
Epigenetic Regulation in Oral Squamous Cell Carcinoma Microenvironment: A Comprehensive Review
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Microenvironment of OSCC
3. Epigenetic Alterations in OSCC
3.1. DNA Methylation
3.1.1. Hypomethylated Genes Involving OSCC
Cyclins
Epidermal Growth Factor Receptor (EGFR)
(Absent in Melanoma 2) AIM2
CEACAM1
LINE-1
PI3
PTHLH
Survivin/BIRC5
3.1.2. Aberrant Hypermethylated Genes Involving OSCC
CDKN2A
MGMT and DAPK1
TIMP3
TFPI2, SOX17, and GATA4
3.1.3. Hypermethylated Genes Involving OSCC
CDKN2A
E-Cadherin and N-Cadherin
PTEN
P53
DAPK1
MGMT
RARβ2
RASSF
3.2. Histone Modifications in OSCC
3.3. Non-Coding RNAs
4. Therapeutic Targeting of Epigenetic Mechanisms in OSCC
4.1. DNA Methylation Inhibitors as Therapeutic Agents in OSCC
Natural Compounds as DNA Methylation Inhibitors in OSCC
4.2. HDAC Inhibitors as Therapeutic Agents in OSCC
5. Epigenetics behind Drug Resistance
5.1. DNA Methylation and Drug Resistance
5.2. Histone Modifications and Drug Resistance
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bugshan, A.; Farooq, I. Oral squamous cell carcinoma: Metastasis, potentially associated malignant disorders, etiology and recent advancements in diagnosis. F1000Research 2020, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Nicolette Salmon, H.; Jan-MichaÉL, H.; Jamshid, J.; Miranda, M.J.; Lars, S. Viral and Molecular Aspects of Oral Cancer. Anticancer Res. 2012, 32, 4201. [Google Scholar]
- Alsaeedi, S.M.; Aggarwal, S. The Holistic Review on Occurrence, Biology, Diagnosis, and Treatment of Oral Squamous Cell Carcinoma. Cureus 2022, 14, e30226. [Google Scholar] [CrossRef]
- Bagan, J.; Sarrion, G. Oral cancer: Clinical features. Oral Oncol. 2010, 46, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Bello, I.O.; Soini, Y.; Salo, T. Prognostic evaluation of oral tongue cancer: Means, markers and perspectives (I). Oral Oncol. 2010, 46, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef]
- Muthu, K.; Vaishnavi, V.; Sivadas, G. Warning Signs and Symptoms of Oral Cancer and its Differential Diagnosis. J. Young Pharm. 2018, 10, 138–143. [Google Scholar] [CrossRef]
- Carreras-Torras, C.; Gay-Escoda, C. Techniques for early diagnosis of oral squamous cell carcinoma: Systematic review. Med. Oral Patol. Oral Cir. Bucal 2015, 20, e305–e315. [Google Scholar] [CrossRef]
- Pałasz, P.; Adamski, Ł.; Górska-Chrząstek, M.; Starzyńska, A.; Studniarek, M. Contemporary Diagnostic Imaging of Oral Squamous Cell Carcinoma—A Review of Literature. Pol. J. Radiol. 2017, 82, 193–202. [Google Scholar] [CrossRef]
- Dunkel, J.; Vaittinen, S.; Grénman, R.; Kinnunen, I.; Irjala, H. Prognostic markers in stage I oral cavity squamous cell carcinoma. Laryngoscope 2013, 123, 2435–2441. [Google Scholar] [CrossRef]
- Oliveira, M.L.; Wagner, V.P.; Sant’ana Filho, M.; Carrard, V.C.; Hugo, F.N.; Martins, M.D. A 10-year analysis of the oral squamous cell carcinoma profile in patients from public health centers in Uruguay. Braz. Oral Res. 2015, 29, 1–8. [Google Scholar] [CrossRef]
- Mohamad, I.; Glaun, M.D.E.; Prabhash, K.; Busheri, A.; Lai, S.Y.; Noronha, V.; Hosni, A. Current Treatment Strategies and Risk Stratification for Oral Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e389810. [Google Scholar] [CrossRef] [PubMed]
- Vissink, A.; Jansma, J.; Spijkervet, F.K.; Burlage, F.R.; Coppes, R.P. Oral sequelae of head and neck radiotherapy. Crit. Rev. Oral Biol. Med. 2003, 14, 199–212. [Google Scholar] [CrossRef]
- Supportive, P.; Board, P.C.E. Oral Complications of Chemotherapy and Head/Neck Radiation (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries [Internet]; National Cancer Institute: Bethesda, MD, USA, 2002. [Google Scholar]
- Mărgăritescu, C.; Pirici, D.; Simionescu, C.; Stepan, A. The utility of CD44, CD117 and CD133 in identification of cancer stem cells (CSC) in oral squamous cell carcinomas (OSCC). Rom. J. Morphol. Embryol. 2011, 52, 985–993. [Google Scholar]
- Saraswathi, T.; Ranganathan, K.; Shanmugam, S.; Sowmya, R.; Narasimhan, P.D.; Gunaseelan, R. Prevalence of oral lesions in relation to habits: Cross-sectional study in South India. Indian J. Dent. Res. 2006, 17, 121. [Google Scholar] [CrossRef]
- Palve, V.; Bagwan, J.; Krishnan, N.M.; Pareek, M.; Chandola, U.; Suresh, A.; Siddappa, G.; James, B.L.; Kekatpure, V.; Kuriakose, M.A.; et al. Detection of High-Risk Human Papillomavirus in Oral Cavity Squamous Cell Carcinoma Using Multiple Analytes and Their Role in Patient Survival. J. Glob. Oncol. 2018, 4, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Ariyawardana, A.; Johnson, N.W. Oral cancer in India continues in epidemic proportions: Evidence base and policy initiatives. Int. Dent. J. 2013, 63, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, A.; Bijai Kumar, L. Actinic cheilosis: Early intervention prevents malignant transformation. BMJ Case Rep. 2017, 2017, bcr2016218654. [Google Scholar] [CrossRef]
- Sathiyasekar, A.C.; Chandrasekar, P.; Pakash, A.; Kumar, K.U.; Jaishlal, M.S. Overview of immunology of oral squamous cell carcinoma. J. Pharm. Bioallied Sci. 2016, 8, S8–S12. [Google Scholar] [CrossRef]
- Mathur, R.; Singhavi, H.R.; Malik, A.; Nair, S.; Chaturvedi, P. Role of Poor Oral Hygiene in Causation of Oral Cancer-a Review of Literature. Indian. J. Surg. Oncol. 2019, 10, 184–195. [Google Scholar] [CrossRef]
- Hema, K.N.; Smitha, T.; Sheethal, H.S.; Mirnalini, S.A. Epigenetics in oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2017, 21, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Alothman, L.; AlSenani, M.A.; Alrabiah, R.; Ras, A.A.; Abulhassan, E.H.; Aldayel, R.; Almutairi, R.; Alsaif, R. Insights into Epigenetics Mechanisms in Oral Squamous Cell Carcinoma. Saudi J. Oral Dent. Res. 2020, 5, 1297–2518. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2008, 66, 596. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.J.; Zhang, S.; Guo, C.; Zhang, S.; Wang, Y.; Zhang, D. Methylation-associated silencing of death-associated protein kinase gene in laryngeal squamous cell cancer. Laryngoscope 2005, 115, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Kim, M.-J.; Hong, S.-P.; Hong, S.-D. Inactivation patterns of p16/INK4A in oral squamous cell carcinomas. Exp. Mol. Med. 2004, 36, 165–171. [Google Scholar] [CrossRef]
- Šupić, G.; Kozomara, R.; Branković-Magić, M.; Jović, N.; Magić, Z. Gene hypermethylation in tumor tissue of advanced oral squamous cell carcinoma patients. Oral Oncol. 2009, 45, 1051–1057. [Google Scholar] [CrossRef]
- Strzelczyk, J.K.; Krakowczyk, Ł.; Owczarek, A.J. Methylation status of SFRP1, SFRP2, RASSF1A, RARβ and DAPK1 genes in patients with oral squamous cell carcinoma. Arch. Oral Biol. 2019, 98, 265–272. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Y.; Liu, Y.; Hang, L.; Yang, J. Expression of Tumor Suppressor SFRP1 Predicts Biological Behaviors and Prognosis: A Potential Target for Oral Squamous Cell Carcinoma. Biomolecules 2022, 12, 1034. [Google Scholar] [CrossRef]
- Viswanathan, M.; Tsuchida, N.; Shanmugam, G. Promoter hypermethylation profile of tumor-associated genes p16, p15, hMLH1, MGMT and E-cadherin in oral squamous cell carcinoma. Int. J. Cancer 2003, 105, 41–46. [Google Scholar] [CrossRef]
- Sawhney, M.; Rohatgi, N.; Kaur, J.; Gupta, S.D.; Deo, S.V.; Shukla, N.K.; Ralhan, R. MGMT expression in oral precancerous and cancerous lesions: Correlation with progression, nodal metastasis and poor prognosis. Oral Oncol. 2007, 43, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Padhi, S.S.; Roy, S.; Kar, M.; Saha, A.; Roy, S.; Adhya, A.; Baisakh, M.; Banerjee, B. Role of CDKN2A/p16 expression in the prognostication of oral squamous cell carcinoma. Oral Oncol. 2017, 73, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-C.; Wang, W.-Y.; Zhou, J.-J.; Wu, L.; Zhang, M.-J.; Yang, Q.-C.; Deng, W.-W.; Sun, Z.-J. Inhibition of DNMT1 potentiates antitumor immunity in oral squamous cell carcinoma. Int. Immunopharmacol. 2022, 111, 109113. [Google Scholar] [CrossRef] [PubMed]
- Tasoulas, J.; Giaginis, C.; Patsouris, E.; Manolis, E.; Theocharis, S. Histone deacetylase inhibitors in oral squamous cell carcinoma treatment. Expert Opin. Investig. Drugs 2015, 24, 69–78. [Google Scholar] [CrossRef]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenet. 2010, 1, 117–136. [Google Scholar] [CrossRef]
- Zhu, W.-G.; Otterson, G.A. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem.-Anti-Cancer Agents 2003, 3, 187–199. [Google Scholar] [CrossRef]
- Osan, C.; Chira, S.; Nutu, A.M.; Braicu, C.; Baciut, M.; Korban, S.S.; Berindan-Neagoe, I. The Connection between MicroRNAs and Oral Cancer Pathogenesis: Emerging Biomarkers in Oral Cancer Management. Genes 2021, 12, 1989. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Arneth, B. Tumor microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Ye, L.; Sanders, A.J.; Lane, J.; Jiang, W.G. Cancer invasion and metastasis: Molecular and cellular perspective. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Bilotta, M.T.; Antignani, A.; Fitzgerald, D.J. Managing the TME to improve the efficacy of cancer therapy. Front. Immunol. 2022, 13, 954992. [Google Scholar] [CrossRef]
- Elmusrati, A.; Wang, J.; Wang, C.-Y. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma. Int. J. Oral Sci. 2021, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via αVβ3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [CrossRef]
- Lin, F.; Ren, X.-D.; Pan, Z.; Macri, L.; Zong, W.-X.; Tonnesen, M.G.; Rafailovich, M.; Bar-Sagi, D.; Clark, R.A. Fibronectin growth factor-binding domains are required for fibroblast survival. J. Investig. Dermatol. 2011, 131, 84–98. [Google Scholar] [CrossRef]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Balazs, E.A.; Laurent, T.C.; Jeanloz, R.W. Nomenclature of hyaluronic acid. Biochem. J. 1986, 235, 903. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Zuidwijk, K.; Verspaget, H.W.; de Jonge-Muller, E.S.; van Duijn, W.; Ferreira, V.; Fontijn, R.D.; David, G.; Hommes, D.W.; Lamers, C.B. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 2008, 44, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Paralkar, V.M.; Vukicevic, S.; Reddi, A. Transforming growth factor β type 1 binds to collagen IV of basement membrane matrix: Implications for development. Dev. Biol. 1991, 143, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.-T.; Yao, J.-Y.; Fang, X.-L.; Di, H.; Ma, Y.-Y. Roles of lncRNAs in cancer: Focusing on angiogenesis. Life Sci. 2020, 252, 117647. [Google Scholar] [CrossRef]
- El Shorbagy, S.; abuTaleb, F.; Labib, H.A.; Ebian, H.; Harb, O.A.; Mohammed, M.S.; Rashied, H.A.; Elbana, K.A.; Haggag, R. Prognostic significance of VEGF and HIF-1 α in hepatocellular carcinoma patients receiving sorafenib versus metformin sorafenib combination. J. Gastrointest. Cancer 2021, 52, 269–279. [Google Scholar] [CrossRef]
- Huang, M.; Huang, B.; Li, G.; Zeng, S. Apatinib affect VEGF-mediated cell proliferation, migration, invasion via blocking VEGFR2/RAF/MEK/ERK and PI3K/AKT pathways in cholangiocarcinoma cell. BMC Gastroenterol. 2018, 18, 169. [Google Scholar] [CrossRef]
