
The Development of p53-Targeted Therapies for Human Cancers
1
Department of Medical Oncology, Key Laboratory of Cancer Prevention and Intervention, Ministry of Education, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310009, China
2
Department of Cardiology, The Second Affiliated Hospital, Cardiovascular Key Lab of Zhejiang Province, School of Medicine, Zhejiang University, Hangzhou 310009, China
3
Division of Biological Sciences, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2023, 15(14), 3560; https://doi.org/10.3390/cancers15143560
Submission received: 3 June 2023
/
Revised: 27 June 2023
/
Accepted: 29 June 2023
/
Published: 10 July 2023
(This article belongs to the Special Issue Targeting Therapies for the p53 Protein in Cancer Treatments)
Abstract
:Simple Summary
Dysfunction of p53 has a significant impact on the resistance of tumor cells to various therapeutic agents. The major mechanisms causing p53 dysfunction include the MDM2/MDMX-mediated destabilization and inactivation of wtp53 and somatic p53 mutations. Despite an intensive search for therapeutic strategies to induce wtp53-like activities in tumor cells, their clinical efficacy and safety remain to be established. In this review, we summarize recent advances in the development of p53-targeting drugs, including their distinct mechanisms of action and potential in tumor therapy.
Abstract
p53 plays a critical role in tumor suppression and is the most frequently mutated gene in human cancers. Most p53 mutants (mutp53) are missense mutations and are thus expressed in human cancers. In human cancers that retain wtp53, the wtp53 activities are downregulated through multiple mechanisms. For example, the overexpression of the negative regulators of p53, MDM2/MDMX, can also efficiently destabilize and inactivate wtp53. Therefore, both wtp53 and mutp53 have become promising and intensively explored therapeutic targets for cancer treatment. Current efforts include the development of small molecule compounds to disrupt the interaction between wtp53 and MDM2/MDMX in human cancers expressing wtp53 and to restore wtp53-like activity to p53 mutants in human cancers expressing mutp53. In addition, a synthetic lethality approach has been applied to identify signaling pathways affected by p53 dysfunction, which, when targeted, can lead to cell death. While an intensive search for p53-targeted cancer therapy has produced potential candidates with encouraging preclinical efficacy data, it remains challenging to develop such drugs with good efficacy and safety profiles. A more in-depth understanding of the mechanisms of action of these p53-targeting drugs will help to overcome these challenges.
1. Introduction
The tumor suppressor p53 plays a critical role in tumor suppression by directly regulating the expression of hundreds of genes important for the cell cycle, apoptosis, senescence, differentiation, and metabolism of normal cells [1]. Therefore, to evade the powerful tumor-suppressive activities of wtp53, the p53 gene has become the most frequently mutated gene in human cancers. The types of p53 mutations in human cancers include mostly missense mutations (70–80%), and less frequently, nonsense mutations (5–7%), frameshift mutations (5–7%), in-frame insertions/deletions (5%), and synonymous mutations (5%) [2]. p53 missense mutants often exhibit the loss of wild-type p53 functions (LOF), dominant-negative (DN) effects, and gain of functions (GOF). Many p53 missense mutants have lost wtp53-like DNA-binding activity while the remaining ones have retained residual activities. It remains unclear how partial LOF affects tumor cell survival in the therapeutic setting. p53 functions as a tetramer [3]. In the heterozygous state, mutp53 retains the ability to form tetramers with wtp53 and interferes with wtp53-dependent functions. The GOFs of p53 mutants promote tumorigenesis mainly through the interaction with other proteins involved in tumor development [4,5]. Considering the extensive and potent roles of p53 mutants in promoting tumorigenesis, p53 mutants have become an ideal therapeutic target for developing treatments for human malignancies (Figure 1).
2. p53 Targeting Therapies
2.1. Development of Drugs to Restore the Activities of wtp53 by Targeting p53–MDM2/MDMX Interaction
Two major negative regulators of wtp53 stability and activity, MDM2 and MDMX (MDM4), are overexpressed in human cancers [6]. MDM2 can negatively regulate the stability and activity of wtp53 through multiple mechanisms. MDM2, an E3 ubiquitin ligase for p53, is transcriptionally activated by p53 and provides a key negative regulatory loop to keep the levels of wtp53 low in normal cells without stresses [7]. In addition to inducing p53 degradation, the transcriptional activity of wtp53 is also inhibited by the interaction between the N-terminus of p53 and MDM2 [8,9,10]. Thirdly, MDM2 promotes the excretion of p53 from the nucleus and thus inhibits the activities of p53 as a transcriptional factor. Like MDM2, MDMX can also suppress the transcriptional activities of p53 by interacting with the N-terminus of p53 [11]. In addition, MDMX can stabilize MDM2, which has a very short half-life, and ultimately promotes p53 degradation [12,13,14]. Therefore, the disruption of the interaction between p53 and MDM2/MDMX in cancer cells overexpressing MDM2/MDMX could reactivate the wtp53-dependent tumor-suppressive activities in human cancer cells.
