
Sorafenib Resistance Contributed by IL7 and MAL2 in Hepatocellular Carcinoma Can Be Overcome by Autophagy-Inducing Stapled Peptides

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Determining the Various Inhibitory Concentrations of Sorafenib in PLC/PRF/5 Cells
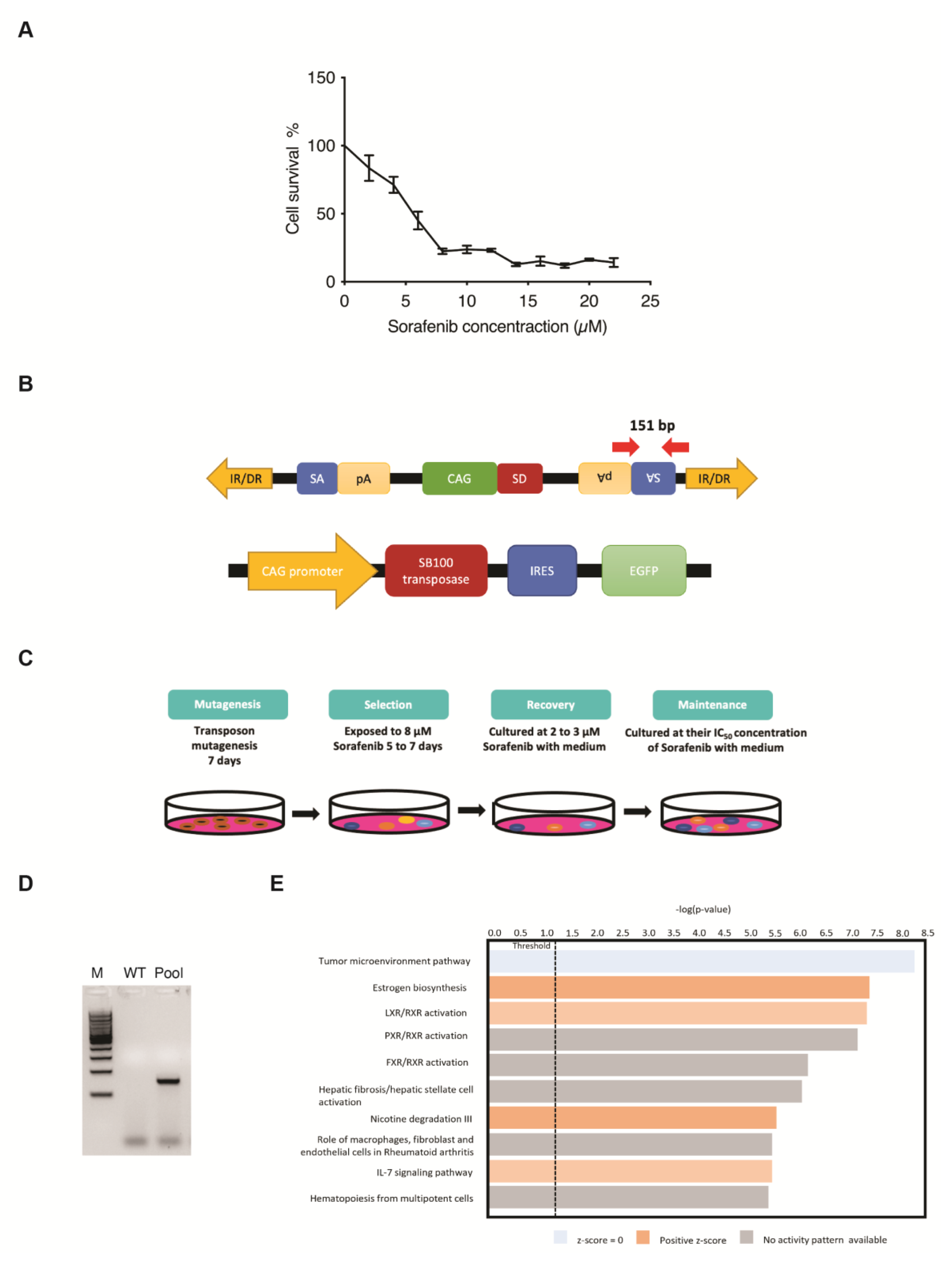
2.3. Establishing Sorafenib-Resistant Cells Using the Sleeping Beauty Transposon Insertional Mutagenesis System
2.4. Chemosensitivity Assay
2.5. Clonogenic Survival Assay
2.6. RNA-Sequencing (RNA-Seq)
2.7. Construction of IL7 and MAL2 Overexpression Plasmids for Transfection into Human Hepatic Cell Line
2.8. Apoptosis Assay
2.9. Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (qPCR)
2.10. Western Blot Analyses
2.11. Semi-Quantitative Analyses
2.12. Immunoblot Analysis for Autophagy
2.13. Cell Viability Assay Using Trypan Blue Dye
2.14. Chemical Synthesis of Stapled Peptides
2.15. Statistical Analyses
3. Results
3.1. SB Transposon Insertional Mutagenesis Induced Drug Resistance in Human HCC Cell Line
3.2. Identification of Candidate Genes Involved with Drug-Resistance
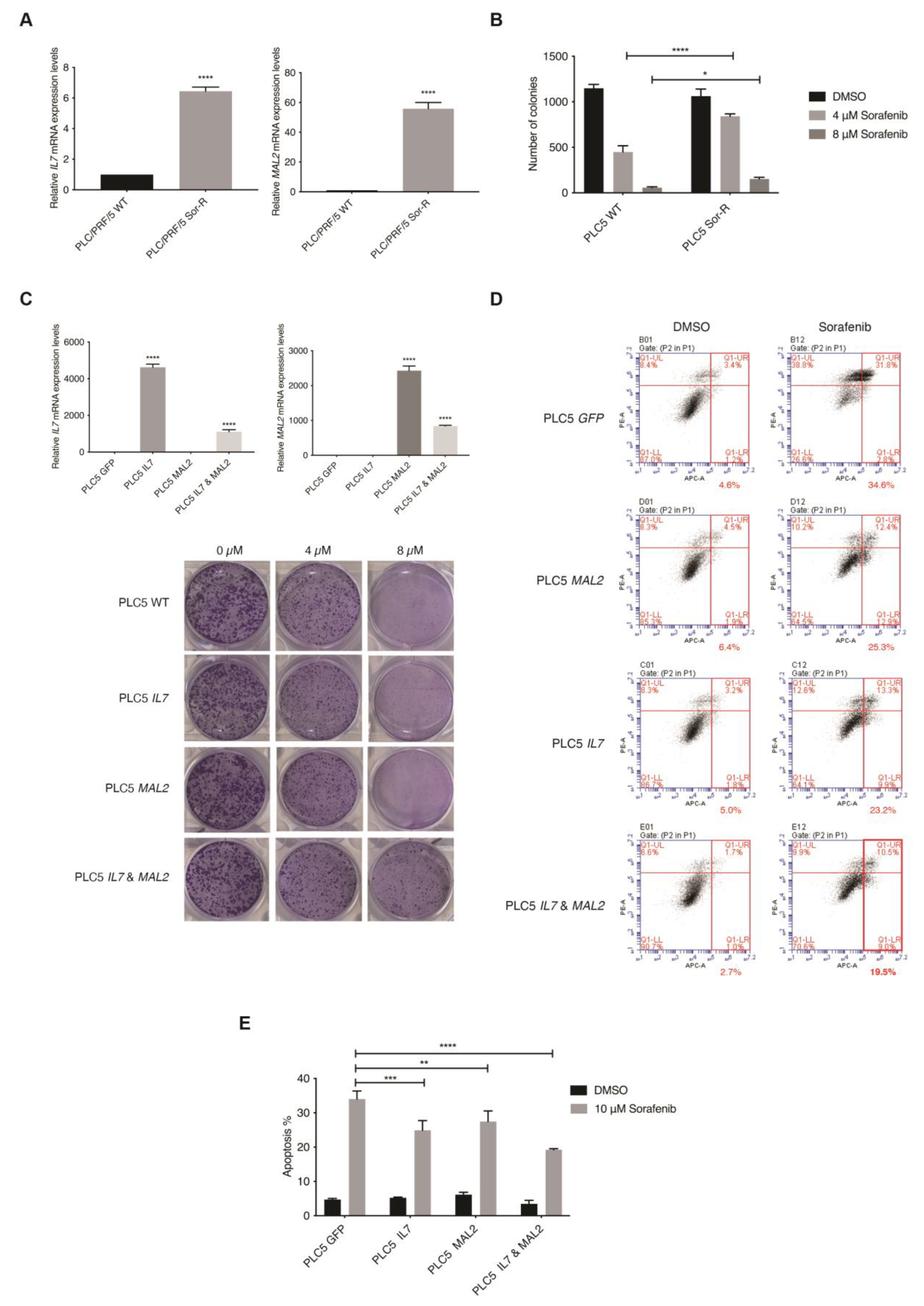
3.3. Clinical Relevance of IL7 and MAL2 in HCC-Associated Drug Resistance
3.4. In Vitro Validation of IL7 and MAL2 in HCC-Associated Drug Resistance
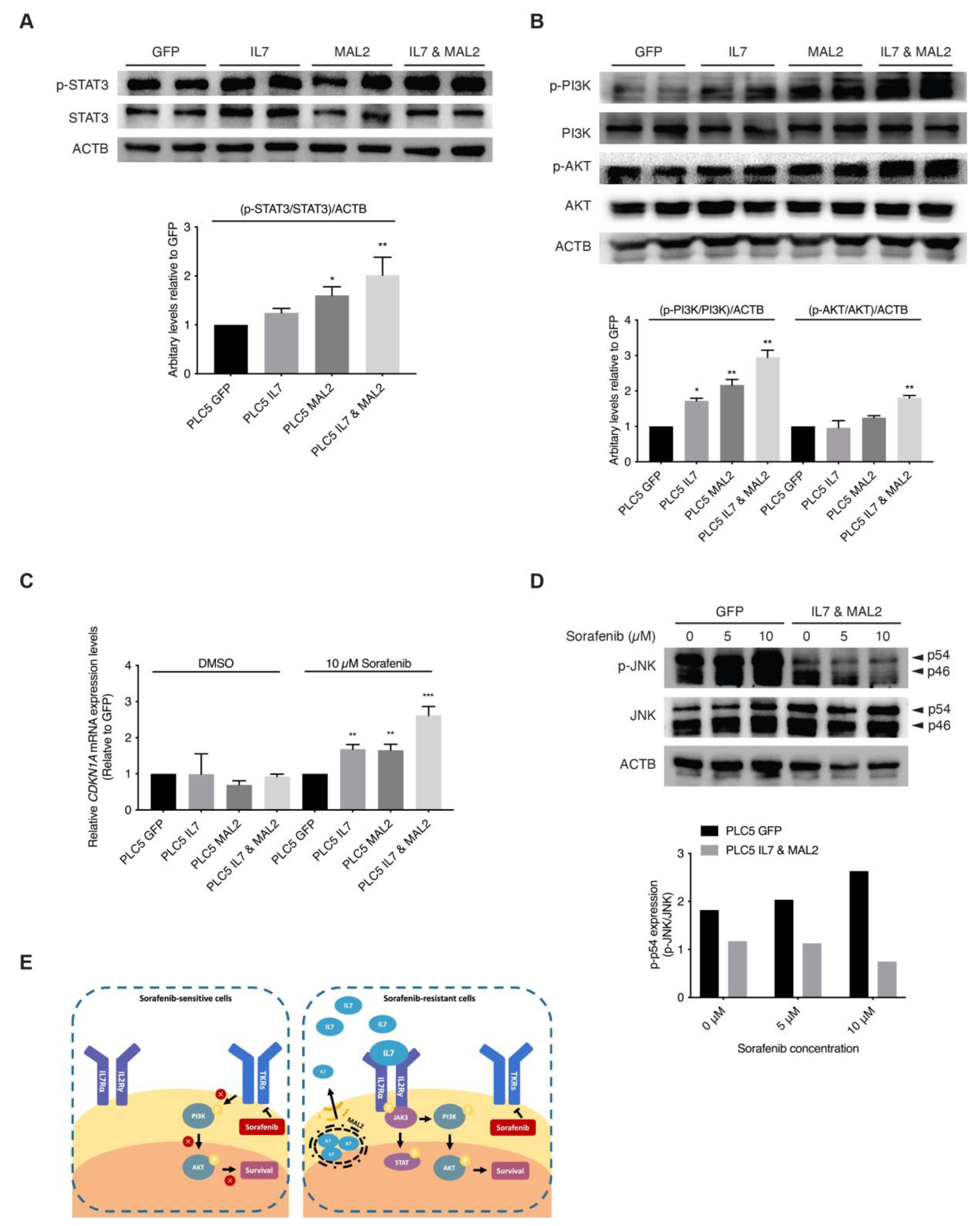
3.5. Validating the Role of IL7 and MAL2 in HCC Drug Resistance by Dysregulating JAK/STAT and PI3K/AKT Signaling Pathways
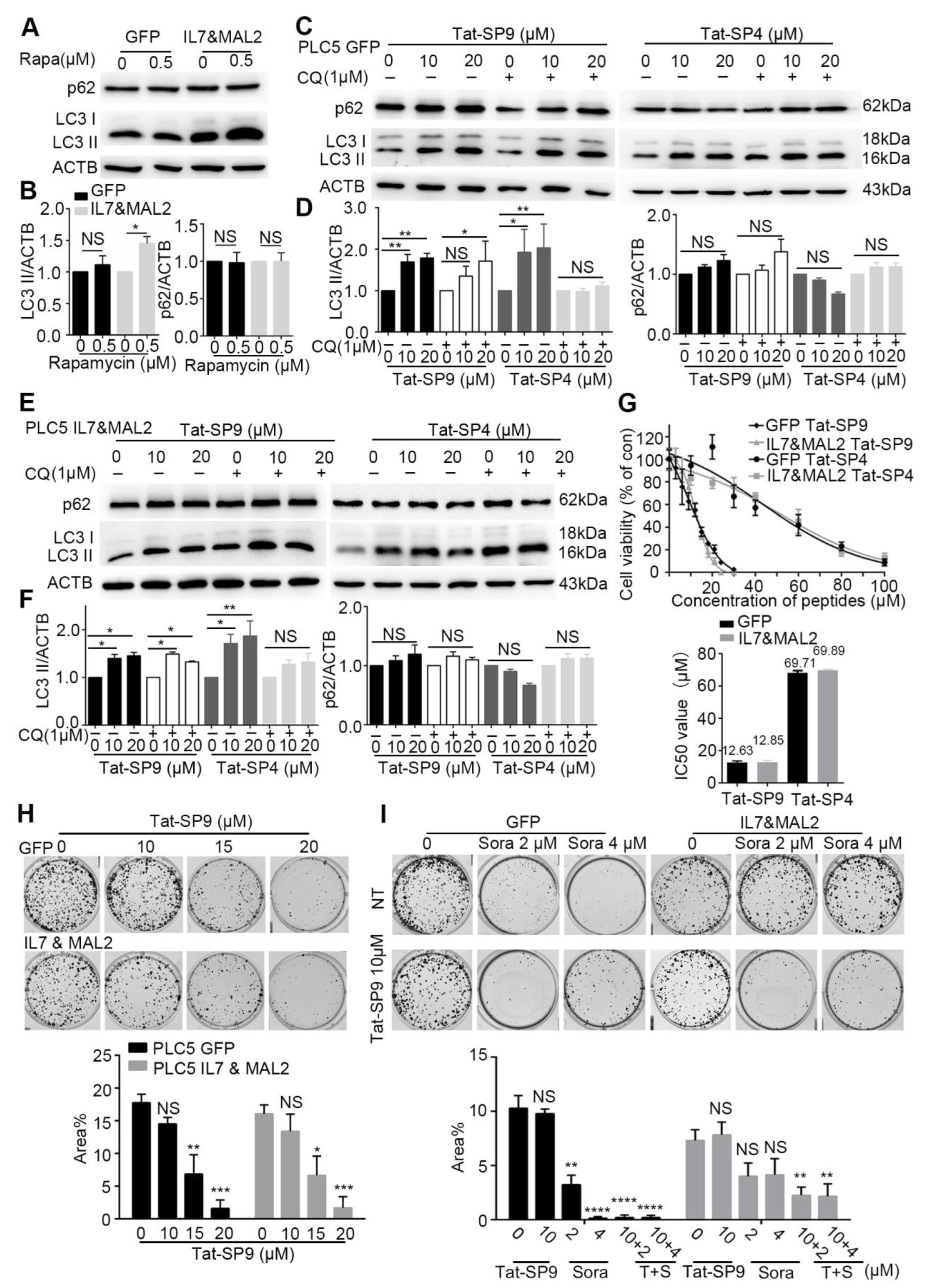
3.6. Autophagy-Inducing Stapled Peptides Readily Enhanced Autophagic Flux in Both Wild-Type and Sorafenib-Resistant HCC Cells Overexpressing IL7 and MAL2
3.7. Autophagy-Inducing Stapled Peptide Showed Synergic Toxicity with Sorafenib in Drug-Resistant IL7&MAL2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Fuster, J.; Bruix, J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: Resection versus transplantation. Hepatology 1999, 30, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Kaseb, A.O.; Carmagnani Pestana, R.; Vence, L.M.; Blando, J.M.; Singh, S.; Ikoma, N.; Vauthey, J.