
Integration of Computational Docking into Anti-Cancer Drug Response Prediction Models
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Outline
2.2. Cell Line, Drug, and Response Data
2.3. Curation of PDB Structures of Anti-Cancer Drug Target Proteins
2.4. Creation of Receptors from Existing Protein-Ligand Complexes
2.5. Preparation of Compound Ligand Library
2.6. High-Throughput Docking Procedure
2.7. LightGBM, FCNN, and DeepTTA Models for Drug Response Prediction
2.8. Performance Evaluation Scheme
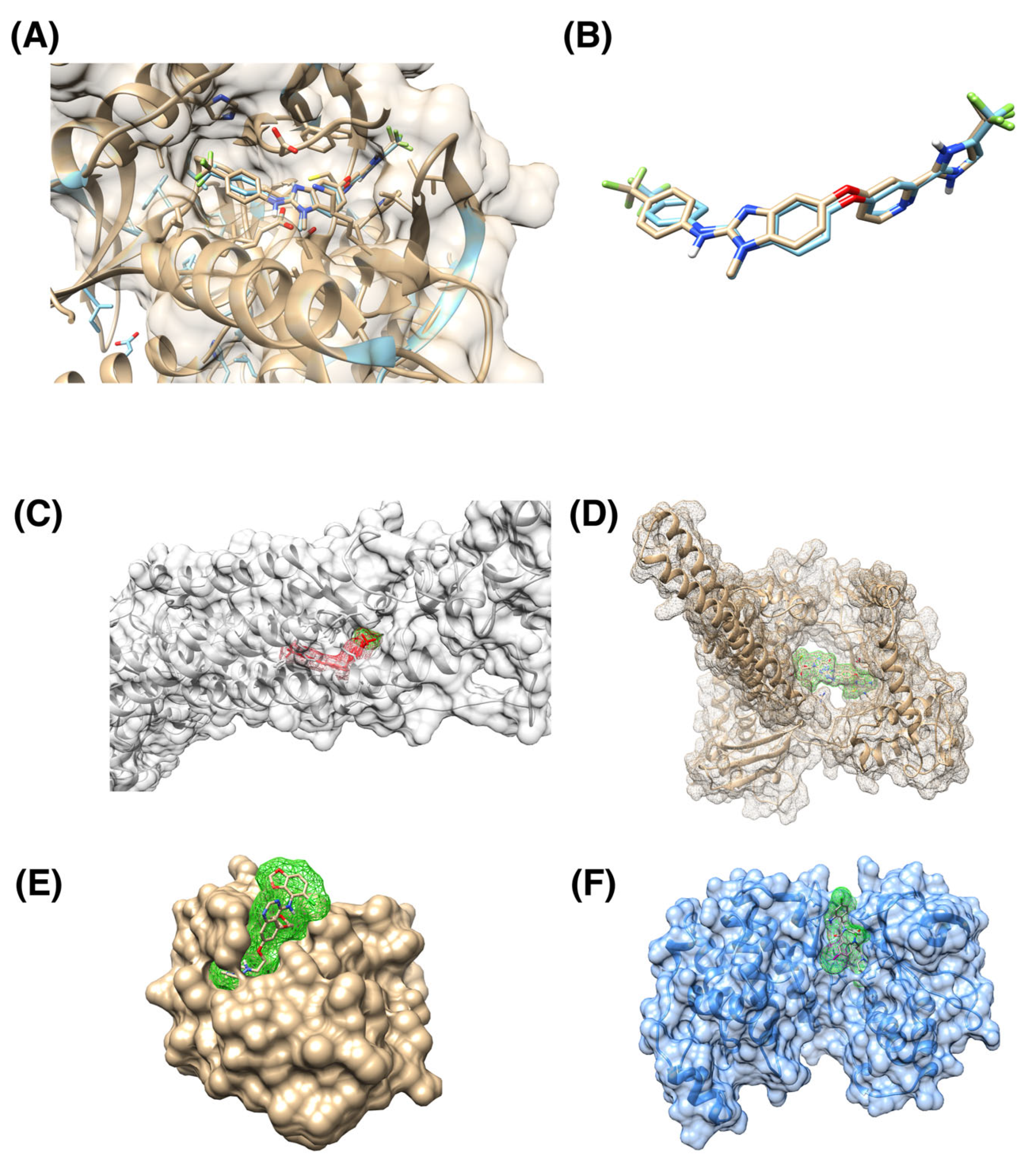
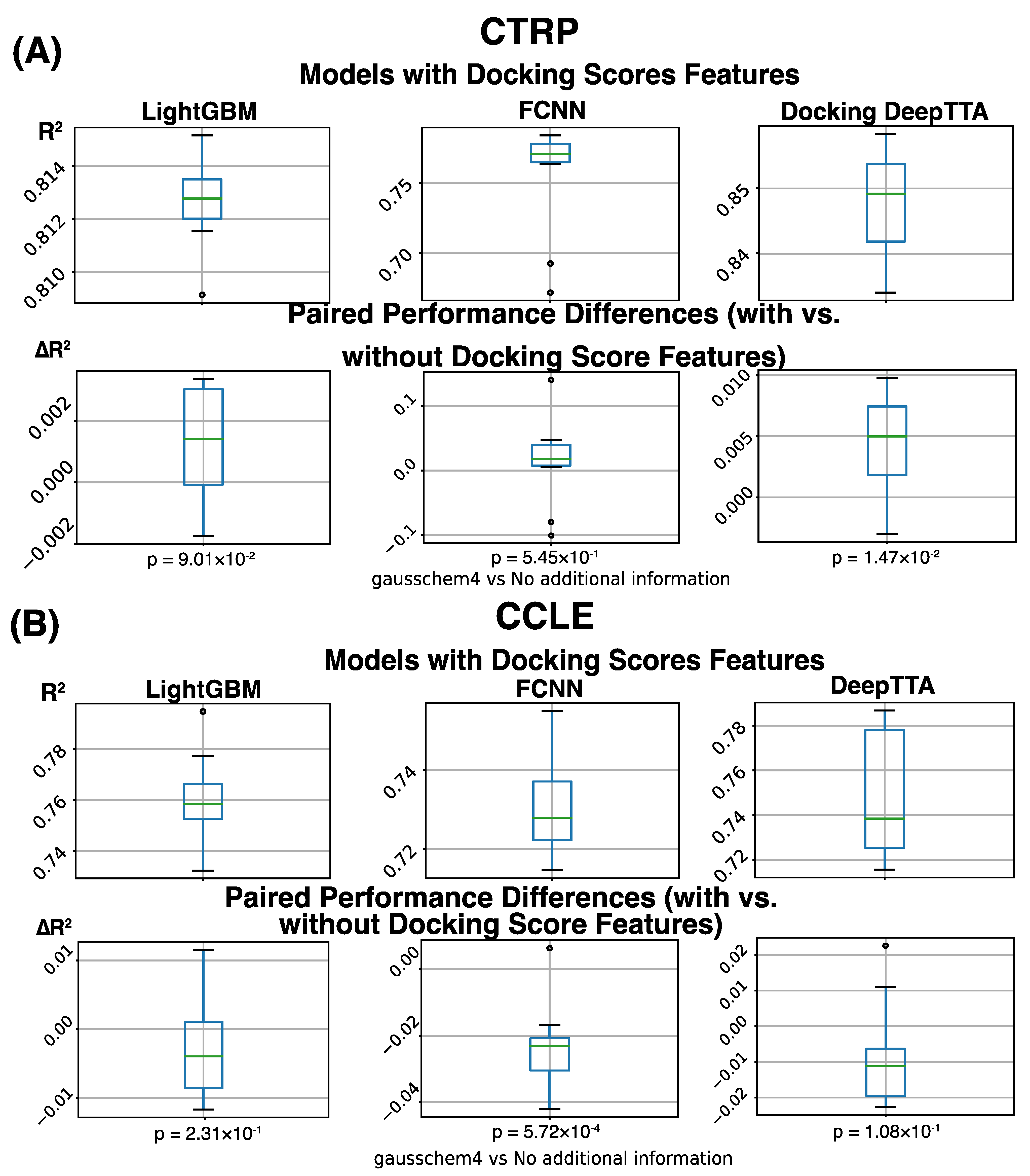
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Performance Metrics
Appendix A.1.1. Coefficient of Determination
Appendix A.1.2. Mean Squared Error (MSE)
Appendix A.1.3. Mean Absolute Error (MAE)
Appendix A.1.4. Pearson Correlation Coefficient (PCC)
Appendix A.1.5. Spearman Correlation Coefficient (SCC)
Appendix B

References
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Sleeman, K.E.; Gomes, B.; de Brito, M.; Shamieh, O.; Harding, R. The burden of serious health-related suffering among cancer decedents: Global projections study to 2060. Palliat. Med. 2021, 35, 231–235. [Google Scholar] [CrossRef]
- Yabroff, K.R.; Mariotto, A.; Tangka, F.; Zhao, J.; Islami, F.; Sung, H.; Sherman, R.L.; Henley, S.J.; Jemal, A.; Ward, E.M. Annual report to the nation on the status of cancer, part 2: Patient economic burden associated with cancer care. JNCI J. Natl. Cancer Inst. 2021, 113, 1670–1682. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Singer, D.S. Progress and potential: The cancer moonshot. Cancer Cell 2021, 39, 889–894. [Google Scholar] [CrossRef]
- Gourd, E. President Biden outlines plans for Cancer Moonshot 2.0. Lancet Oncol. 2022, 23, 335. [Google Scholar] [CrossRef]
- Anderson, B.O.; Ilbawi, A.M.; Fidarova, E.; Weiderpass, E.; Stevens, L.; Abdel-Wahab, M.; Mikkelsen, B. The Global Breast Cancer Initiative: A strategic collaboration to strengthen health care for non-communicable diseases. Lancet Oncol. 2021, 22, 578–581. [Google Scholar] [CrossRef]
- World Health Organization. Seventieth World Health Assembly: Cancer Prevention and Control in the Context of an Integrated Approach; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Vargas, A.J.; Harris, C.C. Biomarker development in the precision medicine era: Lung cancer as a case study. Nat. Rev. Cancer 2016, 16, 525–537. [Google Scholar] [CrossRef]