In the course of developing therapeutic strategies to disrupt the interaction between p53 and MDM2/MDMX, 4,5-dihydroimidazoline (Nutlin; Roche) was the first small molecule compound identified to specifically disrupt the p53–MDM2 interface and thus increase p53 stability and activity, leading to cell-cycle arrest, apoptosis, senescence, and differentiation of cancer cells [15,16]. However, because of its poor solubility, toxicity, and inadequate efficacy in clinical trials, the development of Nutlin as a p53 target drug was halted.
Other small molecule MDM2 inhibitors that have entered into clinical trials include RG7112, Idasanutlin (RG7388), SAR405838 (MI-77301), and Alrizomadlin (APG-115) (Table 1). RG7112 is a Nutlin analog with higher specificity for MDM2 and can induce more robust apoptosis of acute myeloid leukemia (AML) and chronic lymphocytic leukemia (CLL) cells than Nutlin 3a [17]. A Phase I study of RG7112 (NCT00623870) in patients with hematologic malignancies has shown that RG7112 does not affect p53 transcription but can inhibit p53 degradation, thereby activating p53 target genes and promoting cell-cycle arrest and apoptosis in leukemia cells. However, because RG7112 is an oral drug that requires a high dose, it can have serious toxic effects on gastrointestinal and bone marrow, especially on normal progenitors [17]. Among Nutlin derivatives, Idasanutlin is the most powerful so far. It suppresses the growth of SJSA1 human osteosarcoma harboring the wtp53 at a dose about four times lower than RG7112 [18]. Idasanutlin was found to be well tolerated and safe when used alone. However, the MIRROS (NCT02545283) study, a phase III clinical trial evaluating the efficacy of combinational therapy of idasanutlin + cytarabine in patients with relapsed or refractory (R/R) AML, showed disappointing results in its primary and secondary endpoints, overall survival (OS), and complete response (CR) [19]. Further phase I/II clinical trials are presently underway for the treatment of leukemia, glioblastoma, and other solid tumors in combination with immunotherapy, such as atezolizumab and chemotherapy (NCT04029688, NCT04589845, NCT03158389).
SAR405838, a highly specific spiro-oxindole MDM2 antagonist, induces robust activation of wtp53 in vitro and in vivo [20]. While two phase I clinical trials of SAR405838 (NCT01636479, NCT01985191) to treat malignancies have been completed, the findings of these clinical trials have yet to be published. Milademetan, an oral MDM2 inhibitor, has a binding pattern to MDM2 similar to SAR405838. In vivo studies have demonstrated that Milademetan activates p53 and inhibits tumor growth [21]. A first-in-human phase I study (NCT01877382) of Milademetan to treat solid tumors showed that the intermittent administration of the MDM2 inhibitor is beneficial for patients [22]. In addition, the MANTRA-2 trial (NCT05012397) assessed the safety and efficacy of Milademetan monotherapy in patients with metastatic solid tumors harboring wtp53 and MDM2 amplification [23]. The results indicated two unconfirmed partial responses (PRs) in 15 patients treated with Milademetan and a safety profile consistent with the previous phase I trial. The U.S. Food and Drug Administration (FDA) has designated Milademetan as an orphan drug for treating LFS.
Alrizomadlin is a novel oral MDM2-p53 inhibitor that achieves higher chemical stability than spiro-oxindole compounds by adding an ethyl group to the pyrrolidine. At the 2022 ASCO Annual Meeting, the latest data from the phase II clinical study of Alrizomadlin in combination with pembrolizumab to treat solid tumors were shown in a poster, showing the efficacy of the combinational therapy in patients with immuno-oncologic drug-resistant melanoma, including two cases (2/44) of CR, three cases (3/44) of partial response (PR), and a 13% ORR [24]. These data suggest that Alrizomadlin has the potential to treat patients with immunotherapy-resistant tumors.
Further structural optimization targeting a previously underutilized region of MDM2 (G58 “shelf”) led to the discovery of KRT-232, an oral MDM2 inhibitor with increased potency. KRT-232 is currently used in clinical trials to treat advanced malignancies, including AML, multiple myeloma, and glioblastoma. The efficacy of KRT-232 to treat wtp53 Merkel cell carcinoma (MCC) after the failure of anti-PD-1/L1 therapy was reported at the 2022 ASCO Annual Meeting, showing a confirmed ORR of 25%, a disease control rate (DCR) of 63%, and a median remission time of 4.1 months. Notably, one patient achieved CR confirmed by PET/CT after 2 years of treatment with sustained PR [25]. This study suggests that the upregulation of the p53 pathway would be a viable therapeutic strategy for MCC.
NVP-CGM097 is another structurally optimized MDM2 inhibitor. NCT01760525 is the first-in-human phase I study of NVP-CGM097 to treat solid tumors. The DCR was 39%, including one PR and 19 patients in SD with the tolerability of NVP-CGM097 manageable [26].
MK-8242, an oral MDM2 antagonist, showed tumor-suppressive activity in animal tumor models with wild-type p53 by disrupting the interaction between MDM2 and p53 [27]. Two Phase 1 clinical trials evaluating the clinical efficacy of MK-8242 as a monotherapy for advanced solid malignancies or in combination with cytarabine for AML have been completed (NCT01451437, NCT01463696). The results showed 1/24 PR, 1/24 CR, and 1/24 morphologic leukemia-free states [28]. Additionally, in the clinical trial enrolled with advanced solid tumors (NCT01463696), 27 patients with LPS had an mPFS of 237 days. MK-8242 also showed acceptable safety in this study [29].
