
Chimeric Antigen Receptor T Cell and Chimeric Antigen Receptor NK Cell Therapy in Pediatric and Adult High-Grade Glioma—Recent Advances
 and
and
Abstract
:Simple Summary
Abstract
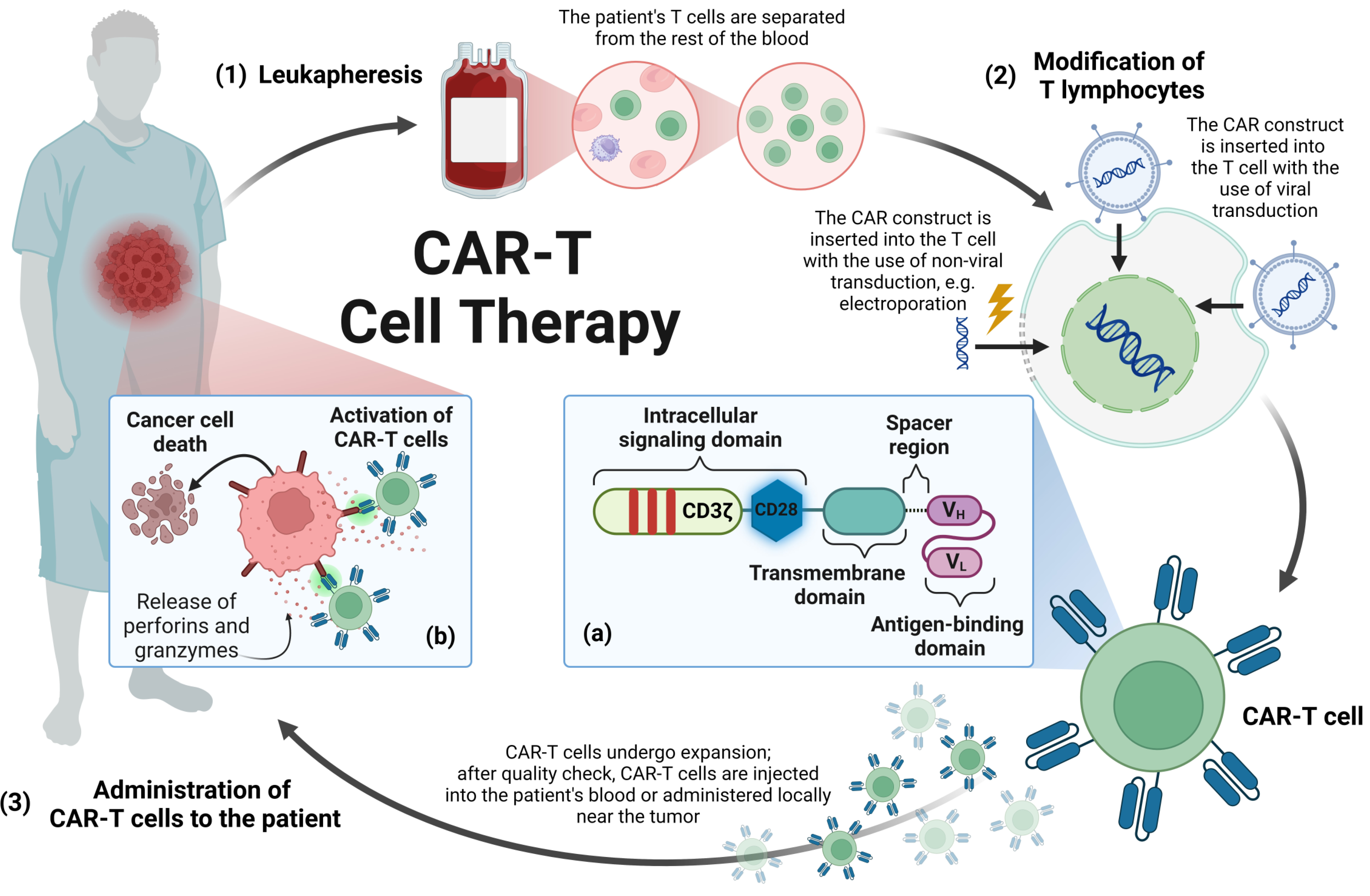
1. Introduction
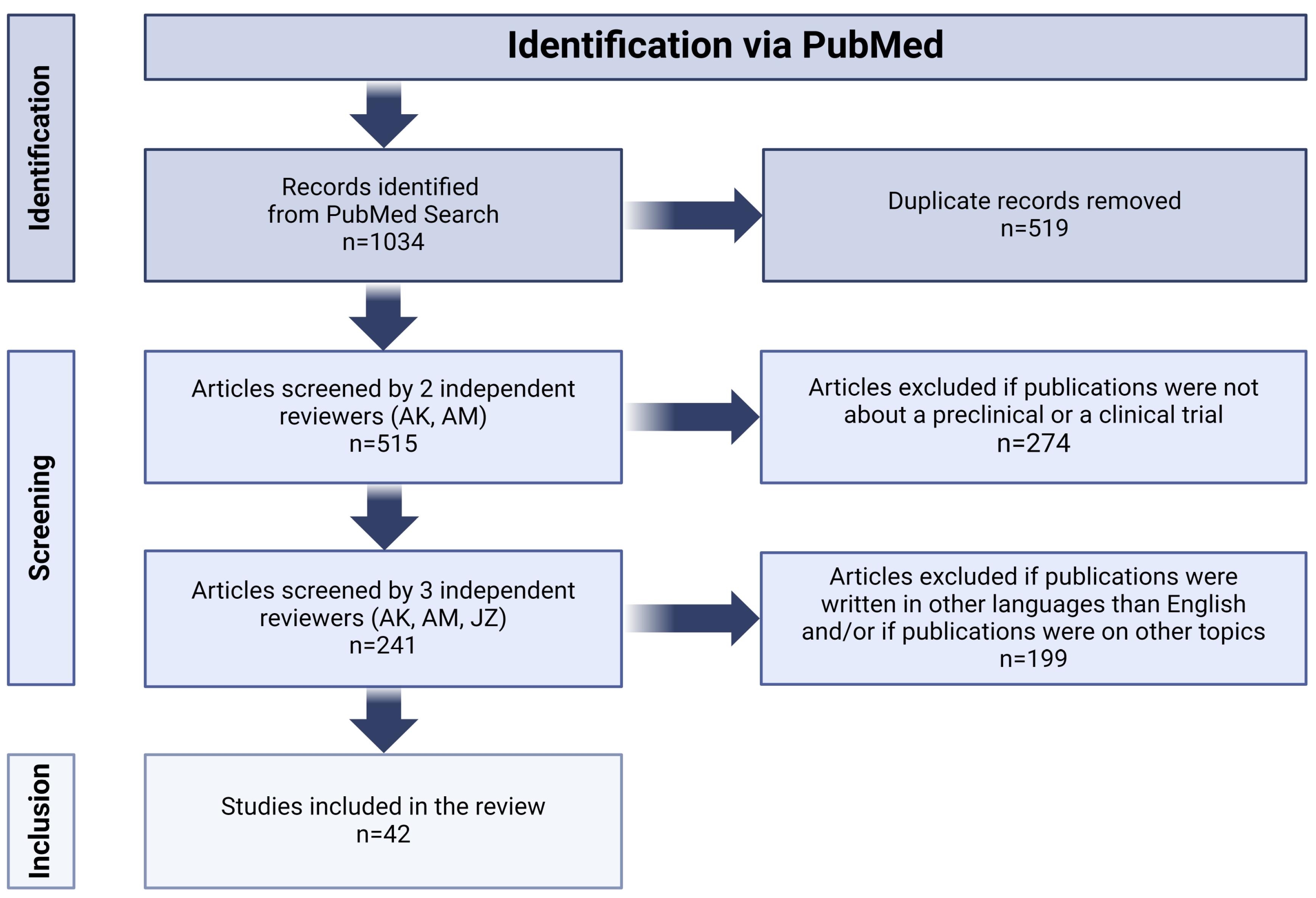
2. Materials and Methods
3. CAR-T Cell Therapy in High-Grade Glioma
3.1. CAR-T Cell Therapy in Pediatric High-Grade Glioma
3.1.1. GD2
3.1.2. B7-H3
3.1.3. Other Antigens
3.2. CAR-T Cell Therapy in Adult High-Grade Glioma
3.2.1. EphA2
3.2.2. IL-13Rα2
3.2.3. EGFRvIII
3.2.4. Other Antigens
3.3. Recruiting Clinical Trials
4. CAR-NK Cell Therapy in High-Grade Glioma
4.1. EGFRvIII
4.2. HER2
4.3. Others
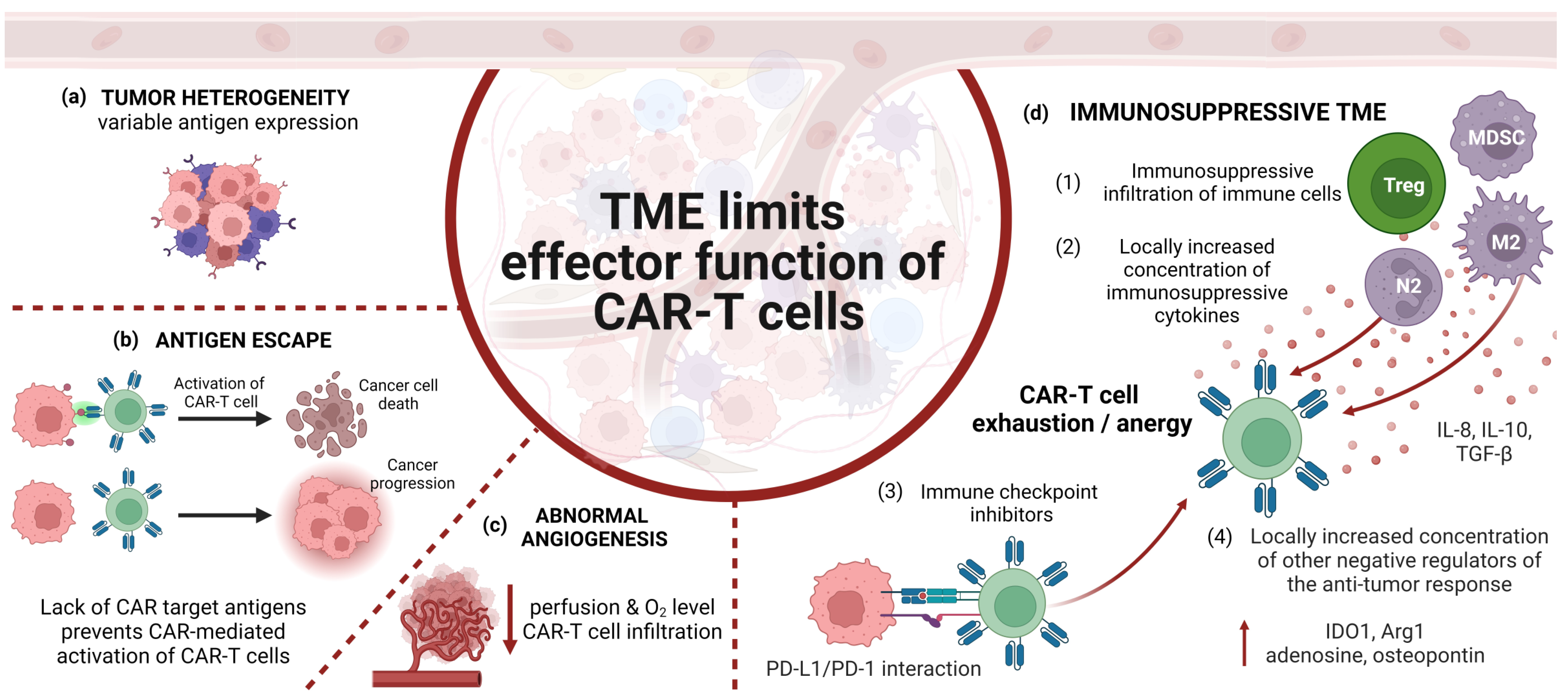
5. Limitations of CAR-Expressing Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Damodharan, S.; Puccetti, D. Pediatric Central Nervous System Tumor Overview and Emerging Treatment Considerations. Brain Sci. 2023, 13, 1106. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Luo, W.; Pehlivan, K.C.; Hoang, H.; Rajappa, P.; Cripe, T.P.; Cassady, K.A.; Lee, D.A.; Cairo, M.S. Pediatric versus adult high grade glioma: Immunotherapeutic and genomic considerations. Front. Immunol. 2022, 13, 1038096. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.G.; Garside, R.; Rogers, G.; Stein, K.; Grant, R. Temozolomide for high grade glioma. Cochrane Database Syst. Rev. 2013, 2013, CD007415. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Cooney, T.M. Epigenetic programming of pediatric high-grade glioma: Pushing beyond proof of concept to clinical benefit. Front. Cell Dev. Biol. 2022, 10, 1089898. [Google Scholar] [CrossRef] [PubMed]
- Napieralska, A.; Krzywon, A.; Mizia-Malarz, A.; Sosna-Zielińska, J.; Pawłowska, E.; Krawczyk, M.A.; Konat-Bąska, K.; Kaczorowska, A.; Dąbrowska, A.; Harat, M. High-Grade Gliomas in Children-A Multi-Institutional Polish Study. Cancers 2021, 13, 2062. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.W.; Vollmuth, P.; Foltyn-Dumitru, M.; Sahm, F.; Ahn, S.S.; Chang, J.H.; Kim, S.H. The 2021 WHO Classification for Gliomas and Implications on Imaging Diagnosis: Part 2-Summary of Imaging Findings on Pediatric-Type Diffuse High-Grade Gliomas, Pediatric-Type Diffuse Low-Grade Gliomas, and Circumscribed Astrocytic Gliomas. J. Magn. Reson. Imaging 2023, 58, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Da-Veiga, M.A.; Rogister, B.; Lombard, A.; Neirinckx, V.; Piette, C. Glioma Stem Cells in Pediatric High-Grade Gliomas: From Current Knowledge to Future Perspectives. Cancers 2022, 14, 2296. [Google Scholar] [CrossRef]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System tumours: A practical update on what neurosurgeons need to know—A minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Sejda, A.; Grajkowska, W.; Trubicka, J.; Szutowicz, E.; Wojdacz, T.; Kloc, W.; Iżycka-Świeszewska, E. WHO CNS5 2021 classification of gliomas: A practical review and road signs for diagnosing pathologists and proper patho-clinical and neuro-oncological cooperation. Folia Neuropathol. 2022, 60, 137–152. [Google Scholar] [CrossRef]