-N.; Allison, J.P.; Sharma, P. Randomized, open-label, perioperative phase II study evaluating nivolumab alone versus nivolumab plus ipilimumab in patients with resectable HCC. J. Clin. Oncol. 2019, 37, 185. [Google Scholar] [CrossRef]
- Tella, S.H.; Mahipal, A.; Kommalapati, A.; Jin, Z. Evaluating the Safety and Efficacy of Nivolumab in Patients with Advanced Hepatocellular Carcinoma: Evidence to Date. Onco Targets Ther. 2019, 12, 10335–10342. [Google Scholar] [CrossRef]
- Finn, R.S.; Zhu, A.X. Targeting angiogenesis in hepatocellular carcinoma: Focus on VEGF and bevacizumab. Expert Rev. Anticancer Ther. 2009, 9, 503–509. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Aggarwal, M.; Arain, A.; Jin, Z. Systemic treatment for hepatocellular carcinoma. Chronic Dis. Transl. Med. 2018, 4, 148–155. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Keng, V.W.; Villanueva, A.; Chiang, D.Y.; Dupuy, A.J.; Ryan, B.J.; Matise, I.; Silverstein, K.A.; Sarver, A.; Starr, T.K.; Akagi, K.; et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat. Biotechnol. 2009, 27, 264–274. [Google Scholar] [CrossRef]
- Rahrmann, E.P.; Watson, A.L.; Keng, V.W.; Choi, K.; Moriarity, B.S.; Beckmann, D.A.; Wolf, N.K.; Sarver, A.; Collins, M.H.; Moertel, C.L.; et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat. Genet. 2013, 45, 756–766. [Google Scholar] [CrossRef]
- Lo, L.H.; Lam, C.Y.; To, J.C.; Chiu, C.H.; Keng, V.W. Sleeping Beauty insertional mutagenesis screen identifies the pro-metastatic roles of CNPY2 and ACTN2 in hepatocellular carcinoma tumor progression. Biochem. Biophys. Res. Commun. 2021, 541, 70–77. [Google Scholar] [CrossRef]