- Marjon, P.L.; Bobrovnikova-Marjon, E.V.; Abcouwer, S.F. Expression of the pro-angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 by human breast carcinomas is responsive to nutrient deprivation and endoplasmic reticulum stress. Mol. Cancer 2004, 3, 4. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Utomo, L.; Bastiaansen-Jenniskens, Y.M.; Verhaar, J.A.; van Osch, G.J. Cartilage inflammation and degeneration is enhanced by pro-inflammatory (M1) macrophages in vitro, but not inhibited directly by anti-inflammatory (M2) macrophages. Osteoarthr. Cartil. 2016, 24, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, E.; Angioni, R.; Molon, B.; Calì, B. Chemokines and chemokine receptors: Orchestrating tumor metastasization. Int. J. Mol. Sci. 2018, 20, 96. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Muenst, S.; Läubli, H.; Soysal, S.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Sullivan, B.M.; Stemmann, C.; Satoskar, A.R.; Sleckman, B.P.; Glimcher, L.H. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science 2002, 295, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Spencer, L.A.; Weller, P.F. Eosinophils and Th2 immunity: Contemporary insights. Immunol. Cell Biol. 2010, 88, 250–256. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, M.; Vermi, W.; Parodi, M.; Pietra, G.; Manzini, C.; Queirolo, P.; Lonardi, S.; Augugliaro, R.; Moretta, A.; Facchetti, F. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur. J. Immunol. 2012, 42, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Ladányi, A. Prognostic and predictive significance of immune cells infiltrating cutaneous melanoma. Pigment. Cell Melanoma Res. 2015, 28, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.; Elsers, D.; Fadel, S.; Omar, A.M. Immunohistological characterisation of tumour infiltrating lymphocytes in melanocytic skin lesions. J. Clin. Pathol. 2006, 59, 316–324. [Google Scholar] [CrossRef] [PubMed]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Rak, J. Extracellular vesicles–biomarkers and effectors of the cellular interactome in cancer. Front. Pharmacol. 2013, 4, 21. [Google Scholar] [CrossRef]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 1–10. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef]
- Razzo, B.M.; Ludwig, N.; Hong, C.-S.; Sharma, P.; Fabian, K.P.; Fecek, R.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived exosomes promote carcinogenesis of murine oral squamous cell carcinoma. Carcinogenesis 2020, 41, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes. Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Board, R.E.; Knight, L.; Greystoke, A.; Blackhall, F.H.; Hughes, A.; Dive, C.; Ranson, M. DNA methylation in circulating tumour DNA as a biomarker for cancer. Biomark. Insights 2007, 2, 117727190700200003. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Foy, J.P.; Pickering, C.R.; Papadimitrakopoulou, V.A.; Jelinek, J.; Lin, S.H.; William, W.N., Jr.; Frederick, M.J.; Wang, J.; Lang, W.; Feng, L.; et al. New DNA methylation markers and global DNA hypomethylation are associated with oral cancer development. Cancer Prev. Res. 2015, 8, 1027–1035. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Viet, C.T.; Yu, G.; Asam, K.; Thomas, C.M.; Yoon, A.J.; Wongworawat, Y.C.; Haghighiabyaneh, M.; Kilkuts, C.A.; McGue, C.M.; Couey, M.A.; et al. The REASON score: An epigenetic and clinicopathologic score to predict risk of poor survival in patients with early stage oral squamous cell carcinoma. Biomark. Res. 2021, 9, 42. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Baba, S.; Yamada, Y.; Hatano, Y.; Miyazaki, Y.; Mori, H.; Shibata, T.; Hara, A. Global DNA hypomethylation suppresses squamous carcinogenesis in the tongue and esophagus. Cancer Sci. 2009, 100, 1186–1191. [Google Scholar] [CrossRef]
- Guerrero-Preston, R.; Baez, A.; Blanco, A.; Berdasco, M.; Fraga, M.; Esteller, M. Global DNA methylation: A common early event in oral cancer cases with exposure to environmental carcinogens or viral agents. Puerto Rico Health Sci. J. 2009, 28, 24–29. [Google Scholar]
- Caliri, A.W.; Tommasi, S.; Besaratinia, A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108365. [Google Scholar] [CrossRef] [PubMed]
- Gasche, J.A.; Goel, A. Epigenetic mechanisms in oral carcinogenesis. Future Oncol. 2012, 8, 1407–1425. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Lukas, J.; Müller, H.; Strauss, M.; Gusterson, B.; Bartek, J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995, 55, 949–956. [Google Scholar]
- Gonzales, C.B.; Jorge, J.; Saikumar, P.; Singha, P.K.; Dybdal-Hargreaves, N.F.; Chavez, J.; Horning, A.M.; Parra, J.; Kirma, N.B. Co-targeting ALK and EGFR parallel signaling in oral squamous cell carcinoma. Oral Oncol. 2016, 59, 12–19. [Google Scholar] [CrossRef]
- Wu, C.-S.; Chang, K.-P.; OuYang, C.-N.; Kao, H.-K.; Hsueh, C.; Chen, L.-C.; Cheng, H.-Y.; Liang, Y.; Liou, W.; Liang, C.-L. ASC contributes to metastasis of oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 50074. [Google Scholar] [CrossRef]
- Nakamura, Y.; Nakahata, S.; Kondo, Y.; Izumi, A.; Yamamoto, K.; Ichikawa, T.; Tamura, T.; Noumi, K.; Yamashita, Y.; Morishita, K. Overexpression of absent in melanoma 2 in oral squamous cell carcinoma contributes to tumor progression. Biochem. Biophys. Res. Commun. 2019, 509, 82–88. [Google Scholar] [CrossRef]
- Ling, Y.; Wang, J.; Wang, L.; Hou, J.; Qian, P.; Xiang-dong, W. Roles of CEACAM1 in cell communication and signaling of lung cancer and other diseases. Cancer Metastasis Rev. 2015, 34, 347–357. [Google Scholar] [CrossRef]
- Wu, B.-H.; Xiong, X.-P.; Jia, J.; Zhang, W.-F. MicroRNAs: New actors in the oral cancer scene. Oral Oncol. 2011, 47, 314–319. [Google Scholar] [CrossRef]
- Facompre, N.D.; Harmeyer, K.H.; Basu, D. Regulation of oncogenic PI3-kinase signaling by JARID1B. Oncotarget 2017, 8, 7218. [Google Scholar] [CrossRef]
- Urosevic, J.; Garcia-Albeniz, X.; Planet, E.; Real, S.; Céspedes, M.V.; Guiu, M.; Fernandez, E.; Bellmunt, A.; Gawrzak, S.; Pavlovic, M. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell Biol. 2014, 16, 685–694. [Google Scholar] [CrossRef]
- Flausino, C.S.; Daniel, F.I.; Modolo, F. DNA methylation in oral squamous cell carcinoma: From its role in carcinogenesis to potential inhibitor drugs. Crit. Rev. Oncol./Hematol. 2021, 164, 103399. [Google Scholar] [CrossRef] [PubMed]
- Motokura, T.; Arnold, A. Cyclin D and oncogenesis. Curr. Opin. Genet. Dev. 1993, 3, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Moharil, R.B.; Khandekar, S.; Dive, A.; Bodhade, A. Cyclin D1 in oral premalignant lesions and oral squamous cell carcinoma: An immunohistochemical study. J. Oral Maxillofac. Pathol. 2020, 24, 397. [Google Scholar] [CrossRef] [PubMed]
- Holley, S.L.; Matthias, C.; Jahnke, V.; Fryer, A.A.; Strange, R.C.; Hoban, P.R. Association of cyclin D1 polymorphism with increased susceptibility to oral squamous cell carcinoma. Oral Oncol. 2005, 41, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Das, S.N.; Khare, P.; Singh, M.K.; Sharma, S.C. Correlation of cyclin D1 expression with aggressive DNA pattern in patients with tobacco-related intraoral squamous cell carcinoma. Indian J. Med. Res. 2011, 133, 381. [Google Scholar]
- Martín-Ezquerra, G.; Salgado, R.; Toll, A.; Gilaberte, M.; Baro, T.; Alameda Quitllet, F.; Yebenes, M.; Sole, F.; Garcia-Muret, M.; Espinet, B. Multiple genetic copy number alterations in oral squamous cell carcinoma: Study of MYC, TP53, CCDN1, EGFR and ERBB2 status in primary and metastatic tumours. Br. J. Dermatol. 2010, 163, 1028–1035. [Google Scholar] [CrossRef]
- Mishra, R.; Das, B.R. Cyclin D1 expression and its possible regulation in chewing tobacco mediated oral squamous cell carcinoma progression. Arch. Oral Biol. 2009, 54, 917–923. [Google Scholar] [CrossRef]
- Huang, S.-F.; Cheng, S.-D.; Chuang, W.-Y.; Chen, I.-H.; Liao, C.-T.; Wang, H.-M.; Hsieh, L.-L. Cyclin D1 overexpression and poor clinical outcomes in Taiwanese oral cavity squamous cell carcinoma. World J. Surg. Oncol. 2012, 10, 40. [Google Scholar] [CrossRef]
- Takes, R.P.; Baatenburg de Jong, R.J.; Wijffels, K.; Schuuring, E.; Litvinov, S.V.; Hermans, J.; Han JM van Krieken, J. Expression of genetic markers in lymph node metastases compared with their primary tumours in head and neck cancer. J. Pathol. 2001, 194, 298–302. [Google Scholar] [CrossRef]
- Myo, K.; Uzawa, N.; Miyamoto, R.; Sonoda, I.; Yuki, Y.; Amagasa, T. Cyclin D1 gene numerical aberration is a predictive marker for occult cervical lymph node metastasis in TNM Stage I and II squamous cell carcinoma of the oral cavity. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2005, 104, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Shpitzer, T.; Hamzany, Y.; Bahar, G.; Feinmesser, R.; Savulescu, D.; Borovoi, I.; Gavish, M.; Nagler, R. Salivary analysis of oral cancer biomarkers. Br. J. Cancer 2009, 101, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.; Hinds, P.; Munger, K.; Rustgi, A.; Opitz, O.; Suliman, Y.; Wong, D. Cell cycle dysregulation in oral cancer. Crit. Rev. Oral Biol. Med. 2002, 13, 51–61. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, Y.; Li, S.; Pan, Y.; Zhu, Y.; Yu, Y. Cyclin E overexpression and centrosome amplification in squamous cell carcinoma of oral cavity. Zhonghua Bing Li Xue Za Zhi=Chin. J. Pathol. 2007, 36, 375–378. [Google Scholar]
- Freier, K.; Knoepfle, K.; Flechtenmacher, C.; Pungs, S.; Devens, F.; Toedt, G.; Hofele, C.; Joos, S.; Lichter, P.; Radlwimmer, B. Recurrent copy number gain of transcription factor SOX2 and corresponding high protein expression in oral squamous cell carcinoma. Genes. Chromosomes Cancer 2010, 49, 9–16. [Google Scholar] [CrossRef]
- Iyoda, M.; Kasamatsu, A.; Ishigami, T.; Nakashima, D.; Endo-Sakamoto, Y.; Ogawara, K.; Shiiba, M.; Tanzawa, H.; Uzawa, K. Epithelial cell transforming sequence 2 in human oral cancer. PLoS ONE 2010, 5, e14082. [Google Scholar] [CrossRef]
- Oliver, R.; MacDonald, D. G1 cyclins in oral epithelial dysplasia. J. Oral Pathol. Med. 2001, 30, 80–86. [Google Scholar] [CrossRef]
- Rodriguez-Pinilla, M.; Rodriguez-Peralto, J.L.; Hitt, R.; Sanchez, J.J.; Ballestin, C.; Diez, A.; Sanchez-Verde, L.; Alameda, F.; Sanchez-Cespedes, M. Cyclin A as a predictive factor for chemotherapy response in advanced head and neck cancer. Clin. Cancer Res. 2004, 10, 8486–8492. [Google Scholar] [CrossRef]
- Ito, R.; Yasui, W.; Ogawa, Y.; Toyosawa, S.; Tahara, E.; Ijuhin, N. Reduced expression of cyclin-dependent kinase inhibitor p27Kip1 in oral malignant tumors. Pathobiology 2000, 67, 169–173. [Google Scholar] [CrossRef]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef]
- Chen, Q.; Zhou, H.; Guo, W.; Samaranayake, L.; Zhou, M.; Li, B. Correlation between the expression of cyclin A protein and p53 activity in oral squamous cell carcinomas. Cytobios 2001, 106, 87–99. [Google Scholar]
- Farhadieh, R.D.; Smee, R.; Rees, C.G.; Salardini, A.; Eggleton, S.; Yang, J.L.; Russell, P.J. Mutant p53 and cyclin A1 protein expression in primary laryngeal squamous cell carcinomas do not correlate to second primary tumours of the head and neck. ANZ J. Surg. 2009, 79, 48–54. [Google Scholar] [CrossRef]
- Kushner, J.; Bradley, G.; Young, B.; Jordan, R.C. Aberrant expression of cyclin A and cyclin B1 proteins in oral carcinoma. J. Oral. Pathol. Med. 1999, 28, 77–81. [Google Scholar] [CrossRef]
- Mihara, M.; Shintani, S.; Nakahara, Y.; Kiyota, A.; Ueyama, Y.; Matsumura, T.; Wong, D.T. Overexpression of CDK2 is a prognostic indicator of oral cancer progression. Jpn. J. Cancer Res. 2001, 92, 352–360. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Yamashita, K.; Osada, M.; Nomoto, S.; Sun, D.-I.; Xiao, Y.; Hoque, M.O.; Westra, W.H.; Califano, J.A.; Sidransky, D. Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res. 2004, 64, 5982–5987. [Google Scholar] [CrossRef]
- Hassan, K.A.; Ang, K.K.; El-Naggar, A.K.; Story, M.D.; Lee, J.I.; Liu, D.; Hong, W.K.; Mao, L. Cyclin B1 overexpression and resistance to radiotherapy in head and neck squamous cell carcinoma. Cancer Res. 2002, 62, 6414–6417. [Google Scholar] [PubMed]
- Nguyen, D.C.; Parsa, B.; Close, A.; Magnusson, B.; Crowe, D.L.; Sinha, U.K. Overexpression of cell cycle regulatory proteins correlates with advanced tumor stage in head and neck squamous cell carcinomas. Int. J. Oncol. 2003, 22, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Omura, K.; Nakajima, Y.; Hasegawa, S.; Mogi, S. Cyclin B1 is useful to predict occult cervical lymph node metastases in tongue carcinoma. J. Exp. Clin. Cancer Res. 2006, 25, 353. [Google Scholar]
- Thomson, P.; Hamadah, O.; Goodson, M.; Cragg, N.; Booth, C. Predicting recurrence after oral precancer treatment: Use of cell cycle analysis. Br. J. Oral Maxillofac. Surg. 2008, 46, 370–375. [Google Scholar] [CrossRef]
- Watanabe, S.; Watanabe, R.; Oton-Leite, A.F.; de CG Alencar, R.; Oliveira, J.C.; Leles, C.R.; Batista, A.C.; Mendonça, E.F. Analysis of cell proliferation and pattern of invasion in oral squamous cell carcinoma. J. Oral Sci. 2010, 52, 417–424. [Google Scholar] [CrossRef]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Renard, T.; Gueydan, C.; Aron, S. DNA methylation and expression of the egfr gene are associated with worker size in monomorphic ants. Sci. Rep. 2022, 12, 21228. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Qin, F.; Yuan, L.; Wei, J.; Sun, Y.; Qin, J.; Deng, K.; Zheng, T.; Li, S. EGFR DNA Methylation Correlates With EGFR Expression, Immune Cell Infiltration, and Overall Survival in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 691915. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.-Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar]