At the 2022 ASCO Annual Meeting, Boehringer Ingelheim presented the results of a phase Ia/Ib, dose-escalation of MDM2 inhibitor BI 907828 to treat patients with solid tumors, including advanced/metastatic LPS. Among 41 enrolled patients with advanced LPS, a response of PR or SD was observed in 88.9% of patients with advanced DDLPS and 92.9% of patients with highly differentiated LPS (WDLPS). In the dose-escalation study, approximately 45% (5/11) of patients with DDLPS and approximately 50% (4/8) of patients with WDLPS had PFS ≥ 10.5 months [30]. In addition, this drug also showed a manageable safety profile, high plasma exposure, target engagement, and encouraging signs of antitumor activity in patients with advanced LPS.
Siremadlin, another MDM2 inhibitor, efficiently inhibited MDM2 and showed antitumor activity in cancer cell lines and wtp53 xenograft tumor models [31]. The drug is currently in 11 clinical trials for the treatment of advanced/metastatic soft-tissue sarcoma, AML, LPS, colorectal cancer, high-risk myelodysplastic syndrome (MDS), hepatic impairment, and other advanced solid and hematological wtp53 tumors. A phase I dose-escalation study (NCT02143635) in patients with TP53 wild-type advanced solid or hematologic cancers established controlled safety and preliminary activity, particularly in AML patients. Overall response rates (ORR) at the recommended dose for expansion (RDEs) were 10.3% in solid tumors and 4.2%, 20%, and 22.2% in AML with increasing concentrations [32]. A recent phase Ib dose-escalation study demonstrated that the combinational therapy with Siremadlin and Ribociclib (CDK4/6 inhibitor) exhibited manageable toxicity and antitumor activity in patients with radiologically progressive advanced WDLPS or DDLPS [33].
Overexpression of MDMX also occurs in several types of cancer, most notably melanoma [34]. To fully reactivate p53 in tumors with MDMX overexpression, dual targeting of MDM2/MDMX may be necessary. Staple peptides have an advantage over small molecule drugs in targeting protein–protein interactions (PPIs) due to enhanced binding affinity and stability for PPI interfaces. ALRN-6924 is the only dual MDM2 and MDMX inhibitor in clinical trials [35]. An open-label trial (NCT05622058) designed to evaluate the safety and efficacy of ALRN-6924 in breast cancer patients was prematurely terminated due to the findings that the first four patients treated had experienced grade 4 neutropenia and alopecia after cycle 1 and, as such, failed to meet the primary endpoint and the main secondary endpoint. VIP116 and its predecessor PM2 are novel stapled peptides targeting the MDM2/X–p53 interaction discovered by Lane’s group [36,37]. PM2 has been shown to avoid Nutlin-3 resistance and increase therapeutic effects by combining radiotherapy [38,39]. Furthermore, VIP116 with PEG-Stabilized Lipodisks promotes p53-mediated apoptosis with increasing peptide accumulation and decreasing toxicity [40]. Other dual MDM2 and MDMX inhibitors are being developed with significant anticancer activities in preclinical studies, including RO-2443 and optimized RO-5963 [41,42].
2.2. Development of Drugs to Target p53 Mutants (Mutp53)
When the p53 mutant protein is expressed, it plays multiple roles in driving tumorigenesis, including dominant-negative (DN) effects and gain of functions (GOF). Therefore, significant effort has been devoted to developing drugs to restore wtp53-dependent activities to mutp53 and inhibit the DN/GOF effects of mutp53. In principle, the drug that restores the wtp53 activities to mutp53 could simultaneously inhibit the DN/GOF effects of mutp53.
2.2.1. Development of Drugs to Restore wtp53-Dependent Activities
Most p53 mutations can affect the transcriptional activities of wtp53, inducing the loss of DNA-binding activity directly or indirectly by the disruption of normal tetramerization of wtp53. Treatments with cysteine-binding compounds, Zn2+-chelating compounds, read-through drugs, and aggregation inhibitors are some approaches taken to restore wtp53 conformation, transcriptional activity, and tumor-suppressive activity.
Cysteine-Binding Compounds
Screening for compounds that suppress mutp53-dependent cell growth led to the discovery of PRIMA-1 [43]. The addition of a methyl group to PRIMA-1 led to APR-246, also known as PRIMA-1MET, which increased the pro-apoptotic activity and membrane permeability of the compound [44]. The proliferation of tumor cells harboring mutp53 is inhibited by the treatment of PRIMA1 and APR-246 both in vitro and in mouse xenograft tumor models [45,46,47]. In syngeneic mice, APR-246 can also suppress the growth of ascites tumors [48].