- Sugii, N.; Ninomiya, Y.; Akimoto, Y.; Tsurubuchi, T.; Ishikawa, E. H3 K27-altered diffuse midline glioma in adults arising from atypical regions: Two case reports and literature review. Radiol. Case Rep. 2023, 19, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.Y.; Won, J.K.; Park, C.K.; Kim, S.K.; Choi, S.H.; Kim, T.; Yun, H.; Park, S.H. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 2021, 38, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.; Kahn, J.; Perez, E.; Ehret, F.; Roohani, S.; Capper, D.; Schmid, S.; Kaul, D. Diffuse paediatric-type high-grade glioma, H3-wildtype and IDH-wildtype: Case series of a new entity. Brain Tumor Pathol. 2023, 40, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, V.; Carai, A.; del Baldo, G.; Vinci, M.; Cacchione, A.; Miele, E.; Rossi, S.; Antonelli, M.; Barresi, S.; Caulo, M.; et al. Molecular Landscape in Infant High-Grade Gliomas: A Single Center Experience. Diagnostics 2022, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Deacu, M.; Docu Axelerad, A.; Popescu, S.; Topliceanu, T.S.; Aschie, M.; Bosoteanu, M.; Cozaru, G.C.; Cretu, A.M.; Voda, R.I.; Orasanu, C.I. Aggressiveness of Grade 4 Gliomas of Adults. Clin. Pract. 2022, 12, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Seker-Polat, F.; Pinarbasi Degirmenci, N.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers 2022, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Zarychta, J.; Kowalczyk, A.; Krawczyk, M.; Lejman, M.; Zawitkowska, J. CAR-T Cells Immunotherapies for the Treatment of Acute Myeloid Leukemia-Recent Advances. Cancers 2023, 15, 2944. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Reppel, L.; Tsahouridis, O.; Akulian, J.; Davis, I.J.; Lee, H.; Fucà, G.; Weiss, J.; Dotti, G.; Pecot, C.V.; Savoldo, B. Targeting disialoganglioside GD2 with chimeric antigen receptor-redirected T cells in lung cancer. J. Immunother. Cancer 2022, 10, e003897. [Google Scholar] [CrossRef]
- Inagaki, F.F.; Kato, T.; Furusawa, A.; Okada, R.; Wakiyama, H.; Furumoto, H.; Okuyama, S.; Choyke, P.L.; Kobayashi, H. Disialoganglioside GD2-Targeted Near-Infrared Photoimmunotherapy (NIR-PIT) in Tumors of Neuroectodermal Origin. Pharmaceutics 2022, 14, 2037. [Google Scholar] [CrossRef]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hung, J.T.; Wang, S.H.; Cheng, J.Y.; Yu, A.L. Targeting glycosphingolipids for cancer immunotherapy. FEBS Lett. 2020, 594, 3602–3618. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Taylor, M.D. CAR T cells for childhood diffuse midline gliomas. Nat. Med. 2018, 24, 534–535. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Furukawa, K. Anti-GD2 CAR T cells could prove transformative for H3-K27M+ diffuse midline gliomas. Transl. Cancer Res. 2019, 8, S87–S93. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Tong, X.; Xu, N.; Zhang, T.; Zhou, F.; Zhu, H. CAR T-Cell Immunotherapy Treating T-ALL: Challenges and Opportunities. Vaccines 2023, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- De Billy, E.; Pellegrino, M.; Orlando, D.; Pericoli, G.; Ferretti, R.; Businaro, P.; Ajmone-Cat, M.A.; Rossi, S.; Petrilli, L.L.; Maestro, N.; et al. Dual IGF1R/IR inhibitors in combination with GD2-CAR T-cells display a potent anti-tumor activity in diffuse midline glioma H3K27M-mutant. Neuro-Oncology 2022, 24, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.T.; Jin, W.L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef]