- Ferrantini, M.; Capone, I.; Belardelli, F. Interferon-α and cancer: Mechanisms of action and new perspectives of clinical use. Biochimie 2007, 89, 884–893. [Google Scholar] [CrossRef]
- Ku, C.-S.; Cooper, D.N.; Wu, M.; Roukos, D.H.; Pawitan, Y.; Soong, R.; Iacopetta, B. Gene discovery in familial cancer syndromes by exome sequencing: Prospects for the elucidation of familial colorectal cancer type X. Mod. Pathol. 2012, 25, 1055–1068. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Zhang, W.; Bai, Y. Whole transcriptome sequencing identifies crucial genes associated with colon cancer and elucidation of their possible mechanisms of action. OncoTargets Ther. 2019, 12, 2737. [Google Scholar] [CrossRef]
- Fais, S.; Overholtzer, M. Cell-in-cell phenomena in cancer. Nat. Rev. Cancer 2018, 18, 758–766. [Google Scholar] [CrossRef]
- Kumar, P.; Kiran, S.; Saha, S.; Su, Z.; Paulsen, T.; Chatrath, A.; Shibata, Y.; Shibata, E.; Dutta, A. ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci. Adv. 2020, 6, eaba2489. [Google Scholar] [CrossRef]
- Palmer, A.C.; Sorger, P.K. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef]
- Chan, R.J.; Cooper, B.; Koczwara, B.; Chan, A.; Tan, C.J.; Paul, S.M.; Dunn, L.B.; Conley, Y.P.; Kober, K.M.; Levine, J.D. A longitudinal analysis of phenotypic and symptom characteristics associated with inter-individual variability in employment interference in patients with breast cancer. Support. Care Cancer 2020, 28, 4677–4686. [Google Scholar] [CrossRef]
- Moschini, M.; D’andrea, D.; Korn, S.; Irmak, Y.; Soria, F.; Compérat, E.; Shariat, S.F. Characteristics and clinical significance of histological variants of bladder cancer. Nat. Rev. Urol. 2017, 14, 651–668. [Google Scholar] [CrossRef]
- Hildebrand, L.A.; Pierce, C.J.; Dennis, M.; Paracha, M.; Maoz, A. Artificial intelligence for histology-based detection of microsatellite instability and prediction of response to immunotherapy in colorectal cancer. Cancers 2021, 13, 391. [Google Scholar] [CrossRef]
- Nagle, P.W.; Plukker, J.T.M.; Muijs, C.T.; van Luijk, P.; Coppes, R.P. Patient-derived tumor organoids for prediction of cancer treatment response. Semin. Cancer Biol. 2018, 53, 258–264. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Zhou, J.; Li, M.; Wang, X.; He, Y.; Xia, Y.; Sweeney, J.A.; Kopp, R.F.; Liu, C.; Chen, C. Drug response-related DNA methylation changes in schizophrenia, bipolar disorder, and major depressive disorder. Front. Neurosci. 2021, 15, 674273. [Google Scholar] [CrossRef]
