
Onward Spread from Liver Metastases Is a Major Cause of Multi-Organ Metastasis in a Mouse Model of Metastatic Colon Cancer
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Organoid Culture
2.2. Ethical Guidelines
2.3. Animals
2.4. Subcutaneous Tumor Initiation
2.5. Microsurgical Implantation of Tumor Organoids into the Caecum and the Liver
2.6. Bioluminescence Imaging
2.7. Immunohistochemistry
2.8. Fluorescent Multiplex Immunohistochemistry
2.9. DNA Isolation and Library Preparation
2.10. Whole Genome Sequencing
2.11. Statistical Analyses
3. Results
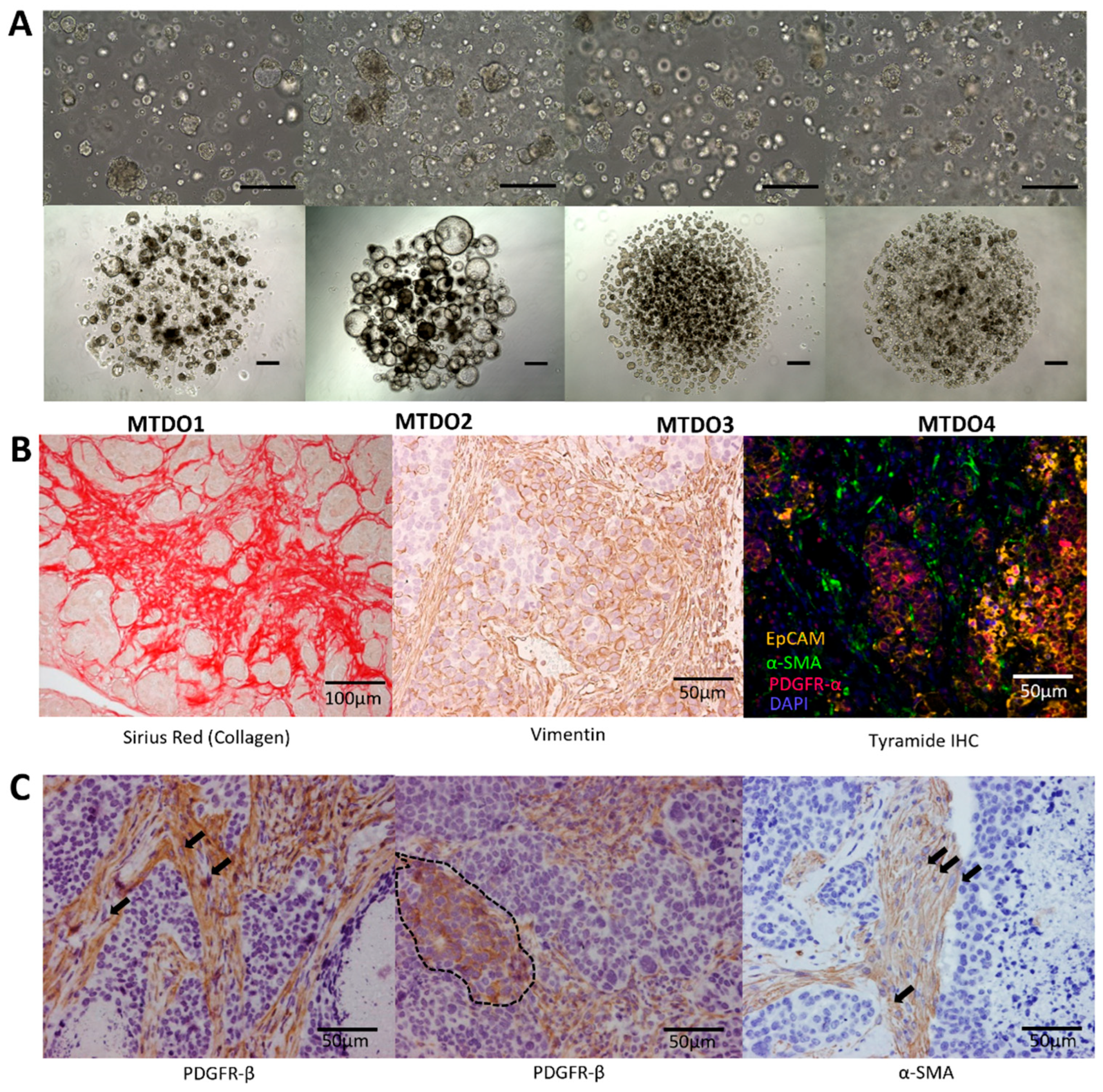
3.1. Generation of Organoids for Modeling Metastatic CRC in Immunocompetent Mice
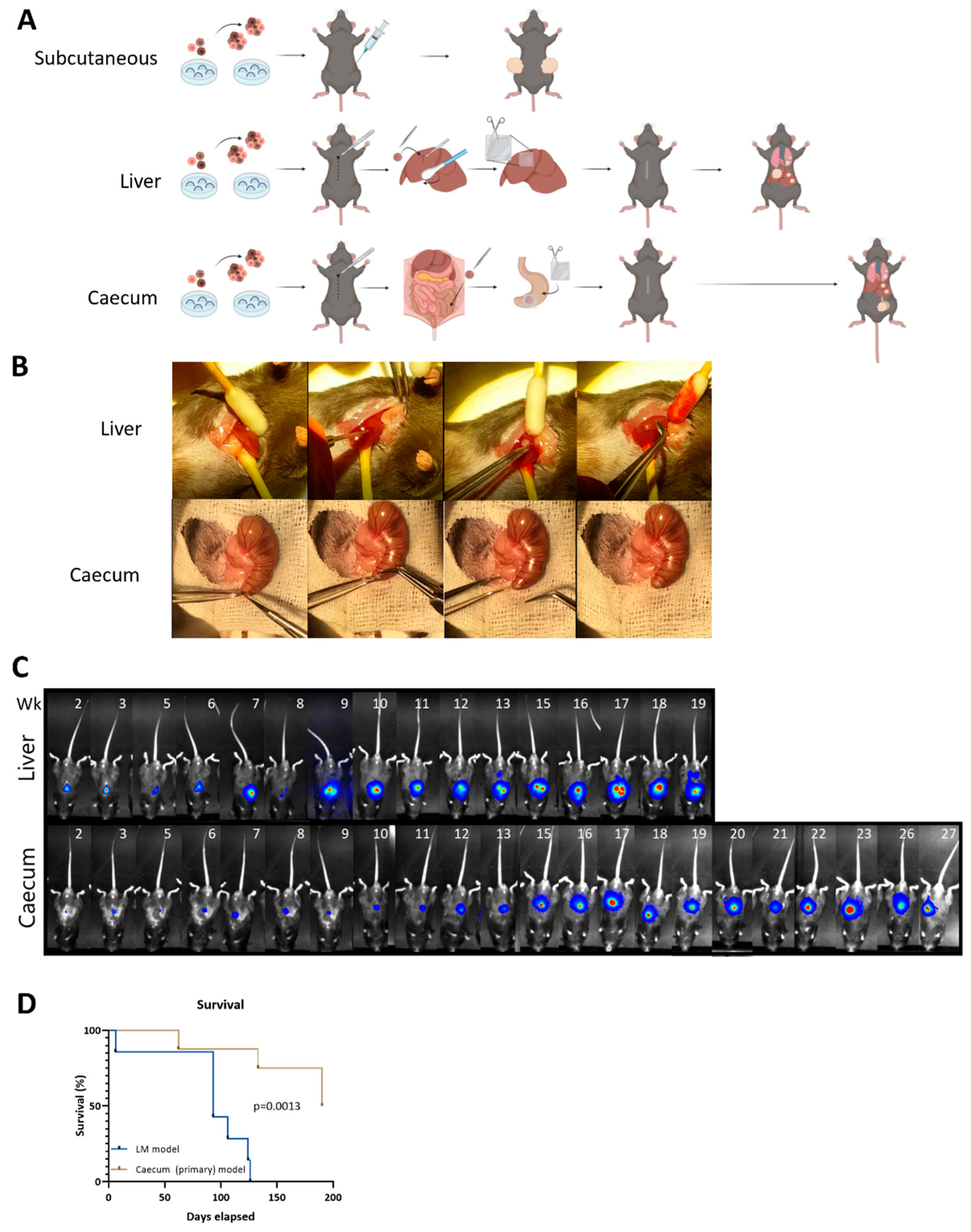
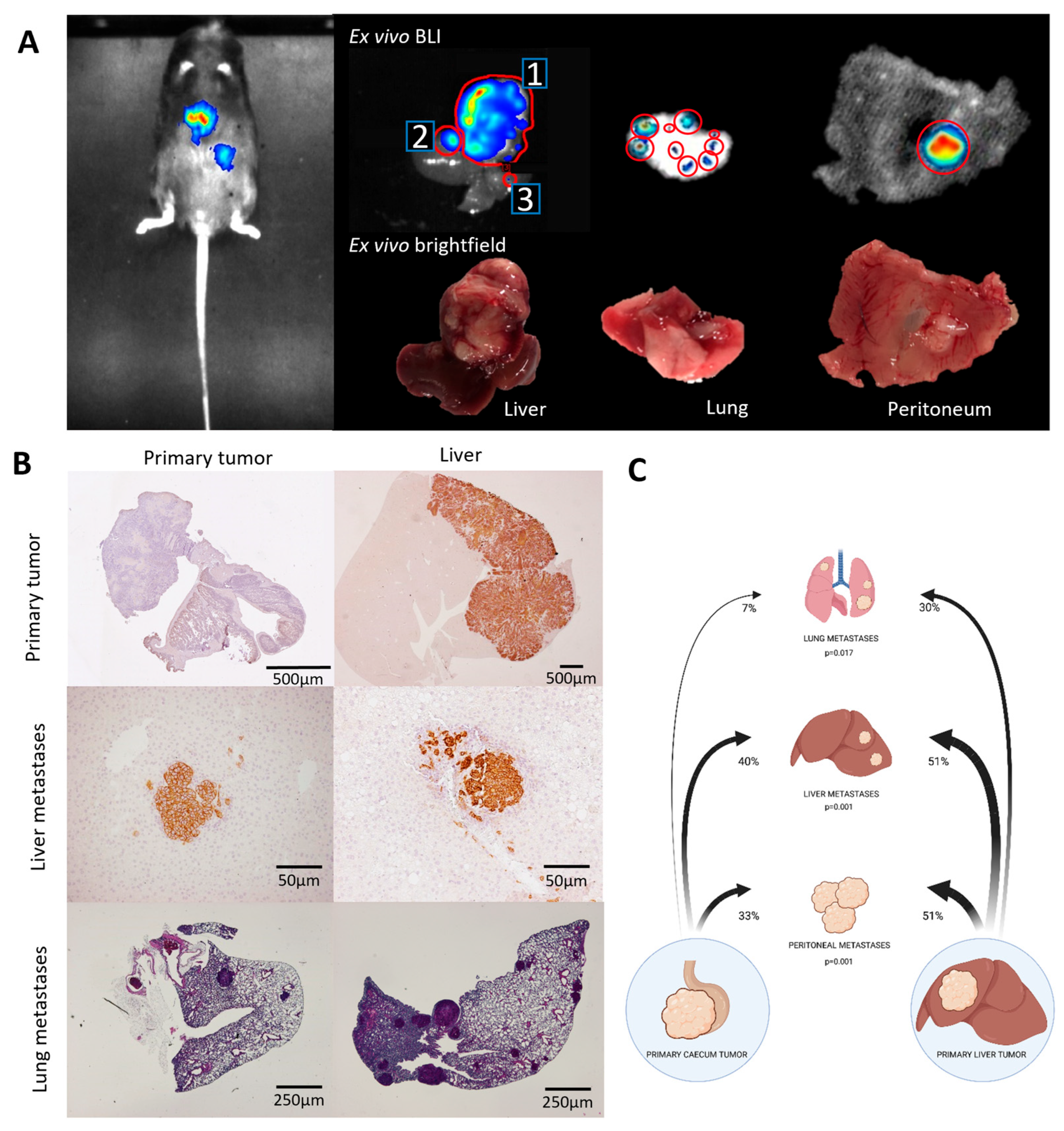
3.2. Metastatic Capacity Is Influenced by Tumor Location
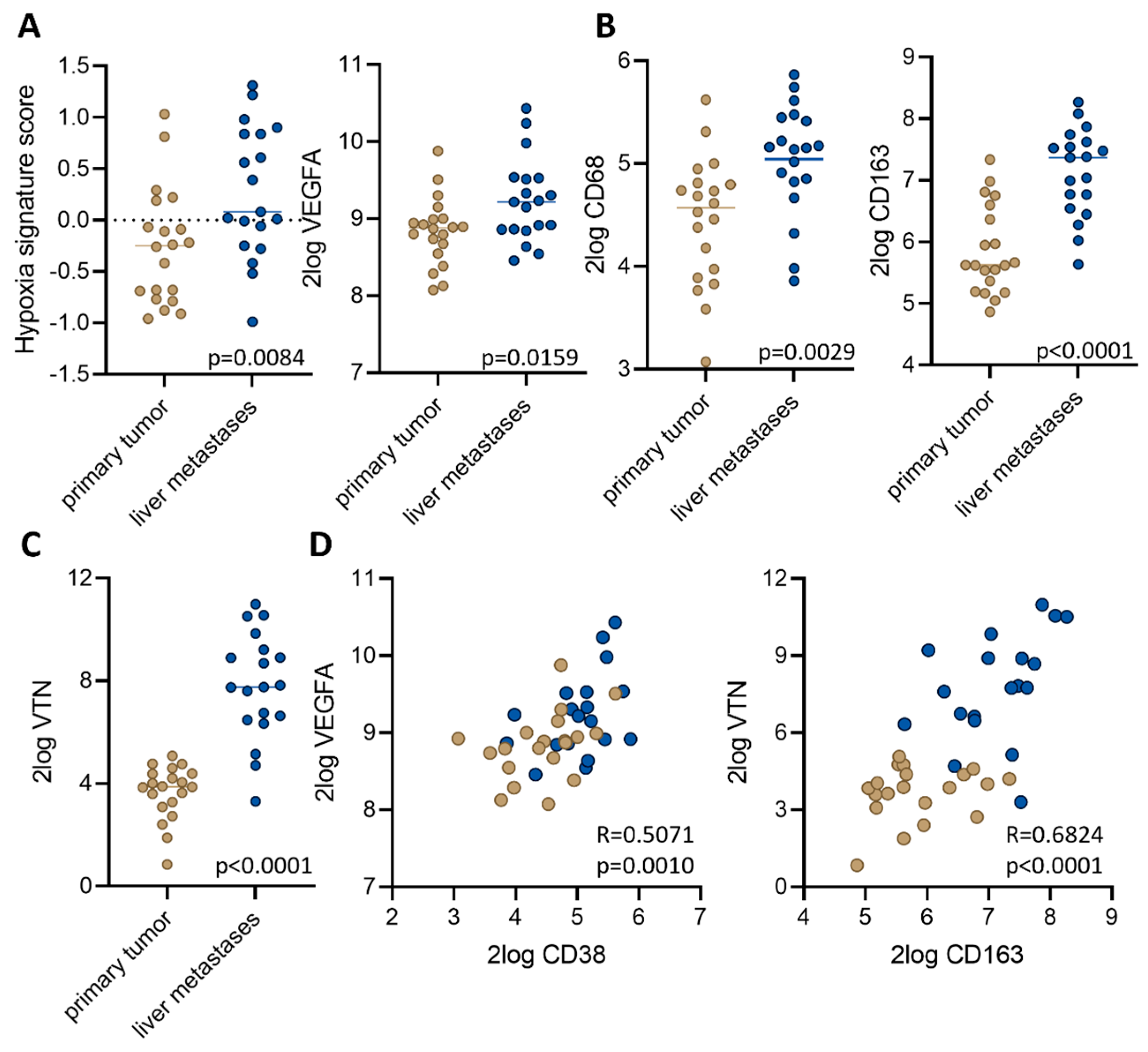
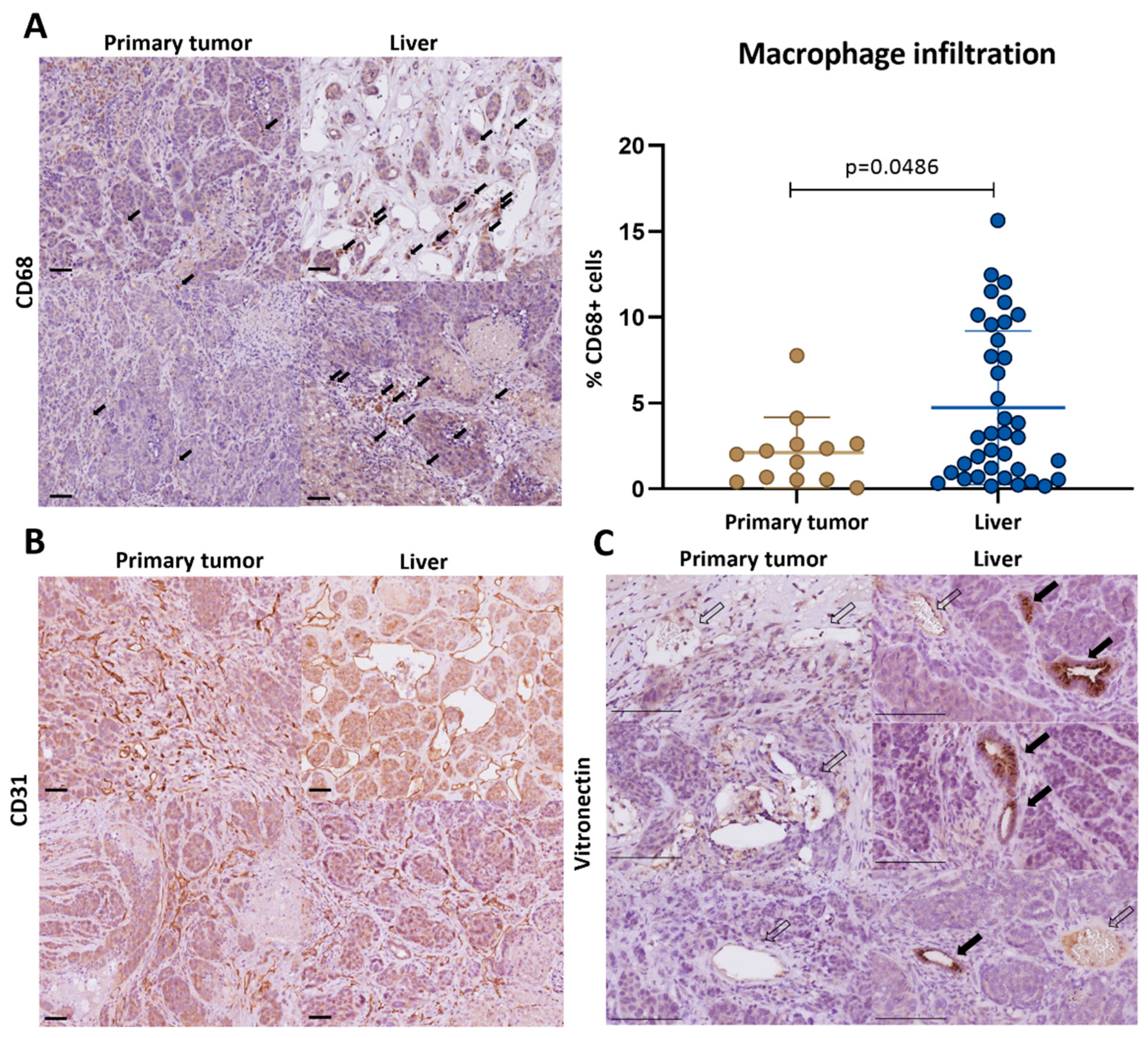