- Saravani, S.; Parsamanesh, N.; Miri-Moghaddam, E. Role of EGFR gene polymorphisms in oral squamous cell carcinoma patients of Southeast Iran: A case-control study. Casp. J. Intern. Med. 2020, 11, 391–397. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef]
- Abdul-Sater, A.A.; Philpott, D.J. Inflammasomes. In Encyclopedia of Immunobiology, Ratcliffe, M.J.H., Ed.; Academic Press: Oxford, UK, 2016; pp. 447–453. [Google Scholar]
- Jin, T.; Perry, A.; Smith, P.; Jiang, J.; Xiao, T.S. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J. Biol. Chem. 2013, 288, 13225–13235. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95. [Google Scholar] [CrossRef]
- Zhu, H.; Zhao, M.; Chang, C.; Chan, V.; Lu, Q.; Wu, H. The complex role of AIM2 in autoimmune diseases and cancers. Immun. Inflamm. Dis. 2021, 9, 649–665. [Google Scholar] [CrossRef]
- Farkas, S.A.; Milutin-Gašperov, N.; Grce, M.; Nilsson, T.K. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 2013, 8, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Nagai, K.; Nakahata, S.; Saito, Y.; Ichikawa, T.; Suekane, A.; Taki, T.; Iwakawa, R.; Enari, M.; Taniwaki, M.; et al. Overexpression of the DNA sensor proteins, absent in melanoma 2 and interferon-inducible 16, contributes to tumorigenesis of oral squamous cell carcinoma with p53 inactivation. Cancer Sci. 2012, 103, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz, K.M.; Gudas, L.J. Altered epigenetic regulation of homeobox genes in human oral squamous cell carcinoma cells. Exp. Cell Res. 2014, 320, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, L.; Byrd, K.M.; Ko, C.-C.; Zhao, Z.; Fang, J. AIM2 inflammasome’s first decade of discovery: Focus on oral diseases. Front. Immunol. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Chew, Z.H.; Cui, J.; Sachaphibulkij, K.; Tan, I.; Kar, S.; Koh, K.K.; Singh, K.; Lim, H.M.; Lee, S.C.; Kumar, A.P.; et al. Macrophage IL-1β contributes to tumorigenesis through paracrine AIM2 inflammasome activation in the tumor microenvironment. Front. Immunol. 2023, 14, 1211730. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Multani, S.; Dabholkar, J.; Saranath, D. Whole genome expression profiling in chewing-tobacco-associated oral cancers: A pilot study. Med. Oncol. 2015, 32, 60. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kanneganti, T.-D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome complexes: Emerging mechanisms and effector functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef]
- Pierini, R.; Juruj, C.; Perret, M.; Jones, C.; Mangeot, P.; Weiss, D.; Henry, T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012, 19, 1709–1721. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers 2019, 11, 1313. [Google Scholar] [CrossRef]
- Ruan, M.; Ji, T.; Yang, W.; Duan, W.; Zhou, X.; He, J.; Zhou, J.; Chen, W.; Zhang, C. Growth inhibition and induction of apoptosis in human oral squamous cell carcinoma Tca-8113 cell lines by shikonin was partly through the inactivation of NF-κB pathway. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2008, 22, 407–415. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Y.; Zhao, W.; Yu, T.; Yu, H. Caspase-8 polymorphisms and risk of oral squamous cell carcinoma. Exp. Ther. Med. 2015, 10, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Rabquer, B.; Mansfield, P.; Ruth, J.; Marotte, H.; Haas, C.; Reamer, E.; Koch, A. Interleukin 18 induces angiogenesis in vitro and in vivo via Src and Jnk kinases. Ann. Rheum. Dis. 2010, 69, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Fahey, E.; Doyle, S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [PubMed]
- Zetter, B.R. Angiogenesis and tumor metastasis. Annu. Rev. Med. 1998, 49, 407–424. [Google Scholar] [CrossRef]
- Lozano-Ruiz, B.; Tzoumpa, A.; Martínez-Cardona, C.; Moreno, D.; Aransay, A.M.; Cortazar, A.R.; Picó, J.; Peiró, G.; Lozano, J.; Zapater, P. Absent in melanoma 2 (AIM2) regulates the stability of regulatory T cells. Int. J. Mol. Sci. 2022, 23, 2230. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X.; Cheng, H.; Zhou, F. AIM2 and Psoriasis. Front. Immunol. 2023, 14, 1085448. [Google Scholar] [CrossRef]
- Koehn, B.H.; Apostolova, P.; Haverkamp, J.M.; Miller, J.S.; McCullar, V.; Tolar, J.; Munn, D.H.; Murphy, W.J.; Brickey, W.J.; Serody, J.S. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood J. Am. Soc. Hematol. 2015, 126, 1621–1628. [Google Scholar] [CrossRef]
- Huan, C.Z. Aim2 Inflammasome Activation in Tumor Associated Macrophages and Its Impact on Cancer Development. Ph.D. Thesis, National University of Singapore, Singapore, 2019. [Google Scholar]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Young, M.R.I. Myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) produce CCL22 which selectively recruits regulatory T-cells (Tregs) to the tumor microenvironment. FASEB J. 2008, 22, 1078–1079. [Google Scholar] [CrossRef]
- Li, S.; Mai, Z.; Gu, W.; Ogbuehi, A.C.; Acharya, A.; Pelekos, G.; Ning, W.; Liu, X.; Deng, Y.; Li, H.; et al. Molecular Subtypes of Oral Squamous Cell Carcinoma Based on Immunosuppression Genes Using a Deep Learning Approach. Front. Cell Dev. Biol. 2021, 9, 687245. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Song, X.; Fan, S.; Deng, R. The role of tumor-associated macrophages in oral squamous cell carcinoma. Front. Physiol. 2022, 13, 959747. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.M.; Huang, Y.H.; Gandhi, A.; Blumberg, R.S. CEACAM1 structure and function in immunity and its therapeutic implications. Semin. Immunol. 2019, 42, 101296. [Google Scholar] [CrossRef]
- Dankner, M.; Gray-Owen, S.D.; Huang, Y.H.; Blumberg, R.S.; Beauchemin, N. CEACAM1 as a multi-purpose target for cancer immunotherapy. Oncoimmunology 2017, 6, e1328336. [Google Scholar] [CrossRef]
- Kirshner, J.; Chen, C.-J.; Liu, P.; Huang, J.; Shively, J.E. CEACAM1-4S, a cell–cell adhesion molecule, mediates apoptosis and reverts mammary carcinoma cells to a normal morphogenic phenotype in a 3D culture. Proc. Natl. Acad. Sci. USA 2003, 100, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Hanssen, T.; Wagener, C.; Obrink, B. Down-regulation of CEACAM1 in human prostate cancer: Correlation with loss of cell polarity, increased proliferation rate, and Gleason grade 3 to 4 transition. Hum. Pathol. 2002, 33, 290–298. [Google Scholar] [CrossRef]
- Lin, S.-H. Tumor Specific Regulation of C-CAM Cell Adhesion Molecule in Prostate Cancer Carcinogenesis; University of Texas M. D. Anderson Cancer Center: Houston, TX, USA, 2000. [Google Scholar]
- Simonetti, O.; Lucarini, G.; Rubini, C.; Zizzi, A.; Aspriello, S.D.; Di Primio, R.; Offidani, A.M. Correlation between immunohistochemical staining of CEACAM1 and clinicopathological findings in oral pre-neoplastic lesions and squamous cell carcinoma. Med. Mol. Morphol. 2018, 51, 41–47. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Z.; Li, C.; Liu, Z.; Zhang, X.; Li, L.; An, F.; Qiao, X. CEACAM1 as a molecular target in oral cancer. Aging 2023, 15, 8137–8154. [Google Scholar] [CrossRef]
- Zang, M.; Zhang, B.; Zhang, Y.; Li, J.; Su, L.; Zhu, Z.; Gu, Q.; Liu, B.; Yan, M. CEACAM6 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition via PI3K/AKT signaling pathway. PLoS ONE 2014, 9, e112908. [Google Scholar] [CrossRef]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef]
- Shinozuka, K.; Uzawa, K.; Fushimi, K.; Yamano, Y.; Shiiba, M.; Bukawa, H.; Yokoe, H.; Tanzawa, H. Downregulation of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 in Oral Squamous Cell Carcinoma: Correlation with Tumor Progression and Poor Prognosis. Oncology 2009, 76, 387–397. [Google Scholar] [CrossRef]
- Weiner, G.M.; Ducruet, A.F. CEACAM1: A novel adhesion molecule that regulates the secretion of matrix metalloproteinase-9 in neutrophils and protects the blood-brain barrier after ischemic stroke. Neurosurgery 2014, 75, N21–N22. [Google Scholar] [CrossRef] [PubMed]
- Reunanen, N.; Kähäri, V. Matrix metalloproteinases in cancer cell invasion. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Hosomi, S.; Chen, Z.; Baker, K.; Chen, L.; Huang, Y.H.; Olszak, T.; Zeissig, S.; Wang, J.H.; Mandelboim, O.; Beauchemin, N.; et al. CEACAM1 on activated NK cells inhibits NKG2D-mediated cytolytic function and signaling. Eur. J. Immunol. 2013, 43, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Tatsumi, T.; Nishio, A.; Kegasawa, T.; Yoshioka, T.; Yamada, R.; Furuta, K.; Kodama, T.; Shigekawa, M.; Hikita, H.; et al. CEACAM1 Is Associated with the Suppression of Natural Killer Cell Function in Patients with Chronic Hepatitis C. Hepatol. Commun. 2018, 2, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; An, N.; Wang, M.; Liu, X.; Mei, Z. Downregulation of AT-rich interaction domain 2 underlies natural killer cell dysfunction in oral squamous cell carcinoma. Immunol. Cell Biol. 2023, 101, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Briggs, E.M.; Ha, S.; Mita, P.; Brittingham, G.; Sciamanna, I.; Spadafora, C.; Logan, S.K. Long interspersed nuclear element-1 expression and retrotransposition in prostate cancer cells. Mob. DNA 2018, 9, 1. [Google Scholar] [CrossRef]
- Saito, K.; Kawakami, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Minamoto, T. Long interspersed nuclear element 1 hypomethylation is a marker of poor prognosis in stage IA non–small cell lung cancer. Clin. Cancer Res. 2010, 16, 2418–2426. [Google Scholar] [CrossRef]
- Beck, C.R.; Garcia-Perez, J.L.; Badge, R.M.; Moran, J.V. LINE-1 elements in structural variation and disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 187–215. [Google Scholar] [CrossRef]
- Kitkumthorn, N.; Mutirangura, A. Long interspersed nuclear element-1 hypomethylation in cancer: Biology and clinical applications. Clin. Epigenet. 2011, 2, 315–330. [Google Scholar] [CrossRef]
- Anwar, S.L.; Hasemeier, B.; Schipper, E.; Vogel, A.; Kreipe, H.; Lehmann, U. LINE-1 hypomethylation in human hepatocellular carcinomas correlates with shorter overall survival and CIMP phenotype. PLoS ONE 2019, 14, e0216374. [Google Scholar] [CrossRef]
- Ponomaryova, A.A.; Rykova, E.Y.; Gervas, P.A.; Cherdyntseva, N.V.; Mamedov, I.Z.; Azhikina, T.L. Aberrant methylation of LINE-1 transposable elements: A search for cancer biomarkers. Cells 2020, 9, 2017. [Google Scholar] [CrossRef]
- Lavasanifar, A.; Sharp, C.N.; Korte, E.A.; Yin, T.; Hosseinnejad, K.; Jortani, S.A. Long interspersed nuclear element-1 mobilization as a target in cancer diagnostics, prognostics and therapeutics. Clin. Chim. Acta 2019, 493, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Grundy, E.E.; Diab, N.; Chiappinelli, K.B. Transposable element regulation and expression in cancer. FEBS J. 2022, 289, 1160–1179. [Google Scholar] [CrossRef] [PubMed]
- Lavia, P.; Sciamanna, I.; Spadafora, C. An Epigenetic LINE-1-Based Mechanism in Cancer. Int. J. Mol. Sci. 2022, 23, 14610. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Yu, J. New Understanding of the Relevant Role of LINE-1 Retrotransposition in Human Disease and Immune Modulation. Front. Cell Dev. Biol. 2020, 8, 657. [Google Scholar] [CrossRef]
- Naufer, M.N.; Furano, A.V.; Williams, M.C. Protein-nucleic acid interactions of LINE-1 ORF1p. In Proceedings of the Seminars in Cell & Developmental Biology, Purwokerto, Indonesia, 5–6 August 2019; pp. 140–149. [Google Scholar]
- Budania, S.; Sur, D.; Nangal, J.; Pilli, S.; Mukherjee, K.; Biswas, M.; Prasad, R.; Saxena, S.; Mandal, P.K. LINE-1 retrotransposon encoded ORF1p expression and promoter methylation in oral squamous cell carcinoma: A pilot study. Cancer Genet. 2020, 244, 21–29. [Google Scholar] [CrossRef]
- Furlan, C.; Polesel, J.; Barzan, L.; Franchin, G.; Sulfaro, S.; Romeo, S.; Colizzi, F.; Rizzo, A.; Baggio, V.; Giacomarra, V.; et al. Prognostic significance of LINE-1 hypomethylation in oropharyngeal squamous cell carcinoma. Clin. Epigenet. 2017, 9, 58. [Google Scholar] [CrossRef]