- Patel, E.S.; Okada, S.; Hachey, K.; Yang, L.J.; Durum, S.K.; Moreb, J.S.; Chang, L.J. Regulation of in vitro human T cell development through interleukin-7 deprivation and anti-CD3 stimulation. BMC Immunol. 2012, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Mertsching, E.; Meyer, V.; Linares, J.; Lombard-Platet, S.; Ceredig, R. Interleukin-7, a non-redundant potent cytokine whose over-expression massively perturbs B-lymphopoiesis. Int. Rev. Immunol. 1998, 16, 285–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Wang, M.H.; Ren, H.J.; Qu, W.; Sun, L.M.; Zhang, Q.F.; Qiu, X.S.; Wang, E.H. Interleukin 7 signaling prevents apoptosis by regulating bcl-2 and bax via the p53 pathway in human non-small cell lung cancer cells. Int. J. Clin. Exp. Pathol. 2014, 7, 870–881. [Google Scholar] [PubMed]
- Ming, J.; Zhang, Q.; Qiu, X.; Wang, E. Interleukin 7/interleukin 7 receptor induce c-Fos/c-Jun-dependent vascular endothelial growth factor-D up-regulation: A mechanism of lymphangiogenesis in lung cancer. Eur. J. Cancer 2009, 45, 866–873. [Google Scholar] [CrossRef] [PubMed]
- de Marco, M.C.; Martín-Belmonte, F.; Kremer, L.; Albar, J.P.; Correas, I.; Vaerman, J.P.; Marazuela, M.; Byrne, J.A.; Alonso, M.A. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J. Cell Biol. 2002, 159, 37–44. [Google Scholar] [CrossRef]
- Dupuy, A.J.; Rogers, L.M.; Kim, J.; Nannapaneni, K.; Starr, T.K.; Liu, P.; Largaespada, D.A.; Scheetz, T.E.; Jenkins, N.A.; Copeland, N.G. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009, 69, 8150–8156. [Google Scholar] [CrossRef]
- Keng, V.W.; Sia, D.; Sarver, A.L.; Tschida, B.R.; Fan, D.; Alsinet, C.; Sole, M.; Lee, W.L.; Kuka, T.P.; Moriarity, B.S.; et al. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology 2013, 57, 120–130. [Google Scholar] [CrossRef]
- Keng, V.W.; Tschida, B.R.; Bell, J.B.; Largaespada, D.A. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology 2011, 53, 781–790. [Google Scholar] [CrossRef]