- Guo, C.; Figueiredo, I.; Gurel, B.; Neeb, A.; Seed, G.; Crespo, M.; Carreira, S.; Rekowski, J.; Buroni, L.; Welti, J.; et al. B7-H3 as a Therapeutic Target in Advanced Prostate Cancer. Eur. Urol. 2023, 83, 224–238. [Google Scholar] [CrossRef]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef]
- Mortezaee, K. B7-H3 immunoregulatory roles in cancer. Biomed. Pharmacother. 2023, 163, 114890. [Google Scholar] [CrossRef]
- Feng, R.; Chen, Y.; Liu, Y.; Zhou, Q.; Zhang, W. The role of B7-H3 in tumors and its potential in clinical application. Int. Immunopharmacol. 2021, 101, 108153. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol. 2021, 23, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04185038 (accessed on 5 November 2023).
- Vitanza, N.A.; Wilson, A.L.; Huang, W.; Seidel, K.; Brown, C.; Gustafson, J.A.; Yokoyama, J.K.; Johnson, A.J.; Baxter, B.A.; Koning, R.W.; et al. Intraventricular B7-H3 CAR T Cells for Diffuse Intrinsic Pontine Glioma: Preliminary First-in-Human Bioactivity and Safety. Cancer Discov. 2023, 13, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Davenport, A.J.; Iliopoulos, M.; Hughes-Parry, H.E.; Watson, K.A.; Arcucci, V.; Mulazzani, M.; Eisenstat, D.D.; Hansford, J.R.; Cross, R.S.; et al. HER2 chimeric antigen receptor T cell immunotherapy is an effective treatment for diffuse intrinsic pontine glioma. Neuro-Oncol. Adv. 2023, 5, vdad024. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, C.; Shuaizhen, Q.; Yu, R.; Zheng, Y. Design of integrin αvβ3 targeting self-assembled protein nanoparticles with RGD peptide. Biomed. Pharmacother. 2020, 128, 110236. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zhao, Y.; Sun, J.; Yang, T.; Zhi, D.; Zhang, E.; Zhong, F.; Zhen, Y.; Zhang, S.; Zhang, S. Integrin αvβ3-targeted liposomal drug delivery system for enhanced lung cancer therapy. Colloids Surf. B Biointerfaces 2021, 201, 111623. [Google Scholar] [CrossRef] [PubMed]
- Cobb, D.A.; de Rossi, J.; Liu, L.; An, E.; Lee, D.W. Targeting of the alphav beta3 integrin complex by CAR-T cells leads to rapid regression of diffuse intrinsic pontine glioma and glioblastoma. J. Immunother. Cancer 2022, 10, e003816. [Google Scholar] [CrossRef] [PubMed]
- Shiuan, E.; Chen, J. Eph Receptor Tyrosine Kinases in Tumor Immunity. Cancer Res. 2016, 76, 6452–6457. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, A.; Cornelison, D.D.W. Eph/Ephrin-Based Protein Complexes: The Importance of cis Interactions in Guiding Cellular Processes. Front. Mol. Biosci. 2022, 8, 809364. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Scott, A.M.; Janes, P.W. Eph Receptors in Cancer. Biomedicines 2023, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef]
- Wilson, K.; Shiuan, E.; Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 2021, 40, 2483–2495. [Google Scholar] [CrossRef]
- Yi, Z.; Prinzing, B.L.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar] [CrossRef]