- Leonova, K.I.; Brodsky, L.; Lipchick, B.; Pal, M.; Novototskaya, L.; Chenchik, A.A.; Sen, G.C.; Komarova, E.A.; Gudkov, A.V. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.-J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A. p53 genes function to restrain mobile elements. Genes. Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef]
- Shin, Y.-J.; Kim, Y.; Wen, X.; Cho, N.-Y.; Lee, S.; Kim, W.H.; Kang, G.H. Prognostic implications and interaction of L1 methylation and p53 expression statuses in advanced gastric cancer. Clin. Epigenet. 2019, 11, 77. [Google Scholar] [CrossRef]
- Tiwari, B.; Jones, A.E.; Caillet, C.J.; Das, S.; Royer, S.K.; Abrams, J.M. p53 directly represses human LINE1 transposons. Genes Dev. 2020, 37, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Subbalekha, K.; Pimkhaokham, A.; Pavasant, P.; Chindavijak, S.; Phokaew, C.; Shuangshoti, S.; Matangkasombut, O.; Mutirangura, A. Detection of LINE-1s hypomethylation in oral rinses of oral squamous cell carcinoma patients. Oral Oncol. 2008, 45, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Gasche, J.A.; Hoffmann, J.; Boland, C.R.; Goel, A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int. J. Cancer 2011, 129, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef]
- Alqahtani, A.; Ayesh, H.S.K.; Halawani, H. PIK3CA Gene Mutations in Solid Malignancies: Association with Clinicopathological Parameters and Prognosis. Cancers 2019, 12, 93. [Google Scholar] [CrossRef]
- Starzyńska, A.; Adamska, P.; Sejda, A.; Sakowicz-Burkiewicz, M.; Adamski Ł, J.; Marvaso, G.; Wychowański, P.; Jereczek-Fossa, B.A. Any Role of PIK3CA and PTEN Biomarkers in the Prognosis in Oral Squamous Cell Carcinoma? Life 2020, 10, 325. [Google Scholar] [CrossRef]
- Yang, J.; Ren, X.; Zhang, L.; Li, Y.; Cheng, B.; Xia, J. Oridonin inhibits oral cancer growth and PI3K/Akt signaling pathway. Biomed. Pharmacother. 2018, 100, 226–232. [Google Scholar] [CrossRef]
- Ren, X.; Luo, W. Exploration of pro-apoptotic effect of Thymoquinone on oral squamous cell carcinoma cells through PI3K/Akt signaling pathway. Cell. Mol. Biol. 2019, 65, 61–64. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, C.; Sun, Y.; Xu, H. Licochalcone A inhibits cell proliferation, migration, and invasion through regulating the PI3K/AKT signaling pathway in oral squamous cell carcinoma. OncoTargets Ther. 2019, 12, 4427–4435. [Google Scholar] [CrossRef]
- Faleiro, I.; Roberto, V.P.; Demirkol Canli, S.; Fraunhoffer, N.A.; Iovanna, J.; Gure, A.O.; Link, W.; Castelo-Branco, P. DNA Methylation of PI3K/AKT Pathway-Related Genes Predicts Outcome in Patients with Pancreatic Cancer: A Comprehensive Bioinformatics-Based Study. Cancers 2021, 13, 6354. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA Methylation in the Resistance to Therapy in Solid Tumors. Front. Oncol. 2020, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Wysolmerski, J.J. Parathyroid hormone-related protein: An update. J. Clin. Endocrinol. Metab. 2012, 97, 2947–2956. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J. Parathyroid hormone-related protein, its regulation of cartilage and bone development, and role in treating bone diseases. Physiol. Rev. 2016, 96, 831–871. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Ono, N.; Ono, W. Mesenchymal Progenitor Regulation of Tooth Eruption: A View from PTHrP. J. Dent. Res. 2020, 99, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.K.; Martinez, D. Physiological roles of parathyroid hormone-related protein. Acta Biomed. 2019, 90, 510–516. [Google Scholar] [CrossRef]
- Guasto, A.; Cormier-Daire, V. Signaling Pathways in Bone Development and Their Related Skeletal Dysplasia. Int. J. Mol. Sci. 2021, 22, 4321. [Google Scholar] [CrossRef]
- Lv, Z.; Wu, X.; Cao, W.; Shen, Z.; Wang, L.; Xie, F.; Zhang, J.; Ji, T.; Yan, M.; Chen, W. Parathyroid hormone-related protein serves as a prognostic indicator in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 100. [Google Scholar] [CrossRef]
- Sahibzada, H.A.; Khurshid, Z.; Sannam Khan, R.; Naseem, M.; Mahmood Siddique, K.; Mali, M.; Zafar, M.S. Salivary IL-8, IL-6 and TNF-α as potential diagnostic biomarkers for oral cancer. Diagnostics 2017, 7, 21. [Google Scholar] [CrossRef]
- Hoshi, S.; Morimoto, T.; Saito, H.; Ichizuka, K.; Matsuoka, R.; Yanaihara, A.; Suzuki, M.; Yanaihara, T.; Okai, T. PTHrP and PTH/PTHrP receptor expressions in human endometrium. Endocr. J. 2001, 48, 219–225. [Google Scholar] [CrossRef]
- Park, S.I.; McCauley, L.K. Nuclear localization of parathyroid hormone-related peptide confers resistance to anoikis in prostate cancer cells. Endocr.-Relat. Cancer 2012, 19, 243–254. [Google Scholar] [CrossRef]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tang, Y.; Luo, J.; Yang, Y.; Zang, H.; Ma, J.; Fan, S.; Wen, Q. High expression of HSP60 and survivin predicts poor prognosis for oral squamous cell carcinoma patients. BMC Oral Health 2023, 23, 629. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Uzawa, K.; Shibahara, T.; Yokoe, H.; Noma, H.; Tanzawa, H. Expression of an inhibitor of apoptosis, survivin, in oral carcinogenesis. J. Dent. Res. 2003, 82, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Muzio, L.L.; Staibano, S.; Pannone, G.; Mignogna, M.D.; Mariggiò, A.; Salvatore, G.; Chieffi, P.; Tramontano, D.; De Rosa, G.; Altieri, D.C. Expression of the apoptosis inhibitor survivin in aggressive squamous cell carcinoma. Exp. Mol. Pathol. 2001, 70, 249–254. [Google Scholar] [CrossRef]
- Hsue, S.S.; Wang, W.C.; Chen, Y.K.; Lin, L.M. Expression of inhibitors of apoptosis family protein in 7, 12-dimethylbenz [a] anthracene-induced hamster buccal-pouch squamous-cell carcinogenesis is associated with mutant p53 accumulation and epigenetic changes. Int. J. Exp. Pathol. 2008, 89, 309–320. [Google Scholar] [CrossRef]
- Chen, Y.-K.; Hsue, S.-S.; Lin, L.-M. Survivin expression is regulated by an epigenetic mechanism for DMBA-induced hamster buccal-pouch squamous-cell carcinomas. Arch. Oral Biol. 2005, 50, 593–598. [Google Scholar] [CrossRef]
- Seta, R.; Mascitti, M.; Campagna, R.; Sartini, D.; Fumarola, S.; Santarelli, A.; Giuliani, M.; Cecati, M.; Muzio, L.L.; Emanuelli, M. Overexpression of nicotinamide N-methyltransferase in HSC-2 OSCC cell line: Effect on apoptosis and cell proliferation. Clin. Oral Investig. 2019, 23, 829–838. [Google Scholar] [CrossRef]
- Togni, L.; Mascitti, M.; Sartini, D.; Campagna, R.; Pozzi, V.; Salvolini, E.; Offidani, A.; Santarelli, A.; Emanuelli, M. Nicotinamide N-Methyltransferase in Head and Neck Tumors: A Comprehensive Review. Biomolecules 2021, 11, 1594. [Google Scholar] [CrossRef]
- Esteller, M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 629–656. [Google Scholar] [CrossRef]
- Goot-Heah, K.; Anisah Froemming, G.R.; Zain, R.B.; Abraham, M.T.; Omar, E.; Su-Keng, T.; Kwai-Lin, T. OP007: Aberrant methylation of genes in oral squamous cell carcinoma. Oral Oncol. 2013, 49, S6–S7. [Google Scholar] [CrossRef]
- Kim, S.Y.; Han, Y.K.; Song, J.M.; Lee, C.H.; Kang, K.; Yi, J.M.; Park, H.R. Aberrantly hypermethylated tumor suppressor genes were identified in oral squamous cell carcinoma (OSCC). Clin. Epigenet. 2019, 11, 116. [Google Scholar] [CrossRef]
- Ogi, K.; Toyota, M.; Ohe-Toyota, M.; Tanaka, N.; Noguchi, M.; Sonoda, T.; Kohama, G.; Tokino, T. Aberrant Methylation of Multiple Genes and Clinicopathological Features in Oral Squamous Cell Carcinoma1. Clin. Cancer Res. 2002, 8, 3164–3171. [Google Scholar] [PubMed]
- Alshammari, E.; Zhang, Y.; Sobota, J.; Yang, Z. Aberrant DNA Methylation of Tumor Suppressor Genes and Oncogenes as Cancer Biomarkers. In Genomic and Epigenomic Biomarkers of Toxicology and Disease; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2022; pp. 251–271. [Google Scholar]
- Kaur, J.; Demokan, S.; Tripathi, S.C.; Macha, M.A.; Begum, S.; Califano, J.A.; Ralhan, R. Promoter hypermethylation in Indian primary oral squamous cell carcinoma. Int. J. Cancer 2010, 127, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Choi, B.Y.; Lee, M.-H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16INK4a) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef]
- Lee, M.-H.; Choi, B.Y.; Cho, Y.-Y.; Lee, S.-Y.; Huang, Z.; Kundu, J.K.; Kim, M.O.; Kim, D.J.; Bode, A.M.; Surh, Y.-J. Tumor suppressor p16INK4a inhibits cancer cell growth by downregulating eEF1A2 through a direct interaction. J. Cell Sci. 2013, 126, 1744–1752. [Google Scholar] [CrossRef]
- Russo, A.A.; Tong, L.; Lee, J.-O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Yamakoshi, K.; Takahashi, A.; Hara, E. The p16INK4a-RB pathway: Molecular link between cellular senescence and tumor suppression. J. Med. Investig. 2004, 51, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Strzelczyk, J.K.; Krakowczyk, Ł.; Owczarek, A.J. Aberrant DNA methylation of the p16, APC, MGMT, TIMP3 and CDH1 gene promoters in tumours and the surgical margins of patients with oral cavity cancer. J. Cancer 2018, 9, 1896–1904. [Google Scholar] [CrossRef]
- Jayaprakash, C.; Varghese, V.K.; Bellampalli, R.; Radhakrishnan, R.; Ray, S.; Kabekkodu, S.P.; Satyamoorthy, K. Hypermethylation of Death-Associated Protein Kinase (DAPK1) and its association with oral carcinogenesis—An experimental and meta-analysis study. Arch. Oral Biol. 2017, 80, 117–129. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, Y.; Zhong, W.; Jiang, Y.; Lin, Y. Regulatory non-coding RNAs for death associated protein kinase family. Front. Mol. Biosci. 2021, 8, 649100. [Google Scholar] [CrossRef]
- Wang, Q.; Weng, S.; Sun, Y.; Lin, Y.; Zhong, W.; Kwok, H.F.; Lin, Y. High DAPK1 Expression Promotes Tumor Metastasis of Gastric Cancer. Biology 2022, 11, 1488. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zaboli, D.; Wang, H.; Liu, Y.; Arnaoutakis, D.; Khan, T.; Khan, Z.; Koch, W.M.; Califano, J.A. Detection of TIMP3 promoter hypermethylation in salivary rinse as an independent predictor of local recurrence-free survival in head and neck cancer. Clin. Cancer Res. 2012, 18, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Butler, R.A.; Eliot, M.; Pawlita, M.; Maccani, J.Z.; McClean, M.D.; Kelsey, K.T. Novel DNA methylation targets in oral rinse samples predict survival of patients with oral squamous cell carcinoma. Oral Oncol. 2014, 50, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Su, C.W.; Lin, C.W.; Yang, W.E.; Yang, S.F. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919864247. [Google Scholar] [CrossRef] [PubMed]
- Su, C.W.; Su, B.F.; Chiang, W.L.; Yang, S.F.; Chen, M.K.; Lin, C.W. Plasma levels of the tissue inhibitor matrix metalloproteinase-3 as a potential biomarker in oral cancer progression. Int. J. Med. Sci. 2017, 14, 37–44. [Google Scholar] [CrossRef]
- Glöckner, S.C.; Dhir, M.; Yi, J.M.; McGarvey, K.E.; Van Neste, L.; Louwagie, J.; Chan, T.A.; Kleeberger, W.; De Bruïne, A.P.; Smits, K.M. Methylation of TFPI2 in stool DNA: A potential novel biomarker for the detection of colorectal cancer. Cancer Res. 2009, 69, 4691–4699. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Gao, Y.; Pei, L.; Zhou, J.; Gu, L.; Zhang, L.; Zhu, B.; Hattori, N.; Ji, J. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin. Cancer Res. 2014, 20, 4598–4612. [Google Scholar] [CrossRef]
- Li, Y.-F.; Hsiao, Y.-H.; Lai, Y.-H.; Chen, Y.-C.; Chen, Y.-J.; Chou, J.-L.; Chan, M.W.; Lin, Y.-H.; Tsou, Y.-A.; Tsai, M.-H. DNA methylation profiles and biomarkers of oral squamous cell carcinoma. Epigenetics 2015, 10, 229–236. [Google Scholar] [CrossRef]
- Sohn, J.; Natale, J.; Chew, L.-J.; Belachew, S.; Cheng, Y.; Aguirre, A.; Lytle, J.; Nait-Oumesmar, B.; Kerninon, C.; Kanai-Azuma, M. Identification of Sox17 as a transcription factor that regulates oligodendrocyte development. J. Neurosci. 2006, 26, 9722–9735. [Google Scholar] [CrossRef]
- Matsui, T.; Kanai-Azuma, M.; Hara, K.; Matoba, S.; Hiramatsu, R.; Kawakami, H.; Kurohmaru, M.; Koopman, P.; Kanai, Y. Redundant roles of Sox17 and Sox18 in postnatal angiogenesis in mice. J. Cell Sci. 2006, 119, 3513–3526. [Google Scholar] [CrossRef]