3.3. Macrophage-Associated Vascular Hotspots in Liver Metastases: Potential Portals for Onward Spread
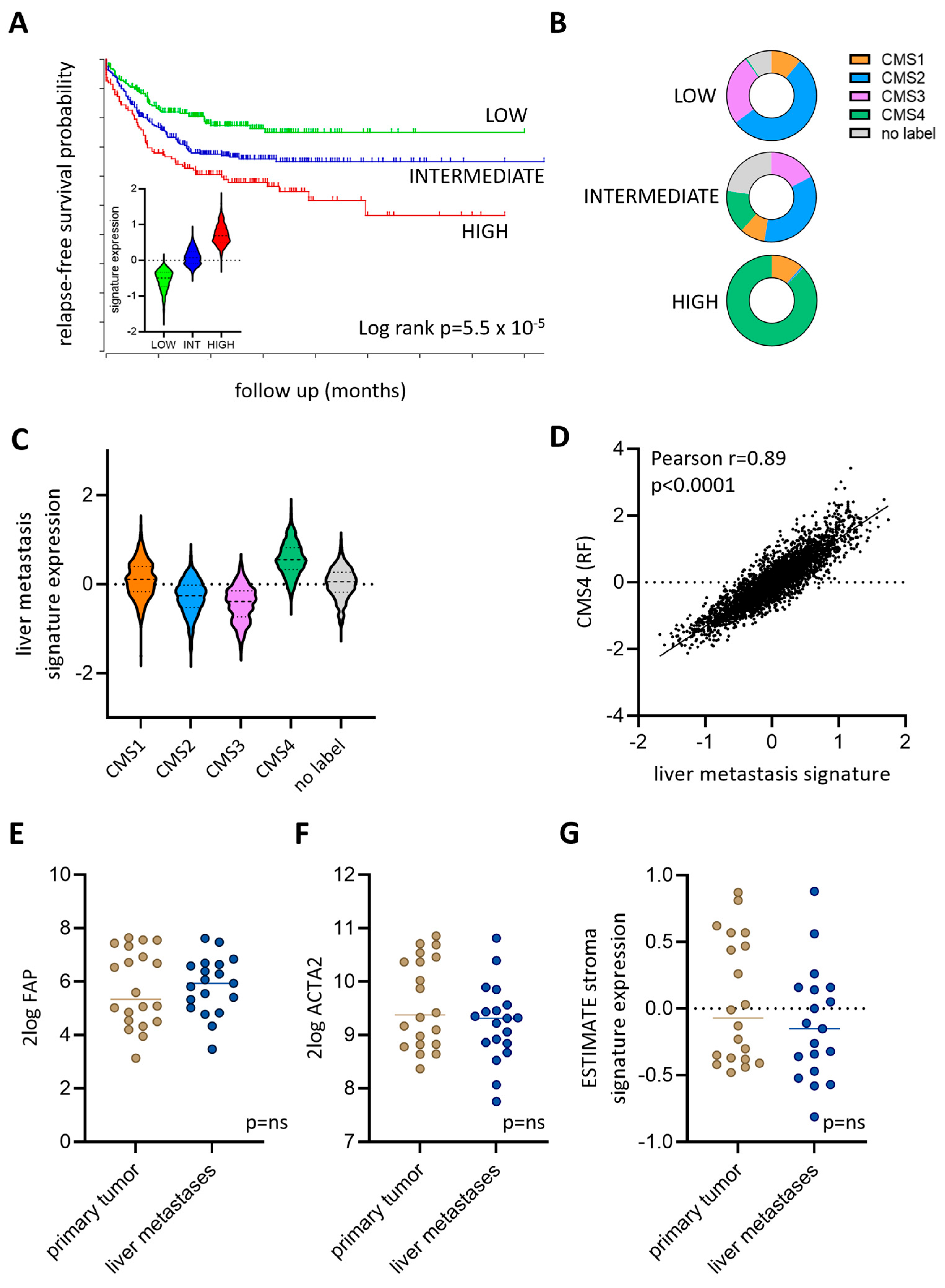
3.4. Expression of a Liver Metastasis Signature in Primary CRC Predicts Distant Metastasis Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Engstrand, J.; Nilsson, H.; Stromberg, C.; Jonas, E.; Freedman, J. Colorectal cancer liver metastases—A population-based study on incidence, management and survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Massague, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Pawlik, T.M. Molecular Mechanisms of Colorectal Liver Metastases. Cells 2023, 12, 1657. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- McDonnell, C.O.; Hill, A.D.; McNamara, D.A.; Walsh, T.N.; Bouchier-Hayes, D.J. Tumour micrometastases: The influence of angiogenesis. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2000, 26, 105–115. [Google Scholar] [CrossRef]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Chanrion, M.; Kuperstein, I.; Barriere, C.; El Marjou, F.; Cohen, D.; Vignjevic, D.; Stimmer, L.; Paul-Gilloteaux, P.; Bieche, I.; Tavares Sdos, R.; et al. Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat. Commun. 2014, 5, 5005. [Google Scholar] [CrossRef] [PubMed]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, M.; Mollevi, C.; Vezzio-Vie, N.; Bibeau, F.; Ychou, M.; Martineau, P. Specific extracellular matrix remodeling signature of colon hepatic metastases. PLoS ONE 2013, 8, e74599. [Google Scholar] [CrossRef] [PubMed]
- Stessels, F.; Van den Eynden, G.; Van der Auwera, I.; Salgado, R.; Van den Heuvel, E.; Harris, A.L.; Jackson, D.G.; Colpaert, C.G.; van Marck, E.A.; Dirix, L.Y.; et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br. J. Cancer 2004, 90, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Lalor, P.F.; Tuncer, C.; Adams, D.H. Vitronectin in human hepatic tumours contributes to the recruitment of lymphocytes in an alpha v beta3-independent manner. Br. J. Cancer 2006, 95, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Abou Khouzam, R.; Rao, S.P.; Venkatesh, G.H.; Zeinelabdin, N.A.; Buart, S.; Meylan, M.; Nimmakayalu, M.; Terry, S.; Chouaib, S. An Eight-Gene Hypoxia Signature Predicts Survival in Pancreatic Cancer and Is Associated With an Immunosuppressed Tumor Microenvironment. Front. Immunol. 2021, 12, 680435. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Strating, E.; Wassenaar, E.; Verhagen, M.; Rauwerdink, P.; van Schelven, S.; de Hingh, I.; Rinkes, I.B.; Boerma, D.; Witkamp, A.; Lacle, M.; et al. Fibroblast activation protein identifies Consensus Molecular Subtype 4 in colorectal cancer and allows its detection by (68)Ga-FAPI-PET imaging. Br. J. Cancer 2022, 127, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Trumpi, K.; Frenkel, N.; Peters, T.; Korthagen, N.M.; Jongen, J.M.J.; Raats, D.; van Grevenstein, H.; Backes, Y.; Moons, L.M.; Lacle, M.M.; et al. Macrophages induce “budding” in aggressive human colon cancer subtypes by protease-mediated disruption of tight junctions. Oncotarget 2018, 9, 19490–19507. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 2023, 23, 238–257. [Google Scholar] [CrossRef] [PubMed]
- Cortese, N.; Soldani, C.; Franceschini, B.; Barbagallo, M.; Marchesi, F.; Torzilli, G.; Donadon, M. Macrophages in Colorectal Cancer Liver Metastases. Cancers 2019, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Green, M.D.; Li, S.; Sun, Y.; Journey, S.N.; Choi, J.E.; Rizvi, S.M.; Qin, A.; Waninger, J.J.; Lang, X.; et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 2021, 27, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Saito, N.; Sugito, M.; Ito, M.; Kobayashi, A.; Nishizawa, Y. Predictive factors for pulmonary metastases after curative resection of rectal cancer without preoperative chemoradiotherapy. Dis. Colon Rectum 2011, 54, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.M.; Hsieh, P.S.; Chen, J.S.; Tang, R.; You, J.F.; Yeh, C.Y. Rectal cancer level significantly affects rates and patterns of distant metastases among rectal cancer patients post curative-intent surgery without neoadjuvant therapy. World J. Surg. Oncol. 2014, 12, 197. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.D.; Zhao, G.; An, Q.; Xiao, G. Pulmonary metastasis in rectal cancer: A retrospective study of clinicopathological characteristics of 404 patients in Chinese cohort. BMJ Open 2018, 8, e019614. [Google Scholar] [CrossRef]