- An, Z.; Hu, Y.; Bai, Y.; Zhang, C.; Xu, C.; Kang, X.; Yang, S.; Li, W.; Zhong, X. Antitumor activity of the third generation EphA2 CAR-T cells against glioblastoma is associated with interferon gamma induced PD-L1. Oncoimmunology 2021, 10, 1960728. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Wang, R.; Li, W.; Zhang, Z.; Chang, Y.; Hu, Y.; Zhao, J.; Zheng, X.; Mao, Q.; Xia, H. A novel TanCAR targeting IL13Rα2 and EphA2 for enhanced glioblastoma therapy. Mol. Ther. Oncolytics 2022, 24, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients with Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.; Middlemiss, S.; Saletta, F.; Gottschalk, S.; McCowage, G.B.; Kramer, B. Chimeric Antigen Receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 2021, 28, 321–334. [Google Scholar] [CrossRef]
- Thaci, B.; Brown, C.E.; Binello, E.; Werbaneth, K.; Sampath, P.; Sengupta, S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro-Oncology 2014, 16, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, 3326. [Google Scholar] [CrossRef] [PubMed]
- Sattiraju, A.; Solingapuram Sai, K.K.; Xuan, A.; Pandya, D.N.; Almaguel, F.G.; Wadas, T.J.; Herpai, D.M.; Debinski, W.; Mintz, A. IL13RA2 targeted alpha particle therapy against glioblastomas. Oncotarget 2017, 8, 42997–43007. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, J.; Yang, Y.Z.; Wang, F.; Jiang, H.; Chen, H.D.; Wu, H.Y.; Sai, K.; Hu, W.M. IL13RA2 is overexpressed in malignant gliomas and related to clinical outcome of patients. Am. J. Transl. Res. 2020, 12, 4702–4714. [Google Scholar]
- Xu, C.; Bai, Y.; An, Z.; Hu, Y.; Zhang, C.; Zhong, X. IL-13Rα2 humanized scFv-based CAR-T cells exhibit therapeutic activity against glioblastoma. Mol. Ther. Oncolytics 2022, 24, 443–451. [Google Scholar] [CrossRef]
- Starr, R.; Aguilar, B.; Gumber, D.; Maker, M.; Huard, S.; Wang, D.; Chang, W.C.; Brito, A.; Chiu, V.; Ostberg, J.R.; et al. Inclusion of 4-1BB Costimulation Enhances Selectivity and Functionality of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells. Cancer Res. Commun. 2023, 3, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.P.; Wang, G.Y.; Arima, K.; Guan, S.P.; Waters, M.R.; Cavenee, W.K.; Pan, E.; Aliwarga, E.; Chong, S.T.; Kok, C.Y.L.; et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat. Commun. 2017, 8, 1913. [Google Scholar] [CrossRef]
- Schmidts, A.; Srivastava, A.A.; Ramapriyan, R.; Bailey, S.R.; Bouffard, A.A.; Cahill, D.P.; Carter, B.S.; Curry, W.T.; Dunn, G.P.; Frigault, M.J.; et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Rα2 are effective against heterogeneous glioblastoma. Neurooncol. Adv. 2022, 5, vdac185. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules. 2021, 26, 6677. [Google Scholar] [CrossRef]
- Kowalski, D.; Krzakowski, M.; Janowicz-Żebrowska, A. Epidermal growth factor receptor (EGFR) inhibition on non-small-cell lung cancer treatment. Onkol. Prak. Klin. 2011, 7, 177–182. [Google Scholar]
- Sierko, E.; Wojtukiewicz, M. Interfering the EGFR activity—New options of therapy for patients with glial neoplasms? Onkol. Prak. Klin. 2011, 7, 215–223. [Google Scholar]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Burgess, A.W.; Clayton, A.H.; Scott, A.M. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 2012, 72, 2924–2930. [Google Scholar] [CrossRef] [PubMed]