- Mates, L.; Chuah, M.K.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Fang, F.; Gould, T.; Li, X.; Kim, C.; Gustafson, C.; Lambert, S.; Weyand, C.M.; Goronzy, J.J. Ecto-NTPDase CD39 is a negative checkpoint that inhibits follicular helper cell generation. J. Clin. Investig. 2020, 130, 3422–3436. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.P.; Tschida, B.R.; Sham, T.T.; Lo, L.H.; Moriarity, B.S.; Li, X.X.; Lo, R.C.; Hinton, D.E.; Rowlands, D.K.; Chan, C.O.; et al. HBx-K130M/V131I Promotes Liver Cancer in Transgenic Mice via AKT/FOXO1 Signaling Pathway and Arachidonic Acid Metabolism. Mol. Cancer Res. 2019, 17, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; He, Y.; Qiu, X.; Yang, W.; Liu, W.; Li, X.; Li, Y.; Shen, H.-M.; Wang, R.; Yue, Z.; et al. Targeting the potent Beclin 1–UVRAG coiled-coil interaction with designed peptides enhances autophagy and endolysosomal trafficking. Proc. Natl. Acad. Sci. USA 2018, 115, E5669–E5678. [Google Scholar] [CrossRef]
- Yang, Q.; Qiu, X.; Zhang, X.; Yu, Y.; Li, N.; Wei, X.; Feng, G.; Li, Y.; Zhao, Y.; Wang, R. Optimization of beclin 1-targeting stapled peptides by staple scanning leads to enhanced antiproliferative potency in cancer cells. J. Med. Chem. 2021, 64, 13475–13486. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; Grossmann, T.N.; Verdine, G.L. Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. [Google Scholar] [CrossRef]
- Shiri, P.; Ramezanpour, S.; Amani, A.M.; Dehaen, W. A patent review on efficient strategies for the total synthesis of pazopanib, regorafenib and lenvatinib as novel anti-angiogenesis receptor tyrosine kinase inhibitors for cancer therapy. Mol. Divers. 2022, 26, 2981–3002. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hao, Y. Analysis of expression profile data identifies key genes and pathways in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 2625–2630. [Google Scholar] [CrossRef]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N.; et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef]