- Oliver, J.; Garcia-Aranda, M.; Chaves, P.; Alba, E.; Cobo-Dols, M.; Onieva, J.L.; Barragan, I. Emerging noninvasive methylation biomarkers of cancer prognosis and drug response prediction. Semin. Cancer Biol. 2022, 83, 584–595. [Google Scholar] [CrossRef]
- Wang, C.-W.; Muzakky, H.; Lee, Y.-C.; Lin, Y.-J.; Chao, T.-K. Annotation-Free Deep Learning-Based Prediction of Thyroid Molecular Cancer Biomarker BRAF (V600E) from Cytological Slides. Int. J. Mol. Sci. 2023, 24, 2521. [Google Scholar] [CrossRef]
- Jiang, L.; Jiang, C.; Yu, X.; Fu, R.; Jin, S.; Liu, X. DeepTTA: A transformer-based model for predicting cancer drug response. Brief. Bioinform. 2022, 23, bbac100. [Google Scholar] [CrossRef]
- Kurilov, R.; Haibe-Kains, B.; Brors, B. Assessment of modelling strategies for drug response prediction in cell lines and xenografts. Sci. Rep. 2020, 10, 2849. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Rajan, R.G.; Fernandez-Vega, V.; Sperry, J.; Nakashima, J.; Do, L.H.; Andrews, W.; Boca, S.; Islam, R.; Chowdhary, S.A.; Seldin, J. In Vitro and In Vivo Drug-Response Profiling Using Patient-Derived High-Grade Glioma. Cancers 2023, 15, 3289. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Bender, R.J.; Rao, S.; Boca, S.M.; Chung, V.; Hendifar, A.E.; Mikhail, S.; Sohal, D.P.; Pohlmann, P.R. A virtual molecular tumor board to improve efficiency and scalability of delivering precision oncology to physicians and their patients. JAMIA Open 2019, 2, 505–515. [Google Scholar] [CrossRef]
- Vidyasagar, M. Identifying predictive features in drug response using machine learning: Opportunities and challenges. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 15–34. [Google Scholar] [CrossRef]
- Yang, J.; Li, A.; Li, Y.; Guo, X.; Wang, M. A novel approach for drug response prediction in cancer cell lines via network representation learning. Bioinformatics 2019, 35, 1527–1535. [Google Scholar] [CrossRef]
- Bienkowska, J.R.; Dalgin, G.S.; Batliwalla, F.; Allaire, N.; Roubenoff, R.; Gregersen, P.K.; Carulli, J.P. Convergent Random Forest predictor: Methodology for predicting drug response from genome-scale data applied to anti-TNF response. Genomics 2009, 94, 423–432. [Google Scholar] [CrossRef]
- Rahman, R.; Matlock, K.; Ghosh, S.; Pal, R. Heterogeneity aware random forest for drug sensitivity prediction. Sci. Rep. 2017, 7, 11347. [Google Scholar] [CrossRef]
