenic cardiomyopathy. Hear. Case Rep. 2020, 6, 15–19. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Rothenberg, E.; Sobreira, N.L.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Simmerman, H.; Jones, L. Phospholamban: Protein Structure, Mechanism of Action, and Role in Cardiac Function. Physiol. Rev. 1998, 78, 921–947. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Alapi, K.Z.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Mavroidis, M.; Psarras, S.; Capetanaki, Y.; Lattanzi, G. Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. Int. J. Mol. Sci. 2021, 22, 4256. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Is a Fully Penetrant, Lethal Arrhythmic Disorder Caused by a Missense Mutation in the TMEM43 Gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef]
- Padrón-Barthe, L.; Villalba-Orero, M.; Gómez-Salinero, J.M.; Domínguez, F.; Román, M.; Larrasa-Alonso, J.; Ortiz-Sánchez, P.; Martínez, F.; López-Olañeta, M.; Bonzón-Kulichenko, E.; et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3β. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef]
- Abdelfatah, N.; Chen, R.; Duff, H.J.; Seifer, C.M.; Buffo, I.; Huculak, C.; Clarke, S.; Clegg, R.; Jassal, D.S.; Gordon, P.M.; et al. Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2. JACC Basic Transl. Sci. 2019, 4, 204–221. [Google Scholar] [CrossRef]
- Ferreira, V.M.; Schulz-Menger, J.; Holmvang, G.; Kramer, C.M.; Carbone, I.; Sechtem, U.; Kindermann, I.; Gutberlet, M.; Cooper, L.T.; Liu, P.; et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation. J. Am. Coll. Cardiol. 2018, 72, 3158–3176. [Google Scholar] [CrossRef]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin Heavy Chain Isoform Expression in the Failing and Nonfailing Human Heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef]
- Mahdavi, V.; Chambers, A.P.; Nadal-Ginard, B. Cardiac alpha- and beta-myosin heavy chain genes are organized in tandem. Proc. Natl. Acad. Sci. USA 1984, 81, 2626–2630. [Google Scholar] [CrossRef]
- England, J.; Loughna, S. Heavy and light roles: Myosin in the morphogenesis of the heart. Cell Mol. Life Sci. 2013, 70, 1221–1239. [Google Scholar] [CrossRef]
- Abu-Daya, A.; Sater, A.K.; Wells, D.E.; Mohun, T.J.; Zimmerman, L.B. Absence of heartbeat in the Xenopus tropicalis mutation muzak is caused by a nonsense mutation in cardiac myosin myh6. Dev. Biol. 2009, 336, 20–29. [Google Scholar] [CrossRef]
- Razmara, E.; Garshasbi, M. Whole-exome sequencing identifies R1279X of MYH6 gene to be associated with congenital heart disease. BMC Cardiovasc. Disord. 2018, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Anfinson, M.; Fitts, R.H.; Lough, J.W.; James, J.M.; Simpson, P.M.; Handler, S.S.; Mitchell, M.E.; Tomita-Mitchell, A. Significance of α-Myosin Heavy Chain (MYH6) Variants in Hypoplastic Left Heart Syndrome and Related Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Jou, C.J.; Arrington, C.B.; Kennedy, B.J.; Earl, A.; Matsunami, N.; Meyers, L.L.; Etheridge, S.P.; Saarel, E.V.; Bleyl, S.B.; et al. Exome analysis of a family with Wolff-Parkinson-White syndrome identifies a novel disease locus. Am. J. Med. Genet. A 2015, 167, 2975–2984. [Google Scholar] [CrossRef]
- Chalazan, B.; Mol, D.; Darbar, F.A.; Ornelas-Loredo, A.; Al-Azzam, B.; Chen, Y.; Tofovic, D.; Sridhar, A.; Alzahrani, Z.; Ellinor, P.; et al. Association of Rare Genetic Variants and Early-Onset Atrial Fibrillation in Ethnic Minority Individuals. JAMA Cardiol. 2021, 6, 811. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.; Ingles, J.; Turner, C.; Kilborn, M.; Bagnall, R.D.; Semsarian, C. Exome sequencing identifies a novel mutation in the MYH6 gene in a family with early-onset sinus node dysfunction, ventricular arrhythmias, and cardiac arrest. Hear. Case Rep. 2015, 1, 141–145. [Google Scholar] [CrossRef]