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Cecchini, M.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Vitanza, N.A.; Orentas, R.J.; et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 2019, 10, 7080–7095. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sun, R.; Shi, B.; Wang, Y.; Di, S.; Luo, H.; Sun, Y.; Li, Z.; Zhou, M.; Jiang, H. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. Biomed. Pharmacother. 2019, 113, 108734. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gao, H.; Kong, J.; Song, B.; Wang, P.; Shi, B.; Wang, H.; Li, Z. Selective Targeting of Glioblastoma with EGFRvIII/EGFR Bitargeted Chimeric Antigen Receptor T Cell. Cancer Immunol. Res. 2018, 6, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.C.; Verdon, D.J.; Gracey, F.M.; Hughes-Parry, H.E.; Iliopoulos, M.; Watson, K.A.; Mulazzani, M.; Luong, K.; D’Arcy, C.; Sullivan, L.C.; et al. Novel high-affinity EGFRvIII-specific chimeric antigen receptor T cells effectively eliminate human glioblastoma. Clin. Transl. Immunol. 2021, 10, e1283. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Tang, O.Y.; Tian, L.; Yoder, T.; Xu, R.; Kulikovskaya, I.; Gupta, M.; Melenhorst, J.J.; Lacey, S.F.; O’Rourke, D.M.; Binder, Z.A. PD1 Expression in EGFRvIII-Directed CAR T Cell Infusion Product for Glioblastoma Is Associated with Clinical Response. Front. Immunol. 2022, 13, 872756. [Google Scholar] [CrossRef]
- Prapa, M.; Chiavelli, C.; Golinelli, G.; Grisendi, G.; Bestagno, M.; Di Tinco, R.; Dall’Ora, M.; Neri, G.; Candini, O.; Spano, C.; et al. GD2 CAR T cells against human glioblastoma. NPJ Precis. Oncol. 2021, 5, 93. [Google Scholar] [CrossRef]
- Gargett, T.; Ebert, L.M.; Truong, N.T.H.; Kollis, P.M.; Sedivakova, K.; Yu, W.; Yeo, E.C.F.; Wittwer, N.L.; Gliddon, B.L.; Tea, M.N.; et al. GD2-targeting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. J. Immunother. Cancer 2022, 10, e005187. [Google Scholar] [CrossRef] [PubMed]
- Saleh, H.A.; Mitwasi, N.; Ullrich, M.; Kubeil, M.; Toussaint, M.; Deuther-Conrad, W.; Neuber, C.; Arndt, C.; Loureiro, L.R.; Kegler, A.; et al. Specific and safe targeting of glioblastoma using switchable and logic-gated RevCAR T cells. Front. Immunol. 2023, 14, 1166169. [Google Scholar] [CrossRef]
- Liu, K.; Tsung, K.; Attenello, F.J. Characterizing Cell Stress and GRP78 in Glioma to Enhance Tumor Treatment. Front. Oncol. 2020, 10, 608911. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wei, W.; Yuan, Y.; Sun, B.; Yang, D.; Liu, N.; Zhao, X. Chimeric antigen receptor T cells targeting cell surface GRP78 efficiently kill glioblastoma and cancer stem cells. J. Transl. Med. 2023, 21, 493. [Google Scholar] [CrossRef] [PubMed]
- Joyce, T.; Jagasia, S.; Tasci, E.; Camphausen, K.; Krauze, A.V. An Overview of CD133 as a Functional Unit of Prognosis and Treatment Resistance in Glioblastoma. Curr. Oncol. 2023, 30, 8278–8293. [Google Scholar] [CrossRef] [PubMed]
- Cimato, T.R.; Conway, A.; Nichols, J.; Wallace, P.K. CD133 expression in circulating hematopoietic progenitor cells. Cytom. B Clin. Cytom. 2019, 96, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.; Chen, T.; Chang, L.J.; et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef]
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct. Target. Ther. 2021, 6, 125. [Google Scholar] [CrossRef]
- CAR T Cells to Target GD2 for DMG (CARMIGO). U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05544526 (accessed on 5 November 2023).