- Wang, C.; Kong, L.; Kim, S.; Lee, S.; Oh, S.; Jo, S.; Jang, I.; Kim, T.D. The Role of IL-7 and IL-7R in Cancer Pathophysiology and Immunotherapy. Int. J. Mol. Sci. 2022, 23, 412. [Google Scholar] [CrossRef]
- Ribeiro, D.; Melao, A.; van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef] [PubMed]
- Lodewijckx, I.; Cools, J. Deregulation of the Interleukin-7 Signaling Pathway in Lymphoid Malignancies. Pharmaceuticals 2021, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, L.; Wan, C.; Sun, Y.; Van der Jeught, K.; Zhou, Z.; Dong, T.; So, K.M.; Yu, T.; Li, Y.; et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. J. Clin. Investig. 2021, 131, e140837. [Google Scholar] [CrossRef]
- Dersh, D.; Yewdell, J.W. Immune MAL2-practice: Breast cancer immunoevasion via MHC class I degradation. J. Clin. Investig. 2021, 131, e144344. [Google Scholar] [CrossRef]
- Li, X.; He, L.; Che, K.H.; Funderburk, S.F.; Pan, L.; Pan, N.; Zhang, M.; Yue, Z.; Zhao, Y. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat. Commun. 2012, 3, 662. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Gao, S.; Li, N.; Keng, V.; Zhao, Y. A Beclin 1-targeting stapled peptide synergizes with erlotinib to potently inhibit proliferation of non-small-cell lung cancer cells. Biochem. Biophys. Res. Commun. 2022, 636, 125–131. [Google Scholar] [CrossRef]
- Gao, S.; Li, N.; Zhang, X.; Chen, J.; Ko, B.C.; Zhao, Y. An autophagy-inducing stapled peptide promotes c-MET degradation and overrides adaptive resistance to sorafenib in c-MET+ hepatocellular carcinoma. Biochem. Biophys. Rep. 2023, 33, 101412. [Google Scholar] [CrossRef]
- Li, N.; Zhang, X.; Chen, J.; Gao, S.; Wang, L.; Zhao, Y. Perturbation of Autophagy by a Beclin 1-Targeting Stapled Peptide Induces Mitochondria Stress and Inhibits Proliferation of Pancreatic Cancer Cells. Cancers 2023, 15, 953. [Google Scholar] [CrossRef]
- Nishida, N.; Kitano, M.; Sakurai, T.; Kudo, M. Molecular Mechanism and Prediction of Sorafenib Chemoresistance in Human Hepatocellular Carcinoma. Dig. Dis. 2015, 33, 771–779. [Google Scholar] [CrossRef]