- Zhao, T.; Ma, Y.; Zhang, Z.; Xian, J.; Geng, X.; Wang, F.; Huang, J.; Yang, Z.; Luo, Y.; Lin, Y. Young and early-onset dilated cardiomyopathy with malignant ventricular arrhythmia and sudden cardiac death induced by the heterozygous LDB3, MYH6, and SYNE1 missense mutations. Ann. Noninvasive Electrocardiol. 2021, 26, e12840. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Bozzao, C.; Pennacchini, E.; Pagannone, E.; Musumeci, B.M.; Piane, M.; Germani, A.; Savio, C.; Francia, P.; Volpe, M.; et al. A Next-Generation Sequencing Approach to Identify Gene Mutations in Early- and Late-Onset Hypertrophic Cardiomyopathy Patients of an Italian Cohort. Int. J. Mol. Sci. 2016, 17, 1239. [Google Scholar] [CrossRef] [PubMed]
- Carniel, E.; Taylor, M.R.; Sinagra, G.; Di Lenarda, A.; Ku, L.; Fain, P.R.; Boucek, M.M.; Cavanaugh, J.; Miocic, S.; Slavov, D.; et al. α-Myosin Heavy Chain. Circulation 2005, 112, 54–59. [Google Scholar] [CrossRef]
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N.; Hershberger, R.E. Rare Variant Mutations in Pregnancy-Associated or Peripartum Cardiomyopathy. Circulation 2010, 121, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Vershinina, T.; Fomicheva, Y.; Muravyev, A.; Jorholt, J.; Kozyreva, A.; Kiselev, A.; Gordeev, M.; Vasichkina, E.; Segrushichev, A.; Pervunina, T.; et al. Genetic Spectrum of Left Ventricular Non-Compaction in Paediatric Patients. Cardiology 2020, 145, 746–756. [Google Scholar] [CrossRef]
- Murray, B.; Hoorntje, E.T.; Riele, A.S.J.M.T.; Tichnell, C.; van der Heijden, J.F.; Tandri, H.; Berg, M.P.v.D.; Jongbloed, J.D.H.; Wilde, A.A.M.; Hauer, R.N.W.; et al. Identification of sarcomeric variants in probands with a clinical diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC). J. Cardiovasc. Electrophysiol. 2018, 29, 1004–1009. [Google Scholar] [CrossRef]
- Chen, K.; Rao, M.; Guo, G.; Chen, X.; Chen, L.; Song, J. Sarcomere variants in arrhythmogenic cardiomyopathy: Pathogenic factor or bystander? Gene 2019, 687, 82–89. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Saguner, A.M.; Magyar, I.; Bahr, A.; Akdis, D.; Brunckhorst, C.; Duru, F.; Berger, W. Arrhythmogenic right ventricular cardiomyopathy: Implications of next-generation sequencing in appropriate diagnosis. Europace 2017, 19, 1063–1069. [Google Scholar] [CrossRef]
- Huang, X.; Yan, L.; Kou, S.; Meng, J.; Lu, Z.; Lin, C.-P.; Liu, C.; Zhang, H. Generation and characterization of a Myh6-driven Cre knockin mouse line. Transgenic. Res. 2021, 30, 821–835. [Google Scholar] [CrossRef]
- Hao, E.; Zhang, G.; Mu, L.; Ma, N.; Wang, T. Establishment of a human MYH6 compound heterozygous knockout hESC line to model cardiomyopathy and congenital heart defects by CRISPR/Cas9 system. Stem. Cell Res. 2021, 50, 102128. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Šaric, T.; Cakar, H.; et al. The Novel Desmin Mutant p.A120D Impairs Filament Formation, Prevents Intercalated Disk Localization, and Causes Sudden Cardiac Death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia Brás, P.; Cardoso, I.; Viegas, J.; Antunes, D.; Rosa, S.A. Sudden Cardiac Death in Biventricular Arrhythmogenic Cardiomyopathy: A New Undescribed Variant of the MYH6 Gene. Cardiogenetics 2023, 13, 145-153. https://doi.org/10.3390/cardiogenetics13040014
Garcia Brás P, Cardoso I, Viegas J, Antunes D, Rosa SA. Sudden Cardiac Death in Biventricular Arrhythmogenic Cardiomyopathy: A New Undescribed Variant of the MYH6 Gene. Cardiogenetics. 2023; 13(4):145-153. https://doi.org/10.3390/cardiogenetics13040014
Chicago/Turabian StyleGarcia Brás, Pedro, Isabel Cardoso, José Viegas, Diana Antunes, and Sílvia Aguiar Rosa. 2023. "Sudden Cardiac Death in Biventricular Arrhythmogenic Cardiomyopathy: A New Undescribed Variant of the MYH6 Gene" Cardiogenetics 13, no. 4: 145-153. https://doi.org/10.3390/cardiogenetics13040014