- GD2 CAR T Cells in Diffuse Intrinsic Pontine Gliomas(DIPG) & Spinal Diffuse Midline Glioma (DMG). U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04196413 (accessed on 5 November 2023).
- C7R-GD2.CAR T Cells for Patients With GD2-expressing Brain Tumors (GAIL-B). U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04099797 (accessed on 5 November 2023).
- Loc3CAR: Locoregional Delivery of B7-H3-CAR T Cells for Pediatric Patients with Primary CNS Tumors. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05835687 (accessed on 5 November 2023).
- Autologous CAR-T Cells Targeting B7-H3 in Recurrent or Refractory GBM CAR.B7-H3Tc. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05366179 (accessed on 5 November 2023).
- B7-H3 CAR-T for Recurrent or Refractory Glioblastoma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04077866 (accessed on 5 November 2023).
- B7-H3 Chimeric Antigen Receptor T Cells (B7-H3CART) in Recurrent Glioblastoma Multiforme. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05474378 (accessed on 5 November 2023).
- Pilot Study of B7-H3 CAR-T in Treating Patients with Recurrent and Refractory Glioblastoma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04385173 (accessed on 5 November 2023).
- Safety and Efficacy of Targeted IL-13 Rα2 or B7-H3 UCAR-T for Advanced Glioma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05752877 (accessed on 5 November 2023).
- A Clinical Study of IL13Rα2 Targeted CAR-T in Patients with Malignant Glioma (MAGIC-I). U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05540873 (accessed on 5 November 2023).
- Brain Tumor-Specific Immune Cells (IL13Ralpha2-CAR T Cells) for the Treatment of Leptomeningeal Glioblastoma, Ependymoma, or Medulloblastoma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04661384 (accessed on 5 November 2023).
- IL13Ra2-CAR T Cells With or Without Nivolumab and Ipilimumab in Treating Patients with GBM. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04003649 (accessed on 5 November 2023).
- Chimeric Antigen Receptor (CAR) T Cells With a Chlorotoxin Tumor-Targeting Domain for the Treatment of MMP2+ Recurrent or Progressive Glioblastoma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT04214392 (accessed on 5 November 2023).
- CAR T Cells in Patients With MMP2+ Recurrent or Progressive Glioblastoma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05627323 (accessed on 5 November 2023).
- The Safety and Efficacy of SNC-109 CAR-T Cells Therapy the Recurrent Glioblastoma. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05868083 (accessed on 5 November 2023).
- HER2-specific CAR T Cell Locoregional Immunotherapy for HER2-positive Recurrent/Refractory Pediatric CNS Tumors. U.S.National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT03500991 (accessed on 5 November 2023).
- NKG2D-based CAR T-cells Immunotherapy for Patient with r/r NKG2DL+ Solid Tumors. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05131763 (accessed on 5 November 2023).
- Phase I Study of IL-8 Receptor-Modified CD70 CAR T Cell Therapy in CD70+ and MGMT-Unmethylated Adult Glioblastoma (IMPACT). U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05353530 (accessed on 5 November 2023).