- Dittman, D.; Khoshgoftaar, T.M.; Wald, R.; Napolitano, A. Random forest: A reliable tool for patient response prediction. In Proceedings of the 2011 IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW), Atlanta, GA, USA, 12–15 November 2011; pp. 289–296. [Google Scholar]
- Turki, T.; Wang, J.T. Clinical intelligence: New machine learning techniques for predicting clinical drug response. Comput. Biol. Med. 2019, 107, 302–322. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Kaushik, B. A systematic literature review for the prediction of anticancer drug response using various machine-learning and deep-learning techniques. Chem. Biol. Drug Des. 2023, 101, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, M.; Qin, Y. Drug-induced cell viability prediction from LINCS-L1000 through WRFEN-XGBoost algorithm. BMC Bioinform. 2021, 22, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Brettin, T.; Evrard, Y.A.; Xia, F.; Partin, A.; Shukla, M.; Yoo, H.; Doroshow, J.H.; Stevens, R.L. Enhanced co-expression extrapolation (COXEN) gene selection method for building anti-cancer drug response prediction models. Genes 2020, 11, 1070. [Google Scholar] [CrossRef]
- Partin, A.; Brettin, T.S.; Zhu, Y.; Narykov, O.; Clyde, A.; Overbeek, J.; Stevens, R.L. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. arXiv 2022, arXiv:2211.10442. [Google Scholar] [CrossRef]
- Sebaugh, J. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Joo, M.; Park, A.; Kim, K.; Son, W.-J.; Lee, H.S.; Lim, G.; Lee, J.; Lee, D.H.; An, J.; Kim, J.H. A deep learning model for cell growth inhibition IC50 prediction and its application for gastric cancer patients. Int. J. Mol. Sci. 2019, 20, 6276. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Z.; Jiang, R.; Zhou, M. DeepCDR: A hybrid graph convolutional network for predicting cancer drug response. Bioinformatics 2020, 36, i911–i918. [Google Scholar] [CrossRef]
- Bazgir, O.; Zhang, R.; Dhruba, S.R.; Rahman, R.; Ghosh, S.; Pal, R. Representation of features as images with neighborhood dependencies for compatibility with convolutional neural networks. Nat. Commun. 2020, 11, 4391. [Google Scholar] [CrossRef]
- Zhu, Y.; Brettin, T.; Xia, F.; Partin, A.; Shukla, M.; Yoo, H.; Evrard, Y.A.; Doroshow, J.H.; Stevens, R.L. Converting tabular data into images for deep learning with convolutional neural networks. Sci. Rep. 2021, 11, 11325. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Ren, S.; Ding, M.Q.; Schwartz, R.; Lu, X. Predicting drug sensitivity of cancer cell lines via collaborative filtering with contextual attention. In Proceedings of the Machine Learning for Healthcare Conference; Carnegie Mellon University: Pittsburgh, PN, USA, 2020; pp. 660–684. [Google Scholar]