- Mitry, E.; Guiu, B.; Cosconea, S.; Jooste, V.; Faivre, J.; Bouvier, A.M. Epidemiology, management and prognosis of colorectal cancer with lung metastases: A 30-year population-based study. Gut 2010, 59, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- De Gramont, A.; Van Cutsem, E.; Schmoll, H.J.; Tabernero, J.; Clarke, S.; Moore, M.J.; Cunningham, D.; Cartwright, T.H.; Hecht, J.R.; Rivera, F.; et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): A phase 3 randomised controlled trial. Lancet Oncol. 2012, 13, 1225–1233. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| From | To Liver * (p = 0.001) | To Lung (p = 0.017) | To Peritoneum (p = 0.01) |
|---|---|---|---|
| Caecum | 40% | 7% | 33% |
| Liver | 51% | 30% | 51% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijler, L.A.; Viergever, B.J.; Strating, E.; van Schelven, S.J.; Poghosyan, S.; Frenkel, N.C.; te Rietmole, H.; Verheem, A.; Raats, D.A.E.; Borel Rinkes, I.H.M.; et al. Onward Spread from Liver Metastases Is a Major Cause of Multi-Organ Metastasis in a Mouse Model of Metastatic Colon Cancer. Cancers 2024, 16, 1073. https://doi.org/10.3390/cancers16051073
Wijler LA, Viergever BJ, Strating E, van Schelven SJ, Poghosyan S, Frenkel NC, te Rietmole H, Verheem A, Raats DAE, Borel Rinkes IHM, et al. Onward Spread from Liver Metastases Is a Major Cause of Multi-Organ Metastasis in a Mouse Model of Metastatic Colon Cancer. Cancers. 2024; 16(5):1073. https://doi.org/10.3390/cancers16051073
Chicago/Turabian StyleWijler, Liza A., Bastiaan J. Viergever, Esther Strating, Susanne J. van Schelven, Susanna Poghosyan, Nicola C. Frenkel, Hedy te Rietmole, Andre Verheem, Danielle A. E. Raats, Inne H. M. Borel Rinkes, and et al. 2024. "Onward Spread from Liver Metastases Is a Major Cause of Multi-Organ Metastasis in a Mouse Model of Metastatic Colon Cancer" Cancers 16, no. 5: 1073. https://doi.org/10.3390/cancers16051073