- Park, K.-S.; Wells, J.M.; Zorn, A.M.; Wert, S.E.; Whitsett, J.A. Sox17 influences the differentiation of respiratory epithelial cells. Dev. Biol. 2006, 294, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Saunders, T.L.; Morrison, S.J. Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 2007, 130, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.; Jusue-Torres, I.; Stark, A.; Chen, C.; Rodgers, K.; Lee, B.; Griffin, C.; Yang, A.; Huang, P.; Wrangle, J. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin. Cancer Res. 2017, 23, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Watanabe, Y.; Yoshida, Y.; Sato, Y.; Hiraishi, T.; Oikawa, R.; Maehata, T.; Suzuki, H.; Toyota, M.; Niwa, H. Hypermethylation of Sox17 gene is useful as a molecular diagnostic application in early gastric cancer. Tumor Biol. 2012, 33, 383–393. [Google Scholar] [CrossRef]
- Jia, Y.; Yang, Y.; Liu, S.; Liu, S.; Herman, J.G.; Lu, F.; Guo, M. SOX17 antagonizes WNT/β-catenin signaling pathway in hepatocellular carcinoma. Epigenetics 2010, 5, 743–749. [Google Scholar] [CrossRef]
- Fu, D.-Y.; Wang, Z.-M.; Wang, B.-L.; Shen, Z.-Z.; Huang, W.; Shao, Z.-M. Sox17, the canonical Wnt antagonist, is epigenetically inactivated by promoter methylation in human breast cancer. Breast Cancer Res. Treat. 2010, 119, 601–612. [Google Scholar] [CrossRef]
- Zhang, W.; Glöckner, S.C.; Guo, M.; Machida, E.O.; Wang, D.H.; Easwaran, H.; Van Neste, L.; Herman, J.G.; Schuebel, K.E.; Watkins, D.N. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008, 68, 2764–2772. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Menon, R.S. Wnt Signaling in Oral Cancer Initiating Cells. Ph.D. Thesis, Harvard School of Dental Medicine, Boston, MA, USA, 2017. [Google Scholar]
- Soares-Lima, S.C.; Mehanna, H.; Camuzi, D.; de Souza-Santos, P.T.; Simão, T.d.A.; Nicolau-Neto, P.; Almeida Lopes, M.d.S.; Cuenin, C.; Talukdar, F.R.; Batis, N. Upper aerodigestive tract squamous cell carcinomas show distinct overall DNA methylation profiles and different molecular mechanisms behind WNT signaling disruption. Cancers 2021, 13, 3014. [Google Scholar] [CrossRef]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef]
- Jayaprakash, C.; Radhakrishnan, R.; Ray, S.; Satyamoorthy, K. Promoter methylation of MGMT in oral carcinoma: A population-based study and meta-analysis. Arch. Oral Biol. 2017, 80, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Supic, G.; Kozomara, R.; Jovic, N.; Zeljic, K.; Magic, Z. Prognostic significance of tumor-related genes hypermethylation detected in cancer-free surgical margins of oral squamous cell carcinomas. Oral Oncol. 2011, 47, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Soudry, E.; Acero, J.; Orera, M.; Moreno-Lopez, L.; Macía-Colón, G.; Jaffe, A.; Berdasco, M.; Ili-Gangas, C.; Brebi-Mieville, P. NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev. Res. 2011, 4, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, A.M.; Na, T.-Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Pentinmikko, N.; Luopajärvi, K.; Willis, N.J.; Gilroy, K.; Raven, A.P.; Mcgarry, L.; Englund, J.I.; Webb, A.T.; Scharaw, S. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature 2021, 594, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Repenning, A.; Happel, D.; Bouchard, C.; Meixner, M.; Verel-Yilmaz, Y.; Raifer, H.; Holembowski, L.; Krause, E.; Kremmer, E.; Feederle, R. PRMT1 promotes the tumor suppressor function of p14ARF and is indicative for pancreatic cancer prognosis. EMBO J. 2021, 40, e106777. [Google Scholar] [CrossRef]
- Olesen, T.B.; Sand, F.L.; Rasmussen, C.L.; Albieri, V.; Toft, B.G.; Norrild, B.; Munk, C.; Kjær, S.K. Prevalence of human papillomavirus DNA and p16INK4a in penile cancer and penile intraepithelial neoplasia: A systematic review and meta-analysis. Lancet Oncol. 2019, 20, 145–158. [Google Scholar] [CrossRef]
- Nikitakis, N.G.; Pentenero, M.; Georgaki, M.; Poh, C.F.; Peterson, D.E.; Edwards, P.; Lingen, M.; Sauk, J.J. Molecular markers associated with development and progression of potentially premalignant oral epithelial lesions: Current knowledge and future implications. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 125, 650–669. [Google Scholar] [CrossRef]
- Daniel, F.I.; Rivero, E.R.C.; Modolo, F.; Lopes, T.G.; Salum, F.G. Immunohistochemical expression of DNA methyltransferases 1, 3a and 3b in oral leukoplakias and squamous cell carcinomas. Arch. Oral Biol. 2010, 55, 1024–1030. [Google Scholar] [CrossRef]
- Supic, G.; Kozomara, R.; Zeljic, K.; Jovic, N.; Magic, Z. Prognostic value of the DNMTs mRNA expression and genetic polymorphisms on the clinical outcome in oral cancer patients. Clin. Oral Investig. 2017, 21, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Kabekkodu, S.; Satyamoorthy, K. DNA hypermethylation as an epigenetic mark for oral cancer diagnosis. J. Oral Pathol. Med. 2011, 40, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Supic, G.; Jovic, N.; Kozomara, R.; Zeljic, K.; Magic, Z. Interaction between the MTHFR C677T polymorphism and alcohol—Impact on oral cancer risk and multiple DNA methylation of tumor-related genes. J. Dent. Res. 2011, 90, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.Y. Epigenetic alterations in head and neck cancer: Prevalence, clinical significance, and implications. Curr. Oncol. Rep. 2004, 6, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Mydlarz, W.K.; Hennessey, P.T.; Califano, J.A. Advances and Perspectives in the Molecular Diagnosis of Head and Neck Cancer. Expert Opin. Med. Diagn. 2010, 4, 53–65. [Google Scholar] [CrossRef]
- Shaw, R. The epigenetics of oral cancer. Int. J. Oral Maxillofac. Surg. 2006, 35, 101–108. [Google Scholar] [CrossRef]
- Shaw, R.J.; Hall, G.L.; Lowe, D.; Liloglou, T.; Field, J.K.; Sloan, P.; Risk, J.M. The role of pyrosequencing in head and neck cancer epigenetics: Correlation of quantitative methylation data with gene expression. Arch. Otolaryngol. Head. Neck Surg. 2008, 134, 251–256. [Google Scholar] [CrossRef]
- Bennett, K.L.; Hackanson, B.; Smith, L.T.; Morrison, C.D.; Lang, J.C.; Schuller, D.E.; Weber, F.; Eng, C.; Plass, C. Tumor suppressor activity of CCAAT/enhancer binding protein alpha is epigenetically down-regulated in head and neck squamous cell carcinoma. Cancer Res. 2007, 67, 4657–4664. [Google Scholar] [CrossRef]
- Calmon, M.F.; Rodrigues, R.V.; Kaneto, C.M.; Moura, R.P.; Silva, S.D.; Mota, L.D.; Pinheiro, D.G.; Torres, C.; de Carvalho, A.F.; Cury, P.M.; et al. Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia 2009, 11, 1329–1339. [Google Scholar] [CrossRef]
- Glazer, C.A.; Chang, S.S.; Ha, P.K.; Califano, J.A. Applying the molecular biology and epigenetics of head and neck cancer in everyday clinical practice. Oral Oncol. 2009, 45, 440–446. [Google Scholar] [CrossRef]
- Lingen, M.W.; Pinto, A.; Mendes, R.A.; Franchini, R.; Czerninski, R.; Tilakaratne, W.M.; Partridge, M.; Peterson, D.E.; Woo, S.B. Genetics/epigenetics of oral premalignancy: Current status and future research. Oral Dis. 2011, 17 (Suppl. S1), 7–22. [Google Scholar] [CrossRef] [PubMed]
- de Freitas Cordeiro-Silva, M.; Oliveira, Z.F.L.; de Podestá, J.R.V.; Gouvea, S.A.; Von Zeidler, S.V.; Louro, I.D. Methylation analysis of cancer-related genes in non-neoplastic cells from patients with oral squamous cell carcinoma. Mol. Biol. Rep. 2011, 38, 5435–5441. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Saranath, D. Concurrent hypermethylation of multiple regulatory genes in chewing tobacco associated oral squamous cell carcinomas and adjacent normal tissues. Oral Oncol. 2004, 40, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Matassa, D.S.; Conte, M.; Colella, G.; Rana, G.; Fucci, L.; Piscopo, M. H3K4 histone methylation in oral squamous cell carcinoma. Acta Biochim. Pol. 2009, 14, 56. [Google Scholar] [CrossRef]
- Lee, E.-S.; Issa, J.-P.; Roberts, D.B.; Williams, M.D.; Weber, R.S.; Kies, M.S.; El-Naggar, A.K. Quantitative promoter hypermethylation analysis of cancer-related genes in salivary gland carcinomas: Comparison with methylation-specific PCR technique and clinical significance. Clin. Cancer Res. 2008, 14, 2664–2672. [Google Scholar] [CrossRef]
- Kozaki, K.-i.; Imoto, I.; Mogi, S.; Omura, K.; Inazawa, J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008, 68, 2094–2105. [Google Scholar] [CrossRef]
- Lee, J.I.; Soria, J.-C.; Hassan, K.A.; El-Naggar, A.K.; Tang, X.; Liu, D.D.; Hong, W.K.; Mao, L. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch. Otolaryngol. Head. Neck Surg. 2001, 127, 1441–1445. [Google Scholar] [CrossRef]
- Pannone, G.; Bufo, P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G.; Serpico, R.; Cafarelli, B.; Abbruzzese, A. WNT pathway in oral cancer: Epigenetic inactivation of WNT-inhibitors. Oncol. Rep. 2010, 24, 1035–1041. [Google Scholar]
- Sogabe, Y.; Suzuki, H.; Toyota, M.; Ogi, K.; Imai, T.; Nojima, M.; Sasaki, Y.; Hiratsuka, H.; Tokino, T. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int. J. Oncol. 2008, 32, 1253–1261. [Google Scholar] [CrossRef]
- Chan, S.H.; Chiang, J.; Ngeow, J. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hered. Cancer Clin. Pract. 2021, 19, 21. [Google Scholar] [CrossRef]
- Cilluffo, D.; Barra, V.; Di Leonardo, A. P14(ARF): The Absence that Makes the Difference. Genes 2020, 11, 824. [Google Scholar] [CrossRef] [PubMed]
- Karami Fath, M.; Babakhaniyan, K.; Anjomrooz, M.; Jalalifar, M.; Alizadeh, S.D.; Pourghasem, Z.; Abbasi Oshagh, P.; Azargoonjahromi, A.; Almasi, F.; Manzoor, H.Z.; et al. Recent Advances in Glioma Cancer Treatment: Conventional and Epigenetic Realms. Vaccines 2022, 10, 1448. [Google Scholar] [CrossRef] [PubMed]
- Zerrouqi, A.; Pyrzynska, B.; Febbraio, M.; Brat, D.; Van Meir, E. p14ARF inhibits tumor-induced angiogenesis by a p53-independent sp1/TIMP3 mechanism. Cancer Res. 2007, 67, 4169. [Google Scholar]
- Zerrouqi, A.; Pyrzynska, B.; Febbraio, M.; Brat, D.J.; Van Meir, E.G. P14ARF inhibits human glioblastoma–induced angiogenesis by upregulating the expression of TIMP3. J. Clin. Investig. 2012, 122, 1283–1295. [Google Scholar] [CrossRef]
- Wei, W.; Hemmer, R.M.; Sedivy, J.M. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol. Cell. Biol. 2001, 21, 6748–6757. [Google Scholar] [CrossRef]
- Ishida, E.; Nakamura, M.; Ikuta, M.; Shimada, K.; Matsuyoshi, S.; Kirita, T.; Konishi, N. Promotor hypermethylation of p14ARF is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol. 2005, 41, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Sailasree, R.; Abhilash, A.; Sathyan, K.M.; Nalinakumari, K.R.; Thomas, S.; Kannan, S. Differential roles of p16INK4A and p14ARF genes in prognosis of oral carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 414–420. [Google Scholar] [CrossRef]
- Takeshima, M.; Saitoh, M.; Kusano, K.; Nagayasu, H.; Kurashige, Y.; Malsantha, M.; Arakawa, T.; Takuma, T.; Chiba, I.; Kaku, T.; et al. High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in Sri Lanka. J. Oral Pathol. Med. 2008, 37, 475–479. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef]
- McConnell, B.B.; Gregory, F.J.; Stott, F.J.; Hara, E.; Peters, G. Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 1999, 19, 1981–1989. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Cai, W.; Shi, H.; Wang, Y.; Li, M.; Jiao, J.; Chen, M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: A meta-analysis. Br. J. Cancer 2013, 108, 2542–2548. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, C.; Zhou, H.; Bao, T.; Gao, T.; Jiang, X.; Ye, M. The association between methylated CDKN2A and cervical carcinogenesis, and its diagnostic value in cervical cancer: A meta-analysis. Ther. Clin. Risk Manag. 2016, 12, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-J.; Yeh, K.-T.; Shih, H.-C.; Wang, Y.-F.; Lin, T.-H.; Chang, J.-Y.; Shih, M.-C.; Chang, J.-G. The correlation between CpG methylation and protein expression of P16 in oral squamous cell carcinomas. Int. J. Mol. Med. 2002, 10, 551–554. [Google Scholar]
- Nakahara, Y.; Shintani, S.; Mihara, M.; Ueyama, Y.; Matsumura, T. High frequency of homozygous deletion and methylation of p16INK4A gene in oral squamous cell carcinomas. Cancer Lett. 2001, 163, 221–228. [Google Scholar] [CrossRef]