- Rampášek, L.; Hidru, D.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Dr. VAE: Improving drug response prediction via modeling of drug perturbation effects. Bioinformatics 2019, 35, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ouyang, Z.; Chen, W.; Feng, R.; Chen, D.Z.; Cao, J.; Wu, J. TGSA: Protein–protein association-based twin graph neural networks for drug response prediction with similarity augmentation. Bioinformatics 2022, 38, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Piao, Y.; Bang, D.; Kim, S.; Jo, K. DRPreter: Interpretable Anticancer Drug Response Prediction Using Knowledge-Guided Graph Neural Networks and Transformer. Int. J. Mol. Sci. 2022, 23, 13919. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Kaushik, B. CTDN (Convolutional Temporal Based Deep-Neural Network): An Improvised Stacked Hybrid Computational Approach for Anticancer Drug Response Prediction. Comput. Biol. Chem. 2023, 105, 107868. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Huang, X.; Fang, S.; Guo, S.; Liu, Y.; Lin, W.; Xiong, M. Conditional generative Adversarial networks for individualized treatment effect estimation and treatment selection. Front. Genet. 2020, 11, 585804. [Google Scholar] [CrossRef] [PubMed]
- Oskooei, A.; Born, J.; Manica, M.; Subramanian, V.; Sáez-Rodríguez, J.; Martínez, M.R. PaccMann: Prediction of anticancer compound sensitivity with multi-modal attention-based neural networks. arXiv 2018, arXiv:1811.06802. [Google Scholar]
- Cadow, J.; Born, J.; Manica, M.; Oskooei, A.; Rodríguez Martínez, M. PaccMann: A web service for interpretable anticancer compound sensitivity prediction. Nucleic Acids Res. 2020, 48, W502–W508. [Google Scholar] [CrossRef]
- Chu, T.; Nguyen, T.T.; Hai, B.D.; Nguyen, Q.H.; Nguyen, T. Graph Transformer for drug response prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 20, 1065–1072. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Huang, K.; Xiao, C.; Glass, L.; Sun, J. Explainable Substructure Partition Fingerprint for Protein, Drug, and More. NeurIPS Learning Meaningful Representation of Life Workshop. 2019. Available online: https://static1.squarespace.com/static/58f7aae1e6f2e1a0f9a56616/t/5e370e2d12092f15876d5753/1580666413389/paper.pdf (accessed on 30 April 2023).