- Study of B7-H3, EGFR806, HER2, And IL13-Zetakine (Quad) CAR T Cell Locoregional Immunotherapy For Pediatric Diffuse Intrinsic Pontine Glioma, Diffuse Midline Glioma, And Recurrent Or Refractory Central Nervous System Tumors. U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/study/NCT05768880 (accessed on 5 November 2023).
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef]
- Cutmore, L.C.; Marshall, J.F. Current Perspectives on the Use of off the Shelf CART/NK Cells for the Treatment of Cancer. Cancers 2021, 13, 1926. [Google Scholar] [CrossRef] [PubMed]
- Motais, B.; Charvátová, S.; Hrdinka, M.; Šimíček, M.; Jelínek, T.; Ševčíková, T.; Kořístek, Z.; Hájek, R.; Bagó, J.R. A Bird’s-Eye View of Cell Sources for Cell-Based Therapies in Blood Cancers. Cancers 2020, 12, 1333. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, K.; Geiger, A.; Dowlati, E.; Lang, H.; Sohai, D.K.; Hwang, E.I.; Lazarski, C.A.; Yvon, E.; Holdhoff, M.; Jones, R.; et al. Co-transducing B7H3 CAR-NK cells with the DNR preserves their cytolytic function against GBM in the presence of exogenous TGF-β. Mol. Ther. Methods Clin. Dev. 2022, 27, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.S.; et al. Novel Human NK Cell Line Carrying CAR Targeting EGFRvIII Induces Antitumor Effects in Glioblastoma Cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J.; et al. An Oncolytic Virus Expressing IL15/IL15Rα Combined with Off-the-Shelf EGFR-CAR NK Cells Targets Glioblastoma. Cancer Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108, djv375. [Google Scholar] [CrossRef] [PubMed]
- Strecker, M.I.; Wlotzka, K.; Strassheimer, F.; Roller, B.; Ludmirski, G.; König, S.; Röder, J.; Opitz, C.; Alekseeva, T.; Reul, J.; et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as experimental therapy for glioblastoma. Oncoimmunology 2022, 11, 2127508. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Li, Y.; He, C.; Wang, T.; Zheng, X.; Liu, H.; Wu, Z.; Zhang, J.; Liao, X.; Zhang, L. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front. Immunol. 2023, 14, 1145706. [Google Scholar] [CrossRef]
- Burga, R.A.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.Y.; Bollard, C.M. Engineering the TGFβ Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef]
- Hernández, A.; Domènech, M.; Muñoz-Mármol, A.M.; Carrato, C.; Balana, C. Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells. 2021, 10, 3529. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Jakovlevs, A.; Vanags, A.; Gardovskis, J.; Strumfa, I. Molecular classification of diffuse gliomas. Pol. J. Pathol. 2019, 70, 246–258. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.M.; Faust Akl, C.; Wheeler, M.A.; Chiocca, E.A.; Reardon, D.A.; Quintana, F.J. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat. Rev. Cancer 2021, 21, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Xiao, Y.; Tian, J.; Lu, Z. Remodeling metabolic fitness: Strategies for improving the efficacy of chimeric antigen receptor T cell therapy. Cancer Lett. 2022, 529, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Valeri, A.; García-Ortiz, A.; Castellano, E.; Córdoba, L.; Maroto-Martín, E.; Encinas, J.; Leivas, A.; Río, P.; Martínez-López, J. Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front. Immunol. 2022, 13, 953849. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | Molecular Markers of Cancer | Morphological Characteristics of the Tumor | Survival Rate | References | |
|---|---|---|---|---|---|
| pHGG | Diffuse midline glioma, H3 K27-altered | H3-3A, HIST1H3B, HIST1H3BC TP53, ACVR1, PDGFRA, EGFR, EZHIP | Diffuse infiltrative growth, affecting midline structures. Located in the brain stem, thalamus and spinal cord. | 10.5–19.6 months | [7,8,9,10,11] |
| Diffuse hemispheric glioma, H3 G34-mutant | H3-3A, TP53, ATRX, MGMT | Glioblastoma-like or PNET-like histomorphology. Mostly located in cerebral hemispheres. | 23.5 months | [7,8,10,12] | |
| Diffuse pediatric-type HGG, H3-wildtype and IDH-wildtype | H3-wildtype (H3-3A, HIST1H3B, HIST1H3BC), IDH-wildtype (IDH1, IDH2) PDGFRA, MYCN, EGFR | Located supratentorial, in the brain stem or cerebellum. | 17 months | [7,8,10,13] | |
| Infant-type hemispheric glioma | NTRK 1/2/3, ALK, ROS, MET | Located in cerebral hemispheres. | - | [7,8,10,14] | |
| aHGG | Astrocytoma, IDH-mutant, CNS WHO grade 4 | IDH1, IDH2 ATRX, TP53 | Necrosis and/or microvascular proliferation, oligodendroglioma-like components. | 14–18 months | [9,10,15] |
| Glioblastoma, IDH-wildtype, CNS WHO grade 4 | IDH-wildtype (IDH1/2) TERT, EGFR amplification, +7/−10 chromosome copy-number changes | Necrosis and/or microvascular proliferation. | 14–18 months | [9,10,15] | |