- Fritzell, S.; Eberstal, S.; Sanden, E.; Visse, E.; Darabi, A.; Siesjo, P. IFNgamma in combination with IL-7 enhances immunotherapy in two rat glioma models. J. Neuroimmunol. 2013, 258, 91–95. [Google Scholar] [CrossRef]
- Lynch, D.H.; Namen, A.E.; Miller, R.E. In vivo evaluation of the effects of interleukins 2, 4 and 7 on enhancing the immunotherapeutic efficacy of anti-tumor cytotoxic T lymphocytes. Eur. J. Immunol. 1991, 21, 2977–2985. [Google Scholar] [CrossRef]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Lee, E.J.; Kim, W.J.; Moon, S.K. p27KIP1 is involved in ERK1/2-mediated MMP-9 expression via the activation of NF-kappaB binding in the IL-7-induced migration and invasion of 5637 cells. Int. J. Oncol. 2014, 44, 1349–1356. [Google Scholar] [CrossRef]
- Marazuela, M.; Alonso, M.A. Expression of MAL and MAL2, two elements of the protein machinery for raft-mediated transport, in normal and neoplastic human tissue. Histol. Histopathol. 2004, 19, 925–933. [Google Scholar] [CrossRef]
- Jeong, J.; Shin, J.H.; Li, W.; Hong, J.Y.; Lim, J.; Hwang, J.Y.; Chung, J.J.; Yan, Q.; Liu, Y.; Choi, J.; et al. MAL2 mediates the formation of stable HER2 signaling complexes within lipid raft-rich membrane protrusions in breast cancer cells. Cell Rep. 2021, 37, 110160. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy: Thematic Review Series: Biology of Lipid Rafts. J. Lipid. Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, D.; Ohuchida, K.; Kozono, S.; Ikenaga, N.; Shindo, K.; Cui, L.; Fujiwara, K.; Akagawa, S.; Ohtsuka, T.; Takahata, S.; et al. MAL2 expression predicts distant metastasis and short survival in pancreatic cancer. Surgery 2013, 154, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Maleki, S.; Hardy, J.R.; Gloss, B.S.; Murali, R.; Scurry, J.P.; Fanayan, S.; Emmanuel, C.; Hacker, N.F.; Sutherland, R.L.; et al. MAL2 and tumor protein D52 (TPD52) are frequently overexpressed in ovarian carcinoma, but differentially associated with histological subtype and patient outcome. BMC Cancer 2010, 10, 497. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Coral, A.; Del Vecchio, G.J.; Chahine, J.J.; Kallakury, B.V.; Tuma, P.L. MAL2-Induced Actin-Based Protrusion Formation is Anti-Oncogenic in Hepatocellular Carcinoma. Cancers 2020, 12, 422. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, R.; Wu, W.; Lei, L. Comprehensive network analysis of the molecular mechanisms associated with sorafenib resistance in hepatocellular carcinoma. Cancer Genet. 2020, 245, 27–34. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Kreis, N.N.; Friemel, A.; Ritter, A.; Roth, S.; Rolle, U.; Louwen, F.; Yuan, J. Function of p21 (Cip1/Waf1/CDKN1A) in Migration and Invasion of Cancer and Trophoblastic Cells. Cancers 2019, 11, 989. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tan, W.; Li, W.; Li, W.; Zhu, S.; Zhong, J.; Shang, C.; Chen, Y. miR-1226-3p Promotes Sorafenib Sensitivity of Hepatocellular Carcinoma via Downregulation of DUSP4 Expression. J. Cancer 2019, 10, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.Q.; Rong, L.W.; Wang, R.X.; Zheng, X.L.; Zhang, L.; Zhang, L.; Lin, Y.; Wang, X.; Li, Z.P. Luteolin and sorafenib combination kills human hepatocellular carcinoma cells through apoptosis potentiation and JNK activation. Oncol. Lett. 2018, 16, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in human diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | p-Value | Involved Genes |
|---|---|---|
| PI3K-Akt signaling pathway | 4.08 × 10−6 | CDKN1A, G6PC, CSF1, PIK3R3, IL2RG, FGF2, NFKB1, NR4A1, COL3A1, KITLG, BCL2L11, IL7, SPP1, KDR, SGK1, PCK1, MET |
| Steroid hormone biosynthesis | 6.17 × 10−5 | UGT2B11, UGT1A1, AKR1C3, CYP1A1, AKR1C4, CYP3A5, CYP3A7 |
| Complement and coagulation cascades | 7.72 × 10−4 | CPB2, SERPIND1, F12, C5AR1, KNG1, SERPINA5, C2 |