Mechanistically, methylene quinuclidinone (MQ), a Michael acceptor, is formed when the prodrug APR-246 is hydrolyzed in vivo. It can restore the mutp53 antitumor function by forming a covalent connection between the thiol groups of cysteines within the core DNA-binding domain. Experimental evidence suggests that Cys124 is a critical site for restoring the wtp53 activities to mutp53 [49]. MQ is also responsible for reactivating mutp53 by blocking its amyloid-like aggregation [50]. The off-target activities of these mutp53-targeting compounds also contribute to their antitumor effects. APR-246/MQ can form a covalent bond with the cysteine of glutathione, leading to increased levels of reactive oxygen species (ROS) and ferroptosis [51]. APR-246/MQ inhibits oxidoreductase enzyme thioredoxin reductase 1 (TrxR1), thioredoxin, and glutaredoxin, leading to oxidative stress [52,53]. APR-246 also appears capable of restoring the function of p53 homologs, including p63, TAP73a, TAp73b, and TAP63g, enhancing the capability of APR-246 to induce tumor cell death [54,55,56].
The efficacy and safety of APR-246 to treat malignant tumors were evaluated in a phase 1 clinical trial, the first-in-human trial for mutp53-targeting drugs (NCT00900614), showing the upregulation of p53 target genes with good safety, tolerance, and PK properties. In the phase II trial (NCT02098343), patients with recurrent platinum-sensitive high-grade ovarian cancer were treated with APR-246 in combination with carboplatin and pegylated doxorubicin at standard doses or carboplatin and pegylated doxorubicin. Using RECIST criteria, 3 of 21 evaluable patients had a confirmed CR, 10 had a confirmed PR, and 8 had SD [57]. APR-246 in combination with Azacitidine has produced encouraging results in patients with untreated TP53-mutated MDS and AML (NCT03072043). However, the data of the phase III trial (NCT03745716), which enrolled 154 patients with mutp53 MDS treated either with Azacitidine + APR-246 or Azacitidine alone, were disappointing with no significant difference between the two treatment groups [58].
Zn2+-Chelating Compounds
Zinc is essential for the correct folding of the central core domain of p53, and the absence of a zinc molecule in the central core can cause p53 to unfold and aggregate [59,60]. Immuno-precipitation experiments using the PAb1620 antibody showed that Zn2+ facilitated the refolding of mutp53 into the wild-type conformation [61]. By screening the NCI-60 cell line panel, Carpizo and his team found the chemical NSC319726 (ZMC1) that specifically reactivated the p53R175H mutant likely by binding Zn2+ [62]. In addition to binding Zn2+, ZMC1 also binds Fe2+ and Cu2+, leading to increased ROS generation and oxidative stress [63,64].
COTI-2, a third-generation thiosemicarbazone identified by an in silico machine learning system, is active against a variety of malignant cell lines [65,66]. Unlike other thiosemicarbazones, COTI-2 appears capable of acting both dependently and independently of p53, resulting in cell death, restoration of p53 target gene expression, and activation of AMPK, while having no effect on intracellular Zn2+ levels [67]. COTI2 has been evaluated in a phase I trial (NCT02433626) for treating gynecological tumors with no evidence of tumor regression.
p53 Nonsense Mutation Read-Through Drugs
The p53 nonsense mutation accounts for about 10% of all p53 mutations. No tRNA can bind to the premature termination codon (PTC) in the p53 mRNA with nonsense mutation. Nonsense-mediated mRNA decay (NMD) efficiently identifies and degrades mRNAs containing PTC in open reading frames to prevent cell toxicity caused by the accumulation of truncated protein products [68]. Therefore, the strategy to restore the wtp53-like function is not feasible in this context. A more effective strategy is to induce the expression of the full-length p53 protein achieved by simultaneously targeting the induction of translational read-through and the inhibition of NMD.
In a cell-based screen, the read-through inducer Ataluren (PTC124) was discovered. This compound appears to be able to induce the expression of a functionally intact antimyotrophic protein. Insufficient evidence from relevant trials and concerns regarding its mode of action have led to its rejection by the FDA. Antibiotic aminoglycosides such as G418 (geneticin) and its derivative NB124 could induce read-through by elongating polypeptide chains by the insertion of a cognate tRNA. Synergistic effects were observed between NMD14, an NMD inhibitor, and G418 in inducing the expression of p53 target genes CDKN1A, BAX, and PUMA of human cancer cells harboring p53 nonsense mutations [69]. Unfortunately, aminoglycosides’ nephrotoxicity and ototoxicity severely restrict their potential therapeutic application [70].
Dispersion of p53 Aggregates
Mutp53 tends to form aggregates due to the exposure of adhesion sequences originally wrapped inside its hydrophobic core, and structural mutp53, such as R110P, R175H, R248Q, R249S, and R282W, can form amyloid aggregates [71]. In order to reactivate mutant p53, it is important to break up p53 aggregates and shift the folding equilibrium towards a “wild-type-like” state. The cell-penetrating peptide ReACp53 was utilized as an aggregation inhibitor of mutp53, particularly for R175H and R248Q mutants [72,73,74]. With its wild-type conformation and nuclear localization restored, p53 is able to promote apoptosis and cell-cycle arrest in cancer cells harboring p53 mutants in vitro and tumor suppression in vivo [73].