| Drug | ClinicalTrials.gov Identifier | Phase of Clinical Study | Estimated Number of Patients | Studied Patient Population | Method of Administration | Dosage | Reference |
|---|---|---|---|---|---|---|---|
| Anti-GD2 CAR-T cells | NCT05544526 | Phase 1 | 12 | Patients with DMG, up to 16 y.o. | - | - | [92] |
| Anti-GD2 CAR-T cells | NCT04196413 | Phase 1 | 54 | Patients with H3K27M-mutated DIPG, 2–50 y.o. | Intravenously; Intracerebroventricularly | 3 × 105–3 × 106 cells/kg; 10 × 106–100 × 106 cells | [93] |
| GD2.C7R-T cells | NCT04099797 | Phase 1 | 34 | Patients with GD2-expressing newly diagnosed DMG, 1–21 y.o. | Intravenously; Intracerebroventricularly | 3 × 106 cells/m2; 5 × 106–5 × 107 cells | [94] |
| B7-H3-CAR-T cells | NCT05835687 | Phase 1 | 36 | Patients with primary CNS tumor, up to 21 y.o. | Locoregionally | - | [95] |
| B7-H3-specific CAR-T cells | NCT04185038 | Phase 1 | 90 | Patients with refractory or recurrent CNS tumor or with DIPG or DMG, 1–26 y.o. | Locoregionally | - | [35] |
| CAR.B7-H3T cells | NCT05366179 | Phase 1 | 36 | Patients with recurrent supratentorial or infratentorial GBM, above 18 y.o. | Intraventricularly | 2–5 × 106 cells/infusion | [96] |
| B7-H3 CAR-T cells | NCT04077866 | Phase 1, Phase 2 | 40 | Patients with relapsed refractory B7-H3+ GBM, 18–75 y.o. | Intratumorally or intracerebroventricularly | - | [97] |
| B7-H3 CAR-T | NCT05474378 | Phase 1 | 39 | Patients with recurrent/progressive HGG, above 18 y.o. | Locoregionally | 5–100 × 106 cells/dose | [98] |
| B7-H3 CAR-T | NCT04385173 | Phase 1 | 12 | Patients with relapsed/refractory B7-H3+ grade IV GBM, 18–75 y.o. | Intratumorally or intracerebroventricularly | - | [99] |
| Anti-IL-13Rα2 UCAR-T cells; Anti-B7-H3 UCAR-T cells | NCT05752877 | Not applicable | 12 | Patients with advanced, locally advanced or recurrent glioma, 18–70 y.o. | Locoregionally | 1–5 × 107 cells | [100] |
| Anti-IL-13Rα2 CAR-T cells | NCT05540873 | Phase 1 | 18 | Patients with HGG, 19–74 y.o. | Intravenously | - | [101] |
| Anti-IL-13Rα2 CAR-T cells | NCT04661384 | Phase 1 | 30 | Patients with leptomeningeal GBM, ependymoma or medulloblastoma, above 18 y.o. | Intracerebroventricularly | - | [102] |
| Anti-IL-13Rα2 CAR-T cells with or without nivolumab and ipilimumab | NCT04003649 | Phase 1 | 60 | Patients with grade IV GBM, above 18 y.o. | Locoregionally | - | [103] |
| Chlorotoxin (EQ)-CD28-CD3ζ-CD19t-expressing CAR-T lymphocytes | NCT04214392 | Phase 1 | 36 | Patients with MMP-2+ recurrent or progressive GBM, above 18 y.o. | Intracavitary/intratumorally and intraventricularly | - | [104] |
| CHM-1101 CAR-T cells | NCT05627323 | Phase 1 | 42 | Patients with MMP-2+ recurrent or progressive GBM, above 18 y.o. | Intracavitary/intratumorally and intraventricularly | - | [105] |
| SNC-109 CAR-T cells | NCT05868083 | Phase 1 | 16 | Patients with recurrent GBM, 18–70 y.o. | - | 2 × 104 cells | [106] |
| Anti-HER2 CAR-T cells | NCT03500991 | Phase 1 | 48 | Patients with HER2+ CNS tumor, 1–26 y.o. | Locoregionally | - | [107] |
| NKG2D-based CAR-T cells | NCT05131763 | Phase 1 | 3 | Patients with NKG2DL+ tumors including CNS tumors, 18–75 y.o. | Intravenously | 1–10 × 106 cells/kg | [108] |
| IL-8R modified CD70 CAR-T cells | NCT05353530 | Phase 1 | 18 | Patients with CD70+ MGMT-unmethylated GBM, 18–80 y.o. | Intravenously | 1 × 106–1 × 108 cells/kg | [109] |
| SC-CAR4BRAIN (T cells expressing B7-H3, EGFR806, HER2 and IL13-zetakine CARs | NCT05768880 | Phase 1 | 72 | Patients with DIPG, DMG or refractory/recurrent CNS tumor, 1–26 y.o. | Intravenously | - | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, A.; Zarychta, J.; Marszołek, A.; Zawitkowska, J.; Lejman, M. Chimeric Antigen Receptor T Cell and Chimeric Antigen Receptor NK Cell Therapy in Pediatric and Adult High-Grade Glioma—Recent Advances. Cancers 2024, 16, 623. https://doi.org/10.3390/cancers16030623
Kowalczyk A, Zarychta J, Marszołek A, Zawitkowska J, Lejman M. Chimeric Antigen Receptor T Cell and Chimeric Antigen Receptor NK Cell Therapy in Pediatric and Adult High-Grade Glioma—Recent Advances. Cancers. 2024; 16(3):623. https://doi.org/10.3390/cancers16030623
Chicago/Turabian StyleKowalczyk, Adrian, Julia Zarychta, Anna Marszołek, Joanna Zawitkowska, and Monika Lejman. 2024. "Chimeric Antigen Receptor T Cell and Chimeric Antigen Receptor NK Cell Therapy in Pediatric and Adult High-Grade Glioma—Recent Advances" Cancers 16, no. 3: 623. https://doi.org/10.3390/cancers16030623