| Pathways in cancer | 0.001003 | CEBPA, CDKN1A, CXCL8, CXCR4, PIK3R3, TGFA, PTGS2, FGF2, FOXO1, NFKB1, BMP2, KITLG, CXCL12, BIRC5, MET, WNT4 |
| Transcriptional misregulation in cancer | 0.001217 | CEBPA, CDKN1A, CXCL8, IGFBP3, TMPRSS2, HMGA2, PROM1, MET, FOXO1, NFKB1 |
| Chemical carcinogenesis | 0.00139 | UGT2B11, UGT1A1, CHRNA7, CYP1A1, PTGS2, CYP3A5, CYP3A7 |
| NF-kappa B signaling pathway | 0.001556 | CXCL12, TICAM2, CXCL8, LY96, PTGS2, NFKB1, TNFSF13B |
| Arachidonic acid metabolism | 0.001901 | CYP2J2, GPX2, GPX3, AKR1C3, GGT1, PTGS2 |
| Retinol metabolism | 0.002199 | CYP26B1, UGT2B11, UGT1A1, CYP1A1, CYP3A5, CYP3A7 |
| Cytokine-cytokine receptor interaction | 0.002682 | IL22RA1, BMP2, CXCL12, CXCL8, THPO, CSF1, IL7, CXCR4, IL2RG, CXCL5, TNFSF13B |
| Altered Genes | Log2 Fold-Change | p-Value |
|---|---|---|
| EPPIN | 10.920 | 2.99 × 10−182 |
| PTGS2 | 9.926 | 1.33 × 10−212 |
| ALB | 9.351 | 0 |
| KDR | 8.758 | 5.76 × 10−128 |
| FABP1 | 8.011 | 3.88 × 10−7 |
| MAL2 | 7.651 | 7.36 × 10−28 |
| ABCG1 | 7.082 | 2.02 × 10−37 |
| IL7 | 6.833 | 8.29 × 10−12 |
| IQGAP2 | 6.755 | 1.38 × 10−152 |
| SLCO3A1 | 6.219 | 9.21 × 10−27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
To, J.C.; Gao, S.; Li, X.-X.; Zhao, Y.; Keng, V.W. Sorafenib Resistance Contributed by IL7 and MAL2 in Hepatocellular Carcinoma Can Be Overcome by Autophagy-Inducing Stapled Peptides. Cancers 2023, 15, 5280. https://doi.org/10.3390/cancers15215280
To JC, Gao S, Li X-X, Zhao Y, Keng VW. Sorafenib Resistance Contributed by IL7 and MAL2 in Hepatocellular Carcinoma Can Be Overcome by Autophagy-Inducing Stapled Peptides. Cancers. 2023; 15(21):5280. https://doi.org/10.3390/cancers15215280
Chicago/Turabian StyleTo, Jeffrey C., Shan Gao, Xiao-Xiao Li, Yanxiang Zhao, and Vincent W. Keng. 2023. "Sorafenib Resistance Contributed by IL7 and MAL2 in Hepatocellular Carcinoma Can Be Overcome by Autophagy-Inducing Stapled Peptides" Cancers 15, no. 21: 5280. https://doi.org/10.3390/cancers15215280