- Maruya, S.-I.; Issa, J.-P.J.; Weber, R.S.; Rosenthal, D.I.; Haviland, J.C.; Lotan, R.; El-Naggar, A.K. Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: Incidence and potential implications. Clin. Cancer Res. 2004, 10, 3825–3830. [Google Scholar] [CrossRef]
- Kato, K.; Hara, A.; Kuno, T.; Mori, H.; Yamashita, T.; Toida, M.; Shibata, T. Aberrant promoter hypermethylation of p16 and MGMT genes in oral squamous cell carcinomas and the surrounding normal mucosa. J. Cancer Res. Clin. Oncol. 2006, 132, 735–743. [Google Scholar] [CrossRef]
- Kresty, L.A.; Mallery, S.R.; Knobloch, T.J.; Song, H.; Lloyd, M.; Casto, B.C.; Weghorst, C.M. Alterations of p16 INK4a and p14 ARF in patients with severe oral epithelial dysplasia. Cancer Res. 2002, 62, 5295–5300. [Google Scholar]
- Rastogi, V.; Puri, N.; Mishra, S.; Arora, S.; Kaur, G.; Yadav, L. An insight to oral epithelial dysplasia. Int. J. Head. Neck Surg. 2013, 4, 74–82. [Google Scholar] [CrossRef]
- Taioli, E.; Ragin, C.; Wang, X.-H.; Chen, J.; Langevin, S.M.; Brown, A.R.; Gollin, S.M.; Garte, S.; Sobol, R.W. Recurrence in oral and pharyngeal cancer is associated with quantitative MGMT promoter methylation. BMC Cancer 2009, 9, 354. [Google Scholar] [CrossRef]
- Ohta, S.; Uemura, H.; Matsui, Y.; Ishiguro, H.; Fujinami, K.; Kondo, K.; Miyamoto, H.; Yazawa, T.; Danenberg, K.; Danenberg, P.V.; et al. Alterations of p16 and p14ARF genes and their 9p21 locus in oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2009, 107, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, J.; Dong, F.; Wang, X.; Zhang, Y. The correlations between alteration of p16 gene and clinicopathological factors and prognosis in squamous cell carcinomas of the buccal mucosa. J. Oral Pathol. Med. 2012, 41, 463–469. [Google Scholar] [CrossRef]
- Niessen, C.M.; Leckband, D.; Yap, A.S. Tissue organization by cadherin adhesion molecules: Dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev. 2011, 91, 691–731. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Na, T.-Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc. Natl. Acad. Sci. USA 2020, 117, 5931–5937. [Google Scholar] [CrossRef]
- Pannone, G.; Santoro, A.; Feola, A.; Bufo, P.; Papagerakis, P.; Lo Muzio, L.; Staibano, S.; Ionna, F.; Longo, F.; Franco, R.; et al. The role of E-cadherin down-regulation in oral cancer: CDH1 gene expression and epigenetic blockage. Curr. Cancer Drug Targets 2014, 14, 115–127. [Google Scholar] [CrossRef]
- Kordi-Tamandani, D.M.; Moazeni-Roodi, A.-K.; Rigi-Ladiz, M.-A.; Hashemi, M.; Birjandian, E.; Torkamanzehi, A. Promoter hypermethylation and expression profile of MGMT and CDH1 genes in oral cavity cancer. Arch. Oral Biol. 2010, 55, 809–814. [Google Scholar] [CrossRef]
- Wen, G.; Wang, H.; Zhong, Z. Associations of RASSF1A, RARβ, and CDH1 promoter hypermethylation with oral cancer risk: A PRISMA-compliant meta-analysis. Medicine 2018, 97, e9971. [Google Scholar] [CrossRef]
- Kadeh, H.; Parsamanesh, N.; Miri-Moghaddam, E. Effect of CDH1 and CDH2 genes polymorphisms in oral squamous cell carcinoma susceptibility in a sample of Iranian population: A case-control study. Health Sci. Rep. 2023, 6, e1221. [Google Scholar] [CrossRef]
- Vered, M.; Allon, I.; Buchner, A.; Dayan, D. E-cadherin in oral SCC: An analysis of the confusing literature and new insights related to its immunohistochemical expression. Histol. Histopathol. 2012, 27, 141–150. [Google Scholar]
- Di Domenico, M.; Pierantoni, G.; Feola, A.; Esposito, F.; Laino, L.; De Rosa, A.; Rullo, R.; Mazzotta, M.; Martano, M.; Sanguedolce, F. Prognostic significance of N-Cadherin expression in oral squamous cell carcinoma. Anticancer. Res. 2011, 31, 4211–4218. [Google Scholar] [PubMed]
- Martin Torgersen, K.; Kim, S.-A.; Dixon, J.E. Chapter 133—PTEN/MTM Phosphatidylinositol Phosphatases. In Handbook of Cell Signaling, 2nd ed.; Bradshaw, R.A., Dennis, E.A., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 1061–1064. [Google Scholar]
- Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Rocco, E.G.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes. Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, B.; Liu, E.; Zhang, Z. Loss of PTEN expression is associated with PI3K pathway-dependent metabolic reprogramming in hepatocellular carcinoma. Cell Commun. Signal. 2020, 18, 131. [Google Scholar] [CrossRef]
- Karami Fath, M.; Azargoonjahromi, A.; Soofi, A.; Almasi, F.; Hosseinzadeh, S.; Khalili, S.; Sheikhi, K.; Ferdousmakan, S.; Owrangi, S.; Fahimi, M.; et al. Current understanding of epigenetics role in melanoma treatment and resistance. Cancer Cell Int. 2022, 22, 313. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- García, Z.; Kumar, A.; Marques, M.; Cortes, I.; Carrera, A.C. Phosphoinositide 3-kinase controls early and late events in mammalian cell division. EMBO J. 2006, 25, 655–661. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The ras/raf/mapk pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- Kurasawa, Y.; Shiiba, M.; Nakamura, M.; Fushimi, K.; Ishigami, T.; Bukawa, H.; Yokoe, H.; Uzawa, K.; Tanzawa, H. PTEN expression and methylation status in oral squamous cell carcinoma. Oncol. Rep. 2008, 19, 1429–1434. [Google Scholar]
- Mavros, A.; Hahn, M.; Wieland, I.; Koy, S.; Koufaki, O.; Strelocke, K.; Koch, R.; Haroske, G.; Schackert, H.; Eckelt, U. Infrequent genetic alterations of the tumor suppressor gene PTEN/MMAC1 in squamous cell carcinoma of the oral cavity. J. Oral Pathol. Med. 2002, 31, 270–276. [Google Scholar] [CrossRef]
- Chang, H.W.; Chow, V.; Lam, K.Y.; Wei, W.I.; Wing Yuen, A.P. Loss of E-cadherin expression resulting from promoter hypermethylation in oral tongue carcinoma and its prognostic significance. Cancer 2002, 94, 386–392. [Google Scholar] [CrossRef]
- Kudo, Y.; Kitajima, S.; Ogawa, I.; Hiraoka, M.; Sargolzaei, S.; Keikhaee, M.R.; Sato, S.; Miyauchi, M.; Takata, T. Invasion and metastasis of oral cancer cells require methylation of E-cadherin and/or degradation of membranous beta-catenin. Clin. Cancer Res. 2004, 10, 5455–5463. [Google Scholar] [CrossRef] [PubMed]
- Díez-Pérez, R.; Campo-Trapero, J.; Cano-Sánchez, J.; López-Durán, M.; Gonzalez-Moles, M.A.; Bascones-Ilundain, J.; Bascones-Martinez, A. Methylation in oral cancer and pre-cancerous lesions (Review). Oncol. Rep. 2011, 25, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Liloglou, T.; Rogers, S.N.; Brown, J.S.; Vaughan, E.D.; Lowe, D.; Field, J.K.; Risk, J.M. Promoter methylation of P16, RARβ, E-cadherin, cyclin A1 and cytoglobin in oral cancer: Quantitative evaluation using pyrosequencing. Br. J. Cancer 2006, 94, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Squarize, C.H.; Castilho, R.M.; Pinto, D.S., Jr. Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. J. Oral Pathol. Med. 2002, 31, 379–384. [Google Scholar] [CrossRef]
- Chen, Q.; Samaranayake, L.P.; Zhou, H.; Xiao, L. Homozygous deletion of the PTEN tumor-suppressor gene is not a feature in oral squamous cell carcinoma. Oral Oncol. 2000, 36, 95–99. [Google Scholar] [CrossRef]
- McBride, O.W.; Merry, D.; Givol, D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13). Proc. Natl. Acad. Sci. USA 1986, 83, 130–134. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef]
- Baral, R.; Patnaik, S.; Das, B.R. Co-overexpression of p53 and c-myc proteins linked with advanced stages of betel-and tobacco-related oral squamous cell carcinomas from eastern India. Eur. J. Oral Sci. 1998, 106, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Caamano, J.; Zhang, S.; Rosvold, E.; Bauer, B.; Klein-Szanto, A. p53 alterations in human squamous cell carcinomas and carcinoma cell lines. Am. J. Pathol. 1993, 142, 1131. [Google Scholar] [PubMed]
- Boyle, J.O.; Hakim, J.; Koch, W.; Riet, P.v.d.; Hruban, R.H.; Roa, R.A.; Correo, R.; Eby, Y.J.; Ruppert, J.M.; Sidransky, D. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res. 1993, 53, 4477–4480. [Google Scholar] [PubMed]
- Mascolo, M.; Siano, M.; Ilardi, G.; Russo, D.; Merolla, F.; De Rosa, G.; Staibano, S. Epigenetic disregulation in oral cancer. Int. J. Mol. Sci. 2012, 13, 2331–2353. [Google Scholar] [CrossRef]
- Celik, S.; Akcora, D.; Ozkan, T.; Varol, N.; Aydos, S.; Sunguroglu, A. Methylation analysis of the DAPK1 gene in imatinib-resistant chronic myeloid leukemia patients. Oncol. Lett. 2015, 9, 399–404. [Google Scholar] [CrossRef]
- Gade, P.; Kalvakolanu, D.V. Chapter 19—Death-Associated Protein Kinase 1 Suppresses Tumor Growth and Metastasis via Autophagy and Apoptosis. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2014; pp. 277–292. [Google Scholar]
- Rosas, S.L.B.; Koch, W.; Carvalho, M.d.G.d.C.; Wu, L.; Califano, J.; Westra, W.; Jen, J.; Sidransky, D. Promoter hypermethylation patterns of p16, O 6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001, 61, 939–942. [Google Scholar]
- Sanchez-Cespedes, M.; Esteller, M.; Wu, L.; Nawroz-Danish, H.; Yoo, G.H.; Koch, W.M.; Jen, J.; Herman, J.G.; Sidransky, D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000, 60, 892–895. [Google Scholar]
- Hasegawa, M.; Nelson, H.H.; Peters, E.; Ringstrom, E.; Posner, M.; Kelsey, K.T. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene 2002, 21, 4231–4236. [Google Scholar] [CrossRef]
- Don, K.; Ramani, P.; Ramshankar, V.; Sherlin, H.; Premkumar, P.; Natesan, A. Promoter hypermethylation patterns of P16, DAPK and MGMT in Oral Squamous Cell Carcinoma: A systematic review and meta-analysis. Indian J. Dent. Res. 2014, 25, 797–805. [Google Scholar] [CrossRef]
- Wong, Y.-K.; Lee, L.-T.; Liu, C.-J. Hypermethylation of MGMT and DAPK gene promoters is associated with tumorigenesis and metastasis in oral squamous cell carcinoma. J. Dent. Sci. 2011, 6, 158–164. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Hegi, M.E.; Reifenberger, G. MGMT promoter methylation in malignant gliomas. Target. Oncol. 2010, 5, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Fan, T.; Sun, G.; Wang, X.; Zhao, L.; Zhong, R. The dual role of DNA repair protein MGMT in cancer prevention and treatment. DNA Repair. 2023, 123, 103449. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-H.; Lee, H.-S.; Mar, K.; Ji, D.-D.; Huang, M.-S.; Hsia, K.-T. Loss expression of O6-methylguanine DNA methyltransferase by promoter hypermethylation and its relationship to betel quid chewing in oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2010, 109, 883–889. [Google Scholar] [CrossRef]
- Murakami, J.; Asaumi, J.-I.; Maki, Y.; Tsujigiwa, H.; Nagatsuka, H.; Kokeguchi, S.; Inoue, T.; Kawasaki, S.; Tanaka, N.; MacPhee, D. Influence of CpG island methylation status in O6-methylguanine-DNA methyltransferase expression of oral cancer cell lines. Oncol. Rep. 2004, 12, 339–345. [Google Scholar] [CrossRef]
- di Masi, A. Repair Mechanisms☆. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Viet, C.T.; Jordan, R.C.; Schmidt, B.L. DNA promoter hypermethylation in saliva for the early diagnosis of oral cancer. J. Calif. Dent. Assoc. 2007, 35, 844–849. [Google Scholar] [CrossRef]
- Xu, X.C. Tumor-suppressive activity of retinoic acid receptor-beta in cancer. Cancer Lett. 2007, 253, 14–24. [Google Scholar] [CrossRef]
- Rybicki, B.; Mitrache, N.; Do, K.; Jankowski, M.; Tang, D.; Rundle, A.; Bock, C.; Beebe-Dimmer, J.; Belinsky, S. Methylation of Retinoic Acid Receptor, Beta (RARB) Gene Increases Risk for Prostate Cancer in African-American Men. Cancer Epidemiol. Biomark. Prev. 2011, 20, 718. [Google Scholar] [CrossRef]
- Yari, K.; Rahimi, Z. Promoter Methylation Status of the Retinoic Acid Receptor-Beta 2 Gene in Breast Cancer Patients: A Case Control Study and Systematic Review. Breast Care 2019, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Youssef, E.M.; Lotan, D.; Issa, J.P.; Wakasa, K.; Fan, Y.H.; Mao, L.; Hassan, K.; Feng, L.; Lee, J.J.; Lippman, S.M.; et al. Hypermethylation of the retinoic acid receptor-beta(2) gene in head and neck carcinogenesis. Clin. Cancer Res. 2004, 10, 1733–1742. [Google Scholar] [CrossRef]
- van der Weyden, L.; Adams, D.J. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 2007, 1776, 58–85. [Google Scholar] [CrossRef]
- Imai, T.; Toyota, M.; Suzuki, H.; Akino, K.; Ogi, K.; Sogabe, Y.; Kashima, L.; Maruyama, R.; Nojima, M.; Mita, H. Epigenetic inactivation of RASSF2 in oral squamous cell carcinoma. Cancer Sci. 2008, 99, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-H.; Huang, S.-F.; Chen, I.-H.; Liao, C.-T.; Wang, H.-M.; Hsieh, L.-L. Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 4174–4180. [Google Scholar] [CrossRef]