Taking advantage of an oligopyridylamide library known to suppress amyloid formation in Alzheimer’s disease, researchers found a tripyridylamide, denoted ADH-6, which can counteract the self-assembly of the aggregation-nucleating subdomain of mutp53 DBD. ADH-6 specifically recognizes and disrupts mutp53 aggregates in human cancer cells, reviving wtp53’s transcriptional activity and thus inducing cell-cycle arrest and apoptosis. Importantly, ADH-6 suppresses the growth of mutp53 xenografts without causing apparent toxicity to healthy tissues [75]. The clinical efficacy of these compounds is highly anticipated.
Members of the p53 family, such as p73 and p63, share a panel of target genes with wtp53. Compounds that activate these members of the p53 family could restore the Wtp53 activity because p73 and p63 are not frequently mutated in malignancies. However, mutant p53 can co-aggregate with p63 and p73, preventing their entry into the nucleus. Screening for compounds that can induce the expression of p53 target genes in mutp53-expressing cancer cells led to the discovery of the drug RETRA [76]. It activates the p73-dependent transcription and cell death by dismantling the mutp53-p73 aggregates. In a p73-dependent fashion, a small molecule compound, Prodigiosin, also promotes the expression of p53 target genes p21, puma, and DR5 in mutp53 cells [77]. NSC59984 is a first-in-class small molecule drug that disrupts the mutp53-p73 complex and thus releases p73, inducing p73-dependent transcription in mutp53 cells. This compound also promotes the constitutive phosphorylation of ERK2 via ROS and MDM2-mediated ubiquitination and degradation of mutp53, suggesting a combinational therapy of this compound and ROS-inducing agents to treat mutp53 human cancers [78].
Other Compounds
Lu et al. demonstrated that arsenic trioxide (ATO) could restore structural mutp53 and identified 390 wtp53, the functions of which could be restored to various degrees by ATO [79]. ATO promotes the folding of structural mutp53 by functioning as an intramolecular glue that connects the LSH motif and β-sandwich motif [80]. In addition, single-cell sequencing of peripheral blood mononuclear cells (PBMCs) in a non-acute promyelocytic leukemia (APL) AML patient harboring a type 1 mutp53 after ATO monotherapy revealed upregulation of the classical p53 target gene and a significant reduction in minimal residual disease (MRD) in blood samples [79]. While this study supports the feasibility of restoring wtp53 function to mutp53 by ATO in human cancers, the low prevalence of type 1 mutp53 (0.1–0.2% in leukemias) as well as the toxicity and poor efficacy of ATO in solid tumors poses a challenge to larger-scale validation. Another compound, potassium antimony tartrate (PAT), was identified to restore 65 (1–2%) reversible temperature-sensitive (TS) mutp53 in preclinical experiments [81].
While the aforementioned compounds all have multiple mutp53 to target, more recent strategies have been to target either a single mutant or a subset of mutants with similar phenotypes. For example, the p53Y220C mutation is the eighth most prevalent p53 mutation across 12 different types of tumors [82,83]. The small molecule compounds PK083 and PK7080 were identified to target the Y220C mutation and restore wild-type conformation to mutp53, leading to tumor suppression [83,84]. PMV Pharmaceuticals used a similar strategy to develop its p53Y220C-targeting small molecule compound PC14586. Positive preliminary results of a phase I/II trial (NCT04585750) for the orally bioavailable PC14586 were presented at the last ASCO annual meeting. This study evaluated the safety, PK, PD, and preliminary efficacy of PC14586 in patients with advanced solid tumors with p53Y220C mutation. Among 21 patients with evaluable efficacy, 5 achieved PR; 30% of patients in the three highest dose groups achieved PR. In addition, the study found a reduction in the circulating tumor DNA and circulating tumor cells with p53Y220C mutation. In terms of safety, PC14586 was well tolerated. Therefore, PC14586 could represent a promising therapeutic agent to treat tumors with p53Y220C mutation [85]. Prof. Kevan M. Shokat’s team has recently discovered a new covalent inhibitor, KG13, that can restore the thermal stability of p53 y220c mutants to wild-type levels. H1299 (p53−/−) and U2OS (p53+/+) cells stably expressing p53 Y220C, as well as patient-derived NUGC-3 (p53Y220C/+) and BxPC-3 (p53−/−) cells, were treated with KG13 and analyzed for p53 target gene activation, cell growth inhibition, and increased caspase-3/7 activity [86].
Several other small molecule compounds have been identified to restore wtp53 activity in preclinical experiments. One such compound is RITA, a tiny chemical found to reactivate various mutp53, later also shown to interfere with the interaction between p53 and E6 [87,88,89,90]. In a p53 reporter screening assay, the C7-aryl piperlongumine derivatives KSS-9 and chetomin were found to bind to heat shock protein 40 (Hsp40), promoting its interaction with p53R175H to restore its wild-type conformation [91,92].
2.2.2. To Develop Strategies to Eradicate Mutp53
Mounting evidence indicates that chemotherapy and radiotherapy of tumors can stabilize mutp53 and increase its GOF activity in promoting tumorigenesis and drug resistance. Therefore, eliminating mutp53 represents an intensively pursued strategy to inhibit GOF and drug resistance promoted by mutp53.