- Adam, G.; Rampášek, L.; Safikhani, Z.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Machine learning approaches to drug response prediction: Challenges and recent progress. NPJ Precis. Oncol. 2020, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, C.; Ramachandran, M. Computational molecular docking studies on anticancer drugs. Asian Pac. J. Trop. Dis. 2012, 2, S734–S738. [Google Scholar] [CrossRef]
- Gowtham, H.G.; Murali, M.; Singh, S.B.; Shivamallu, C.; Pradeep, S.; Shivakumar, C.; Anandan, S.; Thampy, A.; Achar, R.R.; Silina, E. Phytoconstituents of Withania somnifera unveiled Ashwagandhanolide as a potential drug targeting breast cancer: Investigations through computational, molecular docking and conceptual DFT studies. PLoS ONE 2022, 17, e0275432. [Google Scholar] [CrossRef] [PubMed]
- Mcgann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolym. Orig. Res. Biomol. 2003, 68, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3149–3157. [Google Scholar]
- Popescu, M.-C.; Balas, V.E.; Perescu-Popescu, L.; Mastorakis, N. Multilayer perceptron and neural networks. WSEAS Trans. Circuits Syst. 2009, 8, 579–588. [Google Scholar]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef]
- Zhu, Y.; Brettin, T.; Evrard, Y.A.; Partin, A.; Xia, F.; Shukla, M.; Yoo, H.; Doroshow, J.H.; Stevens, R.L. Ensemble transfer learning for the prediction of anti-cancer drug response. Sci. Rep. 2020, 10, 18040. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, 1, PO.17.00011. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Kode Chemoinformatics. Available online: https://chm.kode-solutions.net/products_dragon.php (accessed on 23 October 2022).
- OpenEye, Cadence Molecular Sciences. Santa Fe, NM, USA. OEDOCKING, 4.2.1.0. Available online: https://www.eyesopen.com/ (accessed on 8 June 2022).
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The Universal Protein knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Do, T.D.; Kelley, B.P.; Nicholls, A.; Warren, S.D. Essential considerations for using protein–ligand structures in drug discovery. Drug Discov. Today 2012, 17, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Wlodek, S. Decisions with Confidence: Application to the Conformation Sampling of Molecules in the Solid State. J. Chem. Inf. Model. 2020, 60, 3518–3533. [Google Scholar] [CrossRef]
- OpenEye, Cadence Molecular Sciences. Santa Fe, NM, USA. QUACPAC, 2.2.2.0. Available online: https://www.eyesopen.com/ (accessed on 8 June 2022).