- Tran, T.N.; Liu, Y.; Takagi, M.; Yamaguchi, A.; Fujii, H. Frequent promoter hypermethylation of RASSF1A and p16INK4a and infrequent allelic loss other than 9p21 in betel-associated oral carcinoma in a Vietnamese non-smoking/non-drinking female population. J. Oral Pathol. Med. 2005, 34, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Völkel, P.; Angrand, P.O. The control of histone lysine methylation in epigenetic regulation. Biochimie 2007, 89, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Piyathilake, C.J.; Bell, W.C.; Jones, J.; Henao, O.L.; Heimburger, D.C.; Niveleau, A.; Grizzle, W.E. Patterns of global DNA and histone methylation appear to be similar in normal, dysplastic and neoplastic oral epithelium of humans. Dis. Markers 2005, 21, 147–151. [Google Scholar] [CrossRef]
- Yamamoto, D.; Shima, K.; Matsuo, K.; Nishioka, T.; Chen, C.Y.; Hu, G.F.; Sasaki, A.; Tsuji, T. Ornithine decarboxylase antizyme induces hypomethylation of genome DNA and histone H3 lysine 9 dimethylation (H3K9me2) in human oral cancer cell line. PLoS ONE 2010, 5, e12554. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, B.R.; Chen, S.T.; Hsieh, T.H.; Li, Y.J.; Kuo, M.Y. HDAC2 promotes cell migration/invasion abilities through HIF-1α stabilization in human oral squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 567–575. [Google Scholar] [CrossRef]
- Sakuma, T.; Uzawa, K.; Onda, T.; Shiiba, M.; Yokoe, H.; Shibahara, T.; Tanzawa, H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006, 29, 117–124. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Arif, M.; Vedamurthy, B.M.; Choudhari, R.; Ostwal, Y.B.; Mantelingu, K.; Kodaganur, G.S.; Kundu, T.K. Nitric oxide-mediated histone hyperacetylation in oral cancer: Target for a water-soluble HAT inhibitor, CTK7A. Chem. Biol. 2010, 17, 903–913. [Google Scholar] [CrossRef]
- Das, B.R. Increased ADP-ribosylation of histones in oral cancer. Cancer Lett. 1993, 73, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, M.; Ilardi, G.; Romano, M.F.; Celetti, A.; Siano, M.; Romano, S.; Luise, C.; Merolla, F.; Rocco, A.; Vecchione, M.L.; et al. Overexpression of chromatin assembly factor-1 p60, poly(ADP-ribose) polymerase 1 and nestin predicts metastasizing behaviour of oral cancer. Histopathology 2012, 61, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Staibano, S.; Mignogna, C.; Lo Muzio, L.; Mascolo, M.; Salvatore, G.; Di Benedetto, M.; Califano, L.; Rubini, C.; De Rosa, G. Chromatin assembly factor-1 (CAF-1)-mediated regulation of cell proliferation and DNA repair: A link with the biological behaviour of squamous cell carcinoma of the tongue? Histopathology 2007, 50, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Staibano, S.; Mascolo, M.; Rocco, A.; Lo Muzio, L.; Ilardi, G.; Siano, M.; Pannone, G.; Vecchione, M.L.; Nugnes, L.; Califano, L.; et al. The proliferation marker Chromatin Assembly Factor-1 is of clinical value in predicting the biological behaviour of salivary gland tumours. Oncol. Rep. 2011, 25, 13–22. [Google Scholar] [CrossRef]
- Chen, Y.W.; Kao, S.Y.; Wang, H.J.; Yang, M.H. Histone modification patterns correlate with patient outcome in oral squamous cell carcinoma. Cancer 2013, 119, 4259–4267. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Rastogi, B.; Raut, S.K.; Panda, N.K.; Rattan, V.; Radotra, B.D.; Khullar, M. Overexpression of HDAC9 promotes oral squamous cell carcinoma growth, regulates cell cycle progression, and inhibits apoptosis. Mol. Cell Biochem. 2016, 415, 183–196. [Google Scholar] [CrossRef]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes. Cancer 2015, 6, 169–181. [Google Scholar] [CrossRef]
- Chang, H.H.; Chiang, C.P.; Hung, H.C.; Lin, C.Y.; Deng, Y.T.; Kuo, M.Y. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009, 45, 610–614. [Google Scholar] [CrossRef]
- Lv, Y.; Lu, J.; Liu, X.; Miao, S.; Mao, X.; Li, B.; Pei, R.; Xiang, C. Histone deacetylase 1 regulates the malignancy of oral cancer cells via miR-154-5p/PCNA axis. Biol. Chem. 2020, 401, 1273–1281. [Google Scholar] [CrossRef]
- Roberti, A.; Fernández, A.F.; Fraga, M.F. Nicotinamide N-methyltransferase: At the crossroads between cellular metabolism and epigenetic regulation. Mol. Metab. 2021, 45, 101165. [Google Scholar] [CrossRef] [PubMed]
- Campagna, R.; Vignini, A. NAD(+) Homeostasis and NAD(+)-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhu, M.; Li, X.; Er, L.; Li, S. NNMT Is an Immune-Related Prognostic Biomarker That Modulates the Tumor Microenvironment in Pan-Cancer. Dis. Markers 2023, 2023, 9226712. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, V.; Sartini, D.; Morganti, S.; Giuliante, R.; Ruscio, G.; Santarelli, A.; Rocchetti, R.; Rubini, C.; Tomasetti, M.; Giannatempo, G.; et al. RNA-Mediated Gene Silencing of Nicotinamide N-Methyltransferase Is Associated with Decreased Tumorigenicity in Human Oral Carcinoma Cells. PLoS ONE 2013, 8, e71272. [Google Scholar] [CrossRef] [PubMed]
- van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; Escudé Martinez de Castilla, P.; et al. Esterase-Sensitive Prodrugs of a Potent Bisubstrate Inhibitor of Nicotinamide N-Methyltransferase (NNMT) Display Cellular Activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef]
- van Haren, M.J.; Zhang, Y.; Thijssen, V.; Buijs, N.; Gao, Y.; Mateuszuk, L.; Fedak, F.A.; Kij, A.; Campagna, R.; Sartini, D.; et al. Macrocyclic peptides as allosteric inhibitors of nicotinamide N-methyltransferase (NNMT). RSC Chem. Biol. 2021, 2, 1546–1555. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Li, D.-Q.; Müller, S.; Knapp, S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016, 35, 4423–4436. [Google Scholar] [CrossRef] [PubMed]
- Glatzel-Plucińska, N.; Piotrowska, A.; Dzięgiel, P.; Podhorska-Okołów, M. The Role of SATB1 in Tumour Progression and Metastasis. Int. J. Mol. Sci. 2019, 20, 4156. [Google Scholar] [CrossRef] [PubMed]
- Panchal, O.; Wichmann, G.; Grenman, R.; Eckhardt, L.; Kunz-Schughart, L.A.; Franke, H.; Dietz, A.; Aigner, A. SATB1 as oncogenic driver and potential therapeutic target in head & neck squamous cell carcinoma (HNSCC). Sci. Rep. 2020, 10, 8615. [Google Scholar] [PubMed]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA damage in stem cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Portney, B.A.; Arad, M.; Gupta, A.; Brown, R.A.; Khatri, R.; Lin, P.N.; Hebert, A.M.; Angster, K.H.; Silipino, L.E.; Meltzer, W.A. ZSCAN4 facilitates chromatin remodeling and promotes the cancer stem cell phenotype. Oncogene 2020, 39, 4970–4982. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.H.; Lee, J.S.; Ma, C.; Diggs, L.P.; Heinrich, S.; Chang, C.W.; Ma, L.; Forgues, M.; Budhu, A.; Chaisaingmongkol, J. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat. Commun. 2021, 12, 1455. [Google Scholar] [CrossRef]
- Aydin, Ö.Z.; Vermeulen, W.; Lans, H. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle 2014, 13, 3016–3025. [Google Scholar] [CrossRef]
- Fang, F.-M.; Li, C.-F.; Huang, H.-Y.; Lai, M.-T.; Chen, C.-M.; Chiu, I.-W.; Wang, T.-L.; Tsai, F.-J.; Shih, I.-M.; Sheu, J.J.-C. Overexpression of a chromatin remodeling factor, RSF-1/HBXAP, correlates with aggressive oral squamous cell carcinoma. Am. J. Pathol. 2011, 178, 2407–2415. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setién, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Cervigne, N.K.; Reis, P.P.; Machado, J.; Sadikovic, B.; Bradley, G.; Galloni, N.N.; Pintilie, M.; Jurisica, I.; Perez-Ordonez, B.; Gilbert, R.; et al. Identification of a microRNA signature associated with progression of leukoplakia to oral carcinoma. Hum. Mol. Genet. 2009, 18, 4818–4829. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Wang, X.-Y.; Gong, R.-G.; Li, A.; Yang, S.; Cao, Y.-T.; Wen, Y.-M.; Wang, C.-M.; Yi, X.-Z. The expression profile of microRNAs in a model of 7, 12-dimethyl-benz [a] anthrance-induced oral carcinogenesis in Syrian hamster. J. Exp. Clin. Cancer Res. 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Chang, K.-W.; Liu, C.-J.; Chu, T.-H.; Cheng, H.-W.; Hung, P.-S.; Hu, W.-Y.; Lin, S.-C. Association between high miR-211 microRNA expression and the poor prognosis of oral carcinoma. J. Dent. Res. 2008, 87, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.; Liu, X.B.; Chung-Wai Ho, A.; Po-Wing Yuen, A.; Wai-Man Ng, R.; Ignace Wei, W. Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling. Int. J. Cancer 2008, 123, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Jakymiw, A.; Patel, R.S.; Deming, N.; Bhattacharyya, I.; Shah, P.; Lamont, R.J.; Stewart, C.M.; Cohen, D.M.; Chan, E.K. Overexpression of dicer as a result of reduced let-7 MicroRNA levels contributes to increased cell proliferation of oral cancer cells. Genes. Chromosomes Cancer 2010, 49, 549–559. [Google Scholar] [CrossRef]
- Olive, V.; Jiang, I.; He, L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 2010, 42, 1348–1354. [Google Scholar] [CrossRef]
- Jevnaker, A.M.; Khuu, C.; Kjøle, E.; Bryne, M.; Osmundsen, H. Expression of members of the miRNA17-92 cluster during development and in carcinogenesis. J. Cell Physiol. 2011, 226, 2257–2266. [Google Scholar] [CrossRef]
- Moreira-Silva, F.; Camilo, V.; Gaspar, V.; Mano, J.F.; Henrique, R.; Jerónimo, C. Repurposing Old Drugs into New Epigenetic Inhibitors: Promising Candidates for Cancer Treatment? Pharmaceutics 2020, 12, 410. [Google Scholar] [CrossRef]
- Suzuki, M.; Shinohara, F.; Nishimura, K.; Echigo, S.; Rikiishi, H. Epigenetic regulation of chemosensitivity to 5-fluorouracil and cisplatin by zebularine in oral squamous cell carcinoma. Int. J. Oncol. 2007, 31, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Shinohara, F.; Endo, M.; Sugazaki, M.; Echigo, S.; Rikiishi, H. Zebularine suppresses the apoptotic potential of 5-fluorouracil via cAMP/PKA/CREB pathway against human oral squamous cell carcinoma cells. Cancer Chemother. Pharmacol. 2009, 64, 223–232. [Google Scholar] [CrossRef]
- Murakami, J.; Lee, Y.-J.; Kokeguchi, S.; Tsujigiwa, H.; Asaumi, J.-I.; Nagatsuka, H.; Fukui, K.; Kuroda, M.; Tanaka, N.; Matsubara, N. Depletion of O6-methylguanine-DNA methyltransferase by O6-benzylguanine enhances 5-FU cytotoxicity in colon and oral cancer cell lines. Oncol. Rep. 2007, 17, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, S.; Hinds, P.W.; MuÈnger, K. Re-expression of endogenous p16ink4a in oral squamous cell carcinoma lines by 5-aza-2′-deoxycytidine treatment induces a senescence-like state. Oncogene 1998, 17, 3445–3453. [Google Scholar] [CrossRef]
- Tsao, A.S.; Liu, D.; Martin, J.; Tang, X.-m.; Lee, J.J.; El-Naggar, A.K.; Wistuba, I.; Culotta, K.S.; Mao, L.; Gillenwater, A. Phase II randomized, placebo-controlled trial of green tea extract in patients with high-risk oral premalignant lesions. Cancer Prev. Res. 2009, 2, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Ide, R.; Fujino, Y.; Hoshiyama, Y.; Mizoue, T.; Kubo, T.; Pham, T.-M.; Shirane, K.; Tokui, N.; Sakata, K.; Tamakoshi, A. A prospective study of green tea consumption and oral cancer incidence in Japan. Ann. Epidemiol. 2007, 17, 821–826. [Google Scholar] [CrossRef]
- Kato, K.; Long, N.K.; Makita, H.; Toida, M.; Yamashita, T.; Hatakeyama, D.; Hara, A.; Mori, H.; Shibata, T. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br. J. Cancer 2008, 99, 647–654. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef]
- Fang, M.; Chen, D.; Yang, C.S. Dietary Polyphenols May Affect DNA Methylation123. J. Nutr. 2007, 137, 223S–228S. [Google Scholar] [CrossRef]
- Jha, M.; Aggarwal, R.; Jha, A.K.; Shrivastava, A. Natural compounds: DNA methyltransferase inhibitors in oral squamous cell carcinoma. Appl. Biochem. Biotechnol. 2015, 177, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Suzuki, M.; Sato, Y.; Echigo, S.; Rikiishi, H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int. J. Oncol. 2006, 28, 1233–1241. [Google Scholar] [CrossRef]