Hsp90 regulates the stability of many interacting proteins including mutp53 and its acetylation will weaken its interaction with target proteins [93]. HDAC is involved in the deacetylation of HSP90, and thus HDAC inhibitors can increase the acetylation levels of HSP90, leading to the destabilization of HSP90 target proteins such as mutp53 [94]. Ganetespib (STA-9090) is a novel resorcinolic triazolone compound targeting Hsp90 and binds to the N-terminal ATP-binding site of Hsp90. It can inactivate Hsp90 and induces cell-cycle arrest and apoptosis of cancer cells [95]. Disappointingly, ganetespib alone or in combination with other drugs (platinum, docetaxel, etc.) failed to improve the clinical outcome of patients. The geldanamycin derivative 17-AAG (Tanespimycin) and the 17-AAG hydroquinone hydrochloride salt derivative (IPI-504) have both shown antitumor activities and a well-tolerated profile in multiple clinical trials, including a phase III trial for multiple myeloma [96].
HSP40 proteins serve as co-chaperones for certain HSP70 proteins and modulate their functions by stimulating ATP hydrolysis in HSP70. Furthermore, HSP40 proteins are essential for protein translation, folding, unfolding, assembly, translocation, and degradation [97,98]. Statins were found to degrade conformational or misfolded mutp53 proteins via the mevalonate pathway-DNAJA1 (an HSP40 family member) axis in a Saos2 cell line expressing a p53R175H protein and in xenografts [99,100]. Trials NCT04767984 and NCT03560882 are currently investigating the antitumor activity of statins in patients stratified according to their p53 mutation status. These findings may lend support to the theory that the p53 status determines how effectively statins inhibit tumor growth. Statins also inhibit the HDAC6/Hsp90-dependent accumulation of mutp53 by inhibiting HDAC6 activity directly and disabling the mevalonate-RhoA axis [101,102]. In recent molecular docking studies, Tomoo Iwakuma’s group identified DNAJA1 inhibitors, PLTFBH and A11, which reduced DNAJA1 levels and then induced conformational mutp53 degradation, thereby inhibiting cancer cell migration [103,104].
Spautin-1, a small molecule compound that inhibits macroautophagy, can induce the degradation of a wide range of mutp53 proteins, including p53R175H/C/D, S241F, G245C, R248Q/W/L, E258K, R273H/L, R280K, and R282W [105]. Hence, targeting mutp53 for degradation by small molecule drugs is a promising strategy to improve the efficacy of therapy for tumors expressing mutp53.
2.3. Drugs Induce Synthetic Lethality with mutp53
Mutp53 frequently demonstrates oncogenic GOF activities by modulating many downstream signaling cascades in cancer cells. Hence, targeting critical mutp53 downstream pathways provides an alternate treatment for human cancers expressing mutp53. Cancer cells may develop a secondary survival reliance due to an oncogenic mutation or tumor suppressor deficiency, when the secondary survival pathway is also mutated, leading to a condition known as synthetic lethality [106]. For example, the inactivation of the ATR/CHK1, ATM/CHK2, or p38MAPK/MK2 pathway has been shown to confer synthetic lethality with p53-deficiency of cancer cells [107].
Preclinical studies of the WEE1 inhibitor MK-1775 (AZD1775) have shown efficacy against head and neck tumors expressing mutp53, either alone or in combination with cisplatin [108]. Currently, MK1775 has been tested in multiple clinical trials to examine its efficacy to treat tumors with p53 deletions or mutations. In the NCT01357161 trial, MK-1775 enhanced the effectiveness of carboplatin/paclitaxel chemotherapy in women with platinum-sensitive ovarian cancer expressing mutp53, with a modest clinical benefit as indicated by PFS (median 34.14 vs. 31.86 weeks) [109]. In the NCT01164995 trial, MK-1775 enhanced the efficacy of carboplatin to treat resistant or refractory ovarian cancer expressing mutp53 [110]. In the NCT02272790 trial, the efficacy and safety of MK-1775 in combination with chemotherapy (gemcitabine, paclitaxel, carboplatin, or pegylated liposomal doxorubicin (PLD)) for platinum-resistant ovarian cancer were examined, showing promising results as reported in the 2019 ASCO Annual Meeting. However, this combinational therapy caused very notable hematologic toxicity among the whole cohort [111]. Therefore, the regimens of MK-1775 need to be optimized to improve the safety of the treatment.
2.4. Cancer Immunotherapy for Mutp53
Adoptive cell transfer therapy (ACT) refers to the isolation of immunologically active cells from tumor patients, their expansion and functional characterization in vitro, and then their infusion back to the patient for the purpose of killing the tumor directly or stimulating the body’s immune response to kill the tumor cells.
The vast variety of P53 missense mutations in human malignancies generates a large number of potential neoantigens that may stimulate mutant p53-specific immune responses. Using novel high-throughput techniques, a recent study has evaluated the response of T cells within tumors to p53 mutations, showing that hotspot mutations can be processed and presented by HLA on the surface of tumor cells. p53 hotspot mutations have a complex immunogenicity, which is influenced by many factors such as the stability of mutp53, HLA alleles expressed on tumor cells or APC, and TCR affinity. In the future, it is anticipated that more in-depth studies can be conducted on larger patient cohorts [112].