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.-F.; Cornell, W.D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 5998–6008. [Google Scholar]
- Chung, P.-J.; Bohme, J.F.; Mecklenbrauker, C.F.; Hero, A.O. Detection of the number of signals using the Benjamini-Hochberg procedure. IEEE Trans. Signal Process. 2007, 55, 2497–2508. [Google Scholar] [CrossRef]
- Dhillon, I.S. Co-clustering documents and words using bipartite spectral graph partitioning. In Proceedings of the Seventh ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 26–29 August 2001; pp. 269–274. [Google Scholar]
- Abbenante, G.; Reid, R.C.; Fairlie, D.P. ‘Clean’or ‘Dirty’—Just how selective do drugs need to be? Aust. J. Chem. 2008, 61, 654–660. [Google Scholar] [CrossRef]
- Cesa, L.C.; Mapp, A.K.; Gestwicki, J.E. Direct and propagated effects of small molecules on protein–protein interaction networks. Front. Bioeng. Biotechnol. 2015, 3, 119. [Google Scholar] [CrossRef] [PubMed]
- Narykov, O.; Johnson, N.T.; Korkin, D. Predicting protein interaction network perturbation by alternative splicing with semi-supervised learning. Cell Rep. 2021, 37, 110045. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Sharma, S.; Norton, L.; Chiosis, G. Targeting stressor-induced dysfunctions in protein–protein interaction networks via epichaperomes. Trends Pharmacol. Sci. 2023, 44, 20–33. [Google Scholar] [CrossRef]
- Batista, J.; Hawkins, P.C.; Tolbert, R.; Geballe, M.T. SiteHopper-a unique tool for binding site comparison. J. Cheminformatics 2014, 6, P57. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Tian, L.; Li, Q.; Luo, J.; Zhang, Y. Analysis of the physicochemical properties of acaricides based on Lipinski’s rule of five. J. Comput. Biol. 2020, 27, 1397–1406. [Google Scholar] [CrossRef]
- Pollastri, M.P. Overview on the Rule of Five. Curr. Protoc. Pharmacol. 2010, 49, 9–12. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Sischka, A.; Toensing, K.; Eckel, R.; Wilking, S.D.; Sewald, N.; Ros, R.; Anselmetti, D. Molecular mechanisms and kinetics between DNA and DNA binding ligands. Biophys. J. 2005, 88, 404–411. [Google Scholar] [CrossRef]
- Bates, D.O.; Morris, J.C.; Oltean, S.; Donaldson, L.F. Pharmacology of modulators of alternative splicing. Pharmacol. Rev. 2017, 69, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Clyde, A.; Liu, X.; Brettin, T.; Yoo, H.; Partin, A.; Babuji, Y.; Blaiszik, B.; Mohd-Yusof, J.; Merzky, A.; Turilli, M. AI-accelerated protein-ligand docking for SARS-CoV-2 is 100-fold faster with no significant change in detection. Sci. Rep. 2023, 13, 2105. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.; Ban, F.; Norinder, U.; Gleave, M.; Cherkasov, A. Deep docking: A deep learning platform for augmentation of structure based drug discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yao, K.; Repasky, M.P.; Leswing, K.; Abel, R.; Shoichet, B.K.; Jerome, S.V. Efficient exploration of chemical space with docking and deep learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. [Google Scholar] [CrossRef]