- Jung, M. Inhibitors of histone deacetylase as new anticancer agents. Curr. Med. Chem. 2001, 8, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Anh, T.D.; Ahn, M.-Y.; Kim, S.-A.; Yoon, J.-H.; Ahn, S.-G. The histone deacetylase inhibitor, Trichostatin A, induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells. Oncol. Rep. 2012, 27, 455–460. [Google Scholar] [PubMed]
- Suzuki, T.; Yokozaki, H.; Kuniyasu, H.; Hayashi, K.; Naka, K.; Ono, S.; Ishikawa, T.; Tahara, E.; Yasui, W. Effect of trichostatin A on cell growth and expression of cell cycle-and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int. J. Cancer 2000, 88, 992–997. [Google Scholar] [CrossRef]
- Yao, J.; Duan, L.; Fan, M.; Wu, X. NF-κB signaling pathway is involved in growth inhibition, G2/M arrest and apoptosis induced by Trichostatin A in human tongue carcinoma cells. Pharmacol. Res. 2006, 54, 406–413. [Google Scholar] [CrossRef]
- Chung, Y.-L.; Lee, M.-Y.; Pui, N.N. Epigenetic therapy using the histone deacetylase inhibitor for increasing therapeutic gain in oral cancer: Prevention of radiation-induced oral mucositis and inhibition of chemical-induced oral carcinogenesis. Carcinogenesis 2009, 30, 1387–1397. [Google Scholar] [CrossRef]
- Miki, Y.; Mukae, S.; Murakami, M.; Ishikawa, Y.; Ishii, T.; Ohki, H.; Matsumoto, M.; Komiyama, K. Butyrate inhibits oral cancer cell proliferation and regulates expression of secretory phospholipase A2-X and COX-2. Anticancer Res. 2007, 27, 1493–1502. [Google Scholar]
- Wang, A.; Zeng, R.; Huang, H. Retinoic acid and sodium butyrate as cell cycle regulators in the treatment of oral squamous carcinoma cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2008, 17, 175–182. [Google Scholar] [CrossRef]
- Bertino, E.M.; Otterson, G.A. Romidepsin: A novel histone deacetylase inhibitor for cancer. Expert Opin. Investig. Drugs 2011, 20, 1151–1158. [Google Scholar] [CrossRef]
- Murakami, J.; Asaumi, J.-i.; Maki, Y.; Tsujigiwa, H.; Kuroda, M.; Nagai, N.; Yanagi, Y.; Inoue, T.; Kawasaki, S.; Tanaka, N. Effects of demethylating agent 5-aza-2′-deoxycytidine and histone deacetylase inhibitor FR901228 on maspin gene expression in oral cancer cell lines. Oral Oncol. 2004, 40, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.; Asaumi, J.-i.; Kawai, N.; Tsujigiwa, H.; Yanagi, Y.; Nagatsuka, H.; Inoue, T.; Kokeguchi, S.; Kawasaki, S.; Kuroda, M. Effects of histone deacetylase inhibitor FR901228 on the expression level of telomerase reverse transcriptase in oral cancer. Cancer Chemother. Pharmacol. 2005, 56, 22–28. [Google Scholar] [CrossRef]
- Perego, P.; Zuco, V.; Gatti, L.; Zunino, F. Sensitization of tumor cells by targeting histone deacetylases. Biochem. Pharmacol. 2012, 83, 987–994. [Google Scholar] [CrossRef]
- Iglesias-Linares, A.; Yañez-Vico, R.M.; González-Moles, M.A. Potential role of HDAC inhibitors in cancer therapy: Insights into oral squamous cell carcinoma. Oral Oncol. 2010, 46, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.E.M.; do Nascimento Filho, C.H.V.; Marinho Bezerra, T.M.; Guerra, E.N.S.; Castilho, R.M.; Squarize, C.H. Entinostat is a novel therapeutic agent to treat oral squamous cell carcinoma. J. Oral Pathol. Med. 2020, 49, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y. HDAC inhibitor apicidin suppresses murine oral squamous cell carcinoma cell growth in vitro and in vivo via inhibiting HDAC8 expression. Oncol. Lett. 2018, 16, 6552–6560. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 2017, 20, 233–246.e237. [Google Scholar] [CrossRef]
- Bell, C.C.; Gilan, O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer 2020, 122, 465–472. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, N.; Yang, T.; Li, Y.; Li, J.; Huang, Q.; Wu, C.; Sun, L.; Zhou, X.; Cheng, X. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 2021, 75, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, Q.; Huang, S.; Liu, Y.; Li, Y.; Xing, Y.; Shi, D.; Xu, W.; Liu, W.; Ji, Z. The Interaction between DNMT1 and High-Mannose CD133 Maintains the Slow-Cycling State and Tumorigenic Potential of Glioma Stem Cell. Adv. Sci. 2022, 9, 2202216. [Google Scholar] [CrossRef]
- Ma, Y.; Chai, N.; Jiang, Q.; Chang, Z.; Chai, Y.; Li, X.; Sun, H.; Hou, J.; Linghu, E. DNA methyltransferase mediates the hypermethylation of the microRNA 34a promoter and enhances the resistance of patient-derived pancreatic cancer cells to molecular targeting agents. Pharmacol. Res. 2020, 160, 105071. [Google Scholar] [CrossRef] [PubMed]
- Camero, S.; Vitali, G.; Pontecorvi, P.; Ceccarelli, S.; Anastasiadou, E.; Cicchetti, F.; Flex, E.; Pomella, S.; Cassandri, M.; Rota, R. DNMT3A and DNMT3B targeting as an effective radiosensitizing strategy in embryonal rhabdomyosarcoma. Cells 2021, 10, 2956. [Google Scholar] [CrossRef]
- Jahangiri, R.; Mosaffa, F.; Emami Razavi, A.; Teimoori-Toolabi, L.; Jamialahmadi, K. Altered DNA methyltransferases promoter methylation and mRNA expression are associated with tamoxifen response in breast tumors. J. Cell. Physiol. 2018, 233, 7305–7319. [Google Scholar] [CrossRef]
- Lai, S.-C.; Su, Y.-T.; Chi, C.-C.; Kuo, Y.-C.; Lee, K.-F.; Wu, Y.-C.; Lan, P.-C.; Yang, M.-H.; Chang, T.-S.; Huang, Y.-H. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J. Exp. Clin. Cancer Res. 2019, 38, 1–18. [Google Scholar] [CrossRef]
- Simó-Riudalbas, L.; Melo, S.A.; Esteller, M. DNMT3B gene amplification predicts resistance to DNA demethylating drugs. Genes. Chromosomes Cancer 2011, 50, 527–534. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Liu, T.; Qin, S.; Zhuang, Y.; Liu, D.; Lu, S.W.; Kalari, K.R.; et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Investig. 2018, 128, 2376–2388. [Google Scholar] [CrossRef]
- Stewart, M.L.; Tamayo, P.; Wilson, A.J.; Wang, S.; Chang, Y.M.; Kim, J.W.; Khabele, D.; Shamji, A.F.; Schreiber, S.L. KRAS Genomic Status Predicts the Sensitivity of Ovarian Cancer Cells to Decitabine. Cancer Res. 2015, 75, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, P.; Wei, Z.; Zhang, C.; Xia, M.; Du, Q.; Chen, Y.; Liu, N.; Li, H.; Yang, X.P. Regulation of T cell differentiation and function by epigenetic modification enzymes. Semin. Immunopathol. 2019, 41, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2011, 13, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Ryu, Y.S.; Piao, M.J.; Shilnikova, K.; Kang, H.K.; Yi, J.M.; Boulanger, M.; Paolillo, R.; Bossis, G.; Yoon, S.Y.; et al. DUOX2-mediated production of reactive oxygen species induces epithelial mesenchymal transition in 5-fluorouracil resistant human colon cancer cells. Redox Biol. 2018, 17, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liang, Y.; Sun, G.; He, Q.; Hou, Z.; Jiang, X.; Gao, P.; Qu, H. Upregulation of CRABP2 by TET1-mediated DNA hydroxymethylation attenuates mitochondrial apoptosis and promotes oxaliplatin resistance in gastric cancer. Cell Death Dis. 2022, 13, 848. [Google Scholar] [CrossRef]
- Forloni, M.; Gupta, R.; Nagarajan, A.; Sun, L.S.; Dong, Y.; Pirazzoli, V.; Toki, M.; Wurtz, A.; Melnick, M.A.; Kobayashi, S.; et al. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Rep. 2016, 16, 457–471. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes. Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Huang, R.; Langdon, S.P.; Tse, M.; Mullen, P.; Um, I.H.; Faratian, D.; Harrison, D.J. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 2016, 7, 4695–4711. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Su, J.; Wang, F.; Cai, Y.; Jin, J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 99. [Google Scholar] [CrossRef]
- Di Martile, M.; Del Bufalo, D.; Trisciuoglio, D. The multifaceted role of lysine acetylation in cancer: Prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, P.; Tang, F.; Xie, C. HDAC6-mediated EGFR stabilization and activation restrict cell response to sorafenib in non-small cell lung cancer cells. Med. Oncol. 2016, 33, 50. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Jeoung, D. Tubulin Beta3 Serves as a Target of HDAC3 and Mediates Resistance to Microtubule-Targeting Drugs. Mol. Cells 2015, 38, 705–714. [Google Scholar] [CrossRef]
- Islam, S.; Abiko, Y.; Uehara, O.; Chiba, I. Sirtuin 1 and oral cancer. Oncol. Lett. 2019, 17, 729–738. [Google Scholar] [CrossRef]
- Yousafzai, N.A.; Jin, H.; Ullah, M.; Wang, X. Recent advances of SIRT1 and implications in chemotherapeutics resistance in cancer. Am. J. Cancer Res. 2021, 11, 5233–5248. [Google Scholar]
- Gao, Y.; van Haren, M.J.; Buijs, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W.; et al. Potent Inhibition of Nicotinamide N-Methyltransferase by Alkene-Linked Bisubstrate Mimics Bearing Electron Deficient Aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Rajagopal, S.; Kadnur, S.V.; Hallur, M.S.; Rani, S.; Kristam, R.; Swaminathan, S.; Zope, B.R.; Gondrala, P.K.; Swamy, I.; et al. Novel tricyclic small molecule inhibitors of Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2022, 12, 15440. [Google Scholar] [CrossRef] [PubMed]






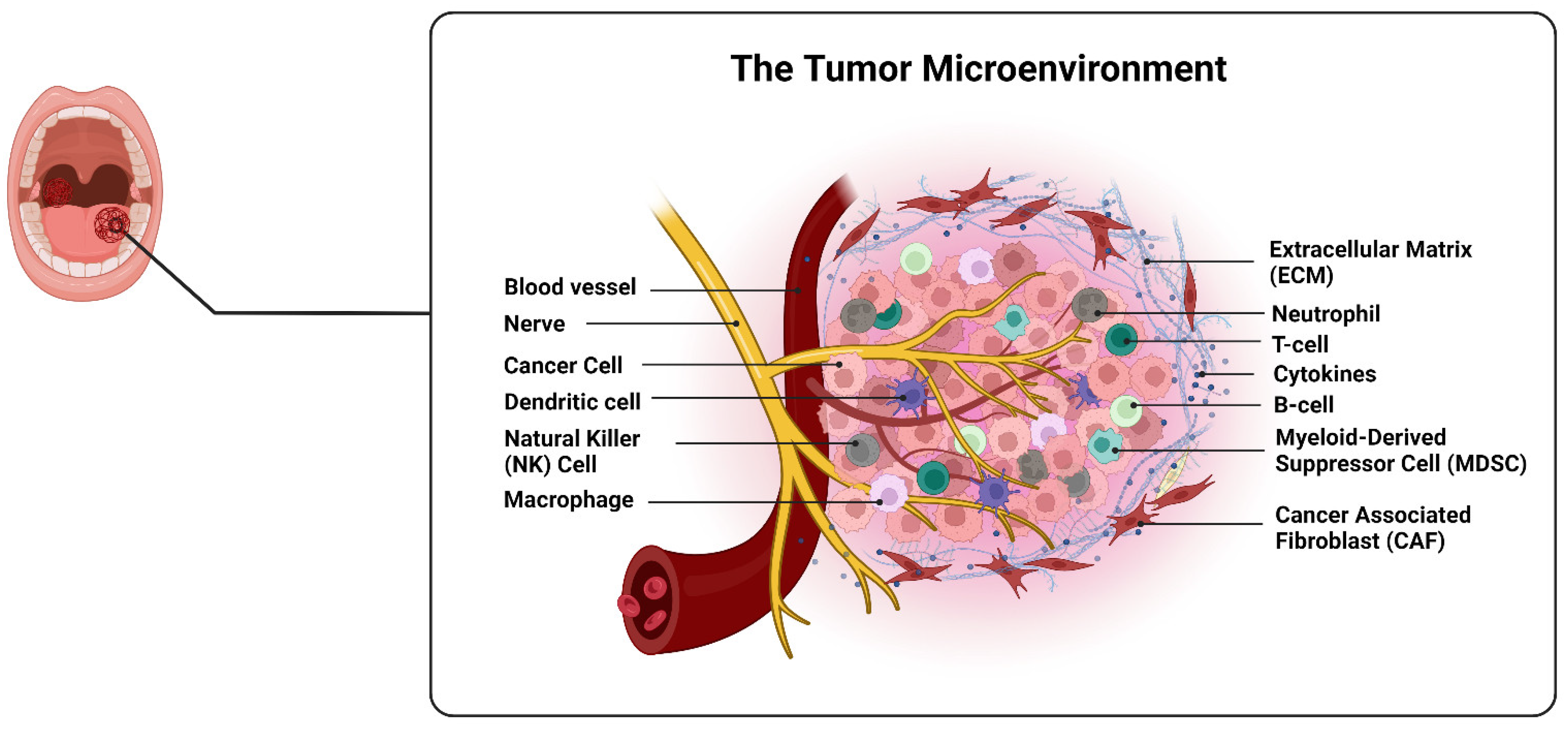{kind=link}
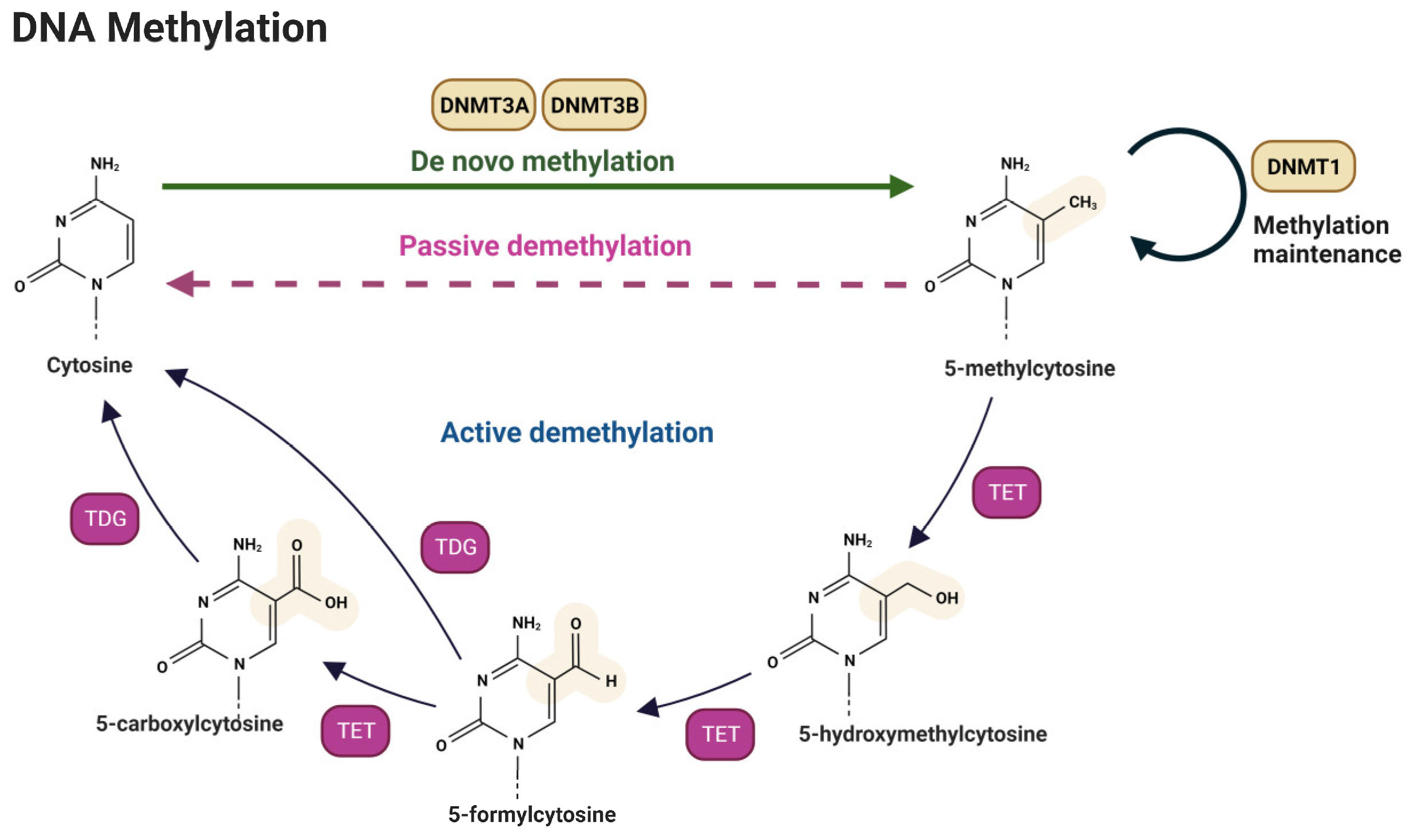{kind=link}
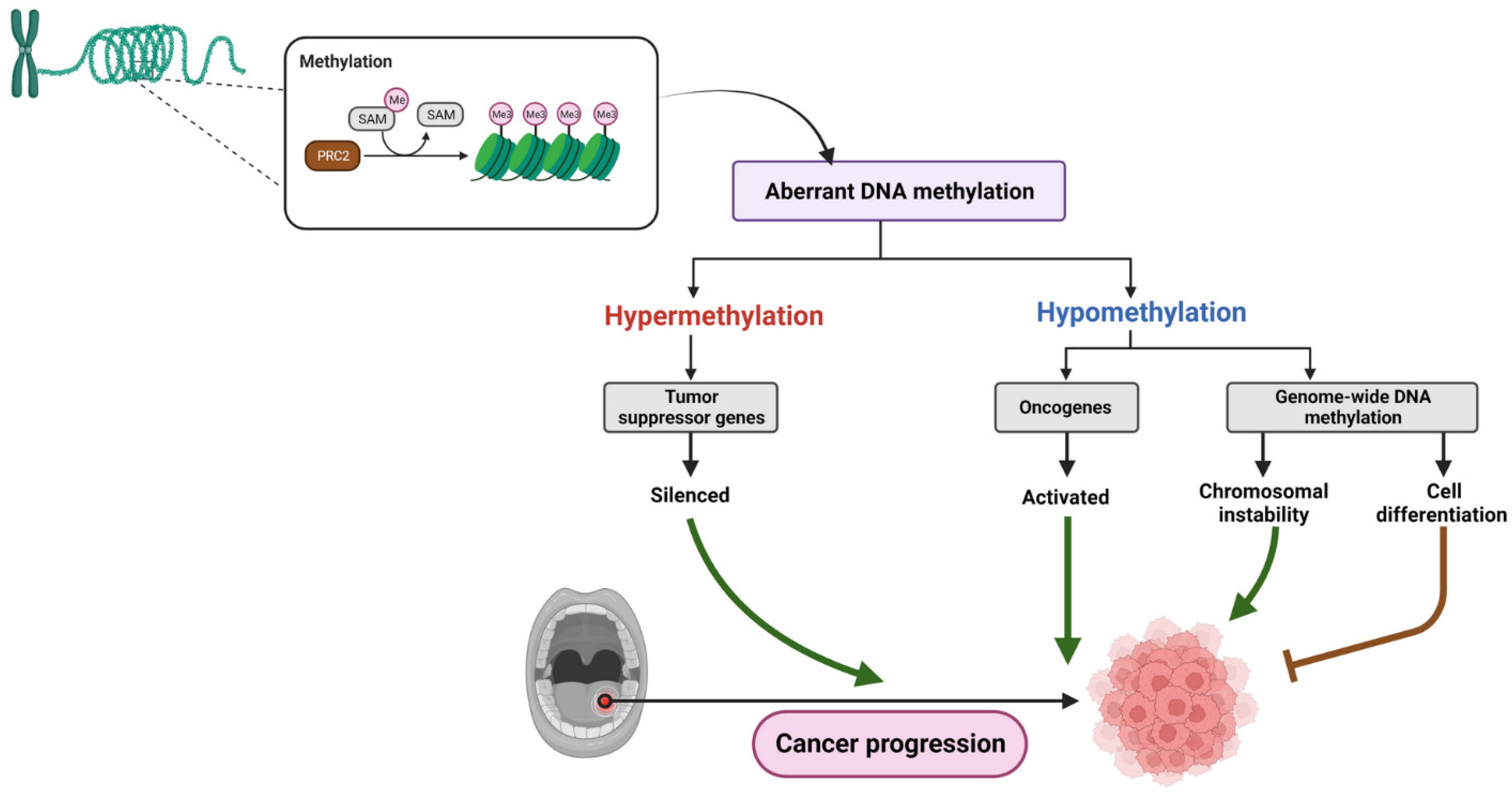{kind=link}
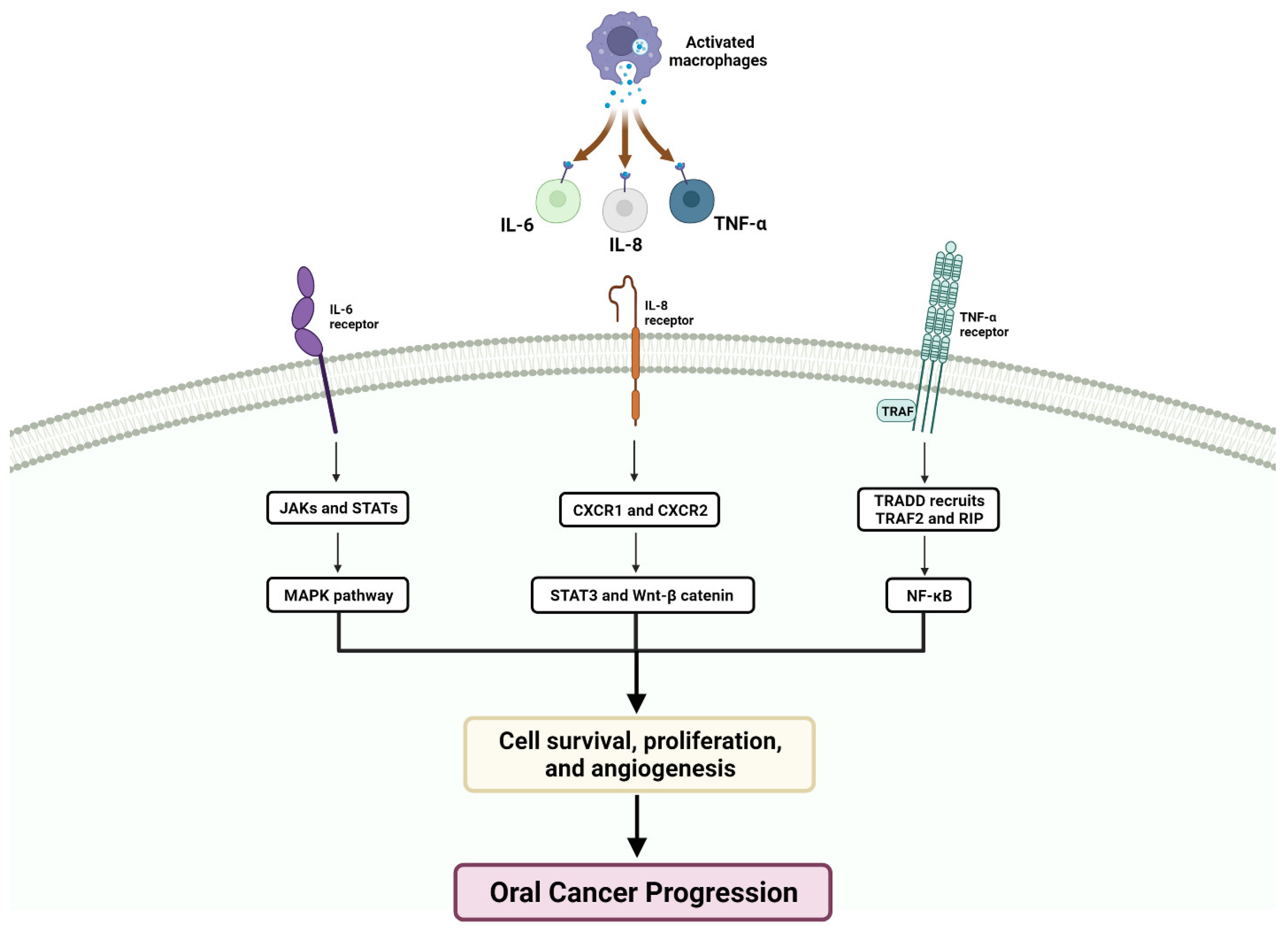{kind=link}
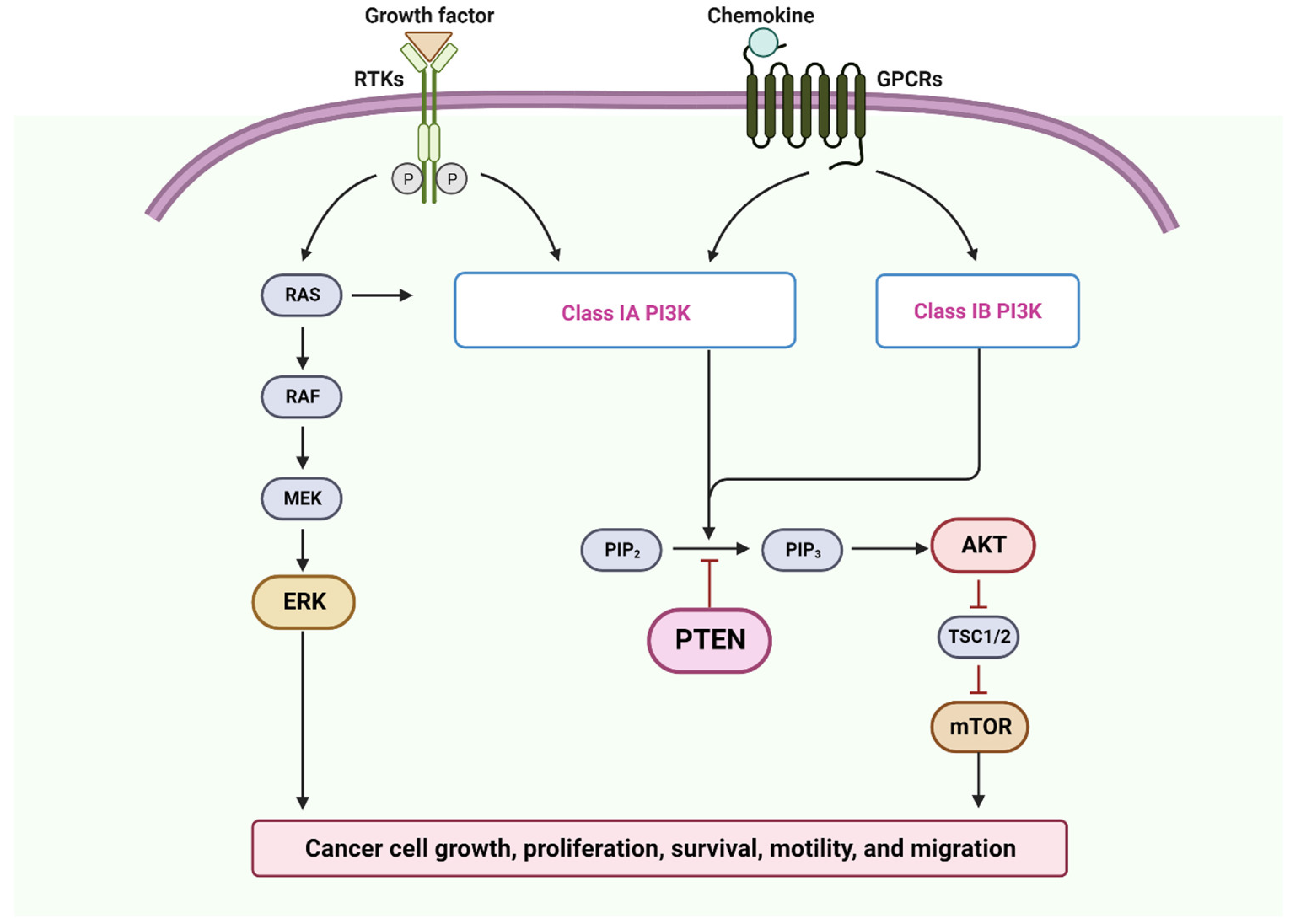{kind=link}
| Gene | Locus | Function | Refs. |
|---|---|---|---|
| ABO | 9q34 | Blood group antigen | [271] |
| APC | 5q21 | Signal transduction | [272,273] |
| ATM | 11q22-q23 | Tumor suppressor | [274,275,276] |
| C/EBPα | 19q13 | Tumor suppressor | [277] |
| CDKN2A | 9p21 | Cell cycle | [274] |
| CRABP2 | 1q21 | Nuclear transcriptional regulator | [278] |
| DAPK | 9q | Apoptosis | [28,271,274,275,279] |
| DCC | 18q21 | Tumor suppressor | [28,271,274,275,279] |
| DKK3 | 11p | Transcriptional regulator | [280] |
| E-cadherin | 16q22 | Signal transduction | [28,32] |
| EDNRB | 13q22 | Signal transduction | [281] |
| GSTP1 | 11q13 | Detoxification of carcinogens | [275,282] |
| H3K4 | 1q21.2 | Histone | [283] |
| HIN1 | 12p13 | Tumor suppressor | [284] |
| Hmlh1 | 3p21 | DNA repair | [275] |
| LHX6 | 9q33 | Transcriptional regulator | [277] |
| MGMT | 10q26 | DNA repair | [275] |
| MINT family | / | / | [275] |
| miR137 | 1p21.3 | Tumor suppressor | [285] |
| miR193a | 17q11.2 | Tumor suppressor | [285] |
| MX1 | 21q22 | / | [278] |
| p14 | 9p21 | Apoptosis | [275] |
| p15 | 9p21 | Cell cycle | [279] |
| p16 | 9p21 | Cell cycle | [275] |
| p53 | 17p13 | Tumor suppressor | [275] |
| p73 | 1p36 | Apoptosis | [275] |
| PTEN | 10q23 | Tumor suppressor | [286] |
| RARB2 | 17q21 | Nuclear transcriptional Regulator | [275] |
| RASSF-1 | 3p21 | Apoptosis | [279] |
| Rb | 13q14 | Tumor suppressor | [273] |
| RUNX3 | 1p36 | Transcriptional regulator | [281] |
| SFRP1 | 8p11.21 | Transcriptional regulator | [287] |
| SFRP1-2-4-5 | 8p11.21, 4q31.3, 7p14.1, 10q24.1 | Transcriptional regulator | [287,288] |
| TCF21 | 6q23-q24 | Epithelial–mesenchymal interactions | [277] |
| THBS1 | 15q15 | cell-to-cell and cell-to-matrix interactions | [273] |
| TIMP3 | 22q12 | Epithelial–mesenchymal interactions | [279] |
| WIF1 | 12q14 | Transcriptional regulator | [287] |
| σ-14-3-3 | 1p36 | Signal transduction | [275] |
| microRNAs | Expression in OSCC | Cellular Function |
|---|---|---|
| miR-193a/miR-137 miR-503/miR-133b/miR-15a/miR-133a | ↓ | Proliferation and apoptosis |
| miR-184/miR-24/miR-21 | ↑ | Proliferation and apoptosis |
| miR-138/miR-222 | ↓ | Metastasis |
| miR-31/miR-211 | ↑ | Metastasis |
| miR-21 | ↓ | Chemoresistance |
| miR-98/miR-23a/miR-214 | ↑ | Chemoresistance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesgari, H.; Esmaelian, S.; Nasiri, K.; Ghasemzadeh, S.; Doroudgar, P.; Payandeh, Z. Epigenetic Regulation in Oral Squamous Cell Carcinoma Microenvironment: A Comprehensive Review. Cancers 2023, 15, 5600. https://doi.org/10.3390/cancers15235600
Mesgari H, Esmaelian S, Nasiri K, Ghasemzadeh S, Doroudgar P, Payandeh Z. Epigenetic Regulation in Oral Squamous Cell Carcinoma Microenvironment: A Comprehensive Review. Cancers. 2023; 15(23):5600. https://doi.org/10.3390/cancers15235600
Chicago/Turabian StyleMesgari, Hassan, Samar Esmaelian, Kamyar Nasiri, Shabnam Ghasemzadeh, Parisa Doroudgar, and Zahra Payandeh. 2023. "Epigenetic Regulation in Oral Squamous Cell Carcinoma Microenvironment: A Comprehensive Review" Cancers 15, no. 23: 5600. https://doi.org/10.3390/cancers15235600