3. Conclusions
p53 is a crucial tumor suppressor and is mutated in about 50% of human cancers, making both wtp53 and mutp53 appealing therapeutic targets for cancer treatment. The strategies to induce wtp53-like activities in human cancer cells are complex and must take into consideration different scenarios such as the loss of function of wtp53, and the dominant-negative and gain of oncogenic functions of mutp53. Apparently, more precision in developing drugs targeting individual mutp53 or mutp53s of similar features is required. Strategies to reverse the DN and GOF effects associated with some types of mutp53 have been shown to boost therapeutic responses and inhibit tumor growth in preclinical models. However, despite extensive clinical testing of these compounds, the efficacy and safety of these drugs remain challenging for their clinical application. Considering the potent roles of p53 in both tumor suppression and aging, in the course of developing p53-targeting drugs, one important balance to maintain is to activate wtp53-dependent tumor suppression without inducing p53-dependent aging. A more in-depth understanding of the mechanism of action of these drugs on various p53-dependent functions is important to improve their efficacy and safety.
Author Contributions
Y.L., M.W., Y.X. and L.Y. all contributed significantly to the writing of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2022R01002), and the National Natural Science Foundation of China (81930084).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression—Potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef]
- Espadinha, M.; Barcherini, V.; Lopes, E.A.; Santos, M.M.M. An Update on MDMX and Dual MDM2/X Inhibitors. Curr. Top. Med. Chem. 2018, 18, 647–660. [Google Scholar] [CrossRef]
- Marine, J.C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.D.; Marechal, V.; Levine, A.J. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol. 1993, 13, 4107–4114. [Google Scholar]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gvuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumor suppressor-p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Lu, W.G.; Pochampally, R.; Chen, L.H.; Traidej, M.; Wang, Y.L.; Chen, J.D. Nuclear exclusion of p53 in a subset of tumors requires MDM2 function. Oncogene 2000, 19, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Chen, J.D. MDM2 promotes ubiquitination and degradation of MDMX. Mol. Cell. Biol. 2003, 23, 5113–5121. [Google Scholar] [CrossRef] [Green Version]
- Sharp, D.A.; Kratowicz, S.A.; Sank, M.J.; George, D.L. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J. Biol. Chem. 1999, 274, 38189–38196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stad, R.; Little, N.A.; Xirodimas, D.P.; Frenk, R.; van der Eb, A.J.; Lane, D.P.; Saville, M.K.; Jochemsen, A.G. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001, 2, 1029–1034. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, T.; Uesugi, M. In-vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Tanpakushitsu Kakusan Koso. Protein Nucleic Acid Enzym. 2007, 52 (Suppl. S13), 1816–1817. [Google Scholar]
- Van Maerken, T.; Speleman, F.; Vermeulen, J.; Lambertz, I.; De Clercq, S.; De Smet, E.; Yigit, N.; Coppens, V.; Philippe, J.; De Paepe, A.; et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006, 66, 9646–9655. [Google Scholar] [CrossRef] [Green Version]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barriere, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [Green Version]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wunschel, J.; Toedling, J.; Deubzer, H.E.; Kunkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Bauer, T.M.; Schwartz, G.K.; LoRusso, P.; Kumar, P.; Kato, K.; Tao, B.; Hong, Y.; Patel, P.; Hong, D. Milademetan, an oral MDM2 inhibitor, in well-differentiated/dedifferentiated liposarcoma: Results from a phase 1 study in patients with solid tumors or lymphomas. Eur. J. Cancer 2020, 138, S3–S4. [Google Scholar] [CrossRef]
- Dumbrava, E.E.; Hanna, G.J.; Cote, G.M.; Stinchcombe, T.; Johnson, M.L.; Chen, C.; Devarakonda, S.; Shah, N.; Xu, F.; Doebele, R.C.; et al. A phase 2 study of the MDM2 inhibitor milademetan in patients with TP53-wild type and MDM2-amplified advanced or metastatic solid tumors (MANTRA-2). J. Clin. Oncol. 2022, 40, 16. [Google Scholar] [CrossRef]
- McKean, M.; Tolcher, A.W.; Reeves, J.A.; Chmielowski, B.; Shaheen, M.F.; Beck, J.T.; Orloff, M.M.; Somaiah, N.; Van Tine, B.A.; Drabick, J.J.; et al. Newly updated activity results of alrizomadlin (APG-115), a novel MDM2/p53 inhibitor, plus pembrolizumab: Phase 2 study in adults and children with various solid tumors. J. Clin. Oncol. 2022, 40, 9517. [Google Scholar] [CrossRef]