- Jin, I.; Nam, H. HiDRA: Hierarchical network for drug response prediction with attention. J. Chem. Inf. Model. 2021, 61, 3858–3867. [Google Scholar] [CrossRef]
- Yin, S.; Biedermannova, L.; Vondrasek, J.; Dokholyan, N.V. MedusaScore: An accurate force field-based scoring function for virtual drug screening. J. Chem. Inf. Model. 2008, 48, 1656–1662. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Information | Not Used | ||||||
|---|---|---|---|---|---|---|---|
| Dataset | CCLE | CTRP | |||||
| Metric | R2 | PCC | SCC | R2 | PCC | SCC | |
| Method | |||||||
| FCNN | 0.753 ± 0.009 | 0.869 ± 0.005 | 0.768 ± 0.008 | 0.742 ± 0.040 | 0.864 ± 0.023 | 0.839 ± 0.006 | |
| LightGBM | 0.764 ± 0.019 | 0.874 ± 0.011 | 0.791 ± 0.018 | 0.811 ± 0.001 | 0.901 ± 0.001 | 0.852 ± 0.001 | |
| DeepTTA | 0.758 ± 0.022 | 0.873 ± 0.012 | 0.779 ± 0.018 | 0.843 ± 0.007 | 0.919 ± 0.004 | 0.878 ± 0.008 | |
| Docking Information | GaussChem4 Scores | ||||||
|---|---|---|---|---|---|---|---|
| Dataset | CCLE | CTRP | |||||
| Metric | R2 | PCC | SCC | R2 | PCC | SCC | |
| Method | |||||||
| FCNN | 0.730 ± 0.012 | 0.856 ± 0.007 | 0.749 ± 0.014 | 0.755 ± 0.039 | 0.871 ± 0.022 | 0.847 ± 0.003 | |
| LightGBM | 0.761 ± 0.017 | 0.873 ± 0.010 | 0.788 ± 0.016 | 0.813 ± 0.002 | 0.902 ± 0.001 | 0.853 ± 0.002 | |
| DeepTTA | 0.749 ± 0.028 | 0.873 ± 0.014 | 0.781 ± 0.022 | 0.848 ± 0.008 | 0.921 ± 0.004 | 0.883 ± 0.007 | |
| Docking Type | Differences | ||||||
|---|---|---|---|---|---|---|---|
| Dataset | CCLE | CTRP | |||||
| Metric | R2 | PCC | SCC | R2 | PCC | SCC | |
| Method | |||||||
| FCNN | −0.0231 (p = 5.72 × 10−4) | −0.0133 (p = 5.20 × 10−4) | −0.0191 (p = 1.21 × 10−4) | 0.0133 (p = 5.45 × 10−1) | 0.0073 (p = 5.54 × 10−1) | 0.0077 (p = 9.26 × 10−3) | |
| LightGBM | −0.0029 (p = 2.31 × 10−1) | −0.0016 (p = 2.38 × 10−1) | −0.0036 (p = 1.52 × 10−1) | 0.0013 (p = 9.01 × 10−2) | 0.0007 (p = 9.51 × 10−2) | 0.0013 (p = 8.38 × 10−3) | |
| DeepTTC | −0.0084 (p = 1.08 × 10−1) | 0.0001 (p = 9.52 × 10−1) | 0.0017 (p = 5.23 × 10−1) | 0.0045 (p = 1.47 × 10−2 | 0.0024 (p = 1.87 × 10−2) | 0.0048 (p = 1.69 × 10−2) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narykov, O.; Zhu, Y.; Brettin, T.; Evrard, Y.A.; Partin, A.; Shukla, M.; Xia, F.; Clyde, A.; Vasanthakumari, P.; Doroshow, J.H.; et al. Integration of Computational Docking into Anti-Cancer Drug Response Prediction Models. Cancers 2024, 16, 50. https://doi.org/10.3390/cancers16010050
Narykov O, Zhu Y, Brettin T, Evrard YA, Partin A, Shukla M, Xia F, Clyde A, Vasanthakumari P, Doroshow JH, et al. Integration of Computational Docking into Anti-Cancer Drug Response Prediction Models. Cancers. 2024; 16(1):50. https://doi.org/10.3390/cancers16010050
Chicago/Turabian StyleNarykov, Oleksandr, Yitan Zhu, Thomas Brettin, Yvonne A. Evrard, Alexander Partin, Maulik Shukla, Fangfang Xia, Austin Clyde, Priyanka Vasanthakumari, James H. Doroshow, and et al. 2024. "Integration of Computational Docking into Anti-Cancer Drug Response Prediction Models" Cancers 16, no. 1: 50. https://doi.org/10.3390/cancers16010050