- Wong, M.K.K.; Burgess, M.A.; Chandra, S.; Schadendorf, D.; Silk, A.W.; Olszanski, A.J.; Grob, J.-J.; Jang, S.; Grewal, J.S.; Lewis, K.D.; et al. Navtemadlin (KRT-232) activity after failure of anti-PD-1/L1 therapy in patients (pts) with TP53WT Merkel cell carcinoma (MCC). J. Clin. Oncol. 2022, 40, 9506. [Google Scholar] [CrossRef]
- Bauer, S.; Demetri, G.D.; Halilovic, E.; Dummer, R.; Meille, C.; Tan, D.S.W.; Guerreiro, N.; Jullion, A.; Ferretti, S.; Jeay, S.; et al. Pharmacokinetic-pharmacodynamic guided optimisation of dose and schedule of CGM097, an HDM2 inhibitor, in preclinical and clinical studies. Br. J. Cancer 2021, 125, 687–698. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.; Keir, S.T.; Maris, J.M.; Wu, J.R.; Lyalin, D.; et al. Initial Testing (Stage 1) of MK-8242A Novel MDM2 Inhibitorby the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2016, 63, 1744–1752. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Gojo, I.; Patnaik, M.M.; Minden, M.D.; Kantarjian, H.; Johnson-Levonas, A.O.; Fancourt, C.; Lam, R.; Jones, M.B.; Knox, C.D.; et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk. Res. 2016, 48, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.; Fancourt, C.; Johnson-Levonas, A.O.; Lam, R.; Meister, A.K.; Russo, G.; et al. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 1304–1311. [Google Scholar] [CrossRef]
- Gounder, M.M.; Yamamoto, N.; Patel, M.R.; Bauer, T.M.; Schoffski, P.; Grempler, R.; Durland-Busbice, S.; Geng, J.; Maerten, A.; LoRusso, P. A phase Ia/Ib, dose-escalation/expansion study of the MDM2-p53 antagonist BI 907828 in patients with solid tumors, including advanced/metastatic liposarcoma (LPS). J. Clin. Oncol. 2022, 40, 3004. [Google Scholar] [CrossRef]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf−/− mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef]
- Abdul Razak, A.R.; Bauer, S.; Suarez, C.; Lin, C.C.; Quek, R.; Hutter-Kronke, M.L.; Cubedo, R.; Ferretti, S.; Guerreiro, N.; Jullion, A.; et al. Co-Targeting of MDM2 and CDK4/6 with Siremadlin and Ribociclib for the Treatment of Patients with Well-Differentiated or Dedifferentiated Liposarcoma: Results from a Proof-of-Concept, Phase Ib Study. Clin. Cancer Res. 2022, 28, 1087–1097. [Google Scholar] [CrossRef]
- Marine, J.C. MDM2 and MDMX in cancer and development. Curr. Top. Dev. Biol. 2011, 94, 45–75. [Google Scholar] [PubMed]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.G.; Santiago, S.; Guerlavais, V.; et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Quah, S.T.; Jong, J.; Goh, A.M.; Chiam, P.C.; Khoo, K.H.; Choong, M.L.; Lee, M.A.; Yurlova, L.; Zolghadr, K.; et al. Stapled Peptides with Improved Potency and Specificity That Activate p53. ACS Chem. Biol. 2013, 8, 506–512. [Google Scholar] [CrossRef]
- Yuen, T.Y.; Brown, C.J.; Xue, Y.Z.; Tan, Y.S.; Gago, F.J.F.; Lee, X.E.; Neo, J.Y.; Thean, D.; Kaan, H.Y.K.; Partridge, A.W.; et al. Stereoisomerism of stapled peptide inhibitors of the p53-Mdm2 interaction: An assessment of synthetic strategies and activity profiles. Chem. Sci. 2019, 10, 6457–6466. [Google Scholar] [CrossRef] [Green Version]
- Spiegelberg, D.; Mortensen, A.C.; Lundsten, S.; Brown, C.J.; Lane, D.P.; Nestor, M. The MDM2/MDMX-p53 Antagonist PM2 Radiosensitizes Wild-Type p53 Tumors. Cancer Res. 2018, 78, 5084–5093. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.J.; Chee, S.; Yurlova, L.; Lane, D.; Verma, C.; Brown, C.; Ghadessy, F. Avoiding drug resistance through extended drug target interfaces: A case for stapled peptides. Oncotarget 2016, 7, 32232–32246. [Google Scholar] [CrossRef]
- Lundsten, S.; Hernandez, V.A.; Gedda, L.; Saren, T.; Brown, C.J.; Lane, D.P.; Edwards, K.; Nestor, M. Tumor-Targeted Delivery of the p53-Activating Peptide VIP116 with PEG-Stabilized Lipodisks. Nanomaterials 2020, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.S.; et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, X.; Yuan, W.; Zou, Y.; Guo, Z.; Chai, Y.; Lu, W. Rapid identification of dual p53-MDM2/MDMX interaction inhibitors through virtual screening and hit-based substructure search. RSC Adv. 2017, 7, 9989–9997. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.S.; Read, M.; Cullinane, C.; Azar, W.J.; Fennell, C.M.; Montgomery, K.G.; Haupt, S.; Haupt, Y.; Wiman, K.G.; Duong, C.P.; et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 2015, 64, 1506–1516. [Google Scholar] [CrossRef]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [Green Version]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.R.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1(MET) inhibits growth of mouse tumors carrying mutant p53. Anal. Cell. Oncol. 2008, 30, 411–418. [Google Scholar]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Rangel, L.P.; Ferretti, G.D.S.; Costa, C.L.; Andrade, S.; Carvalho, R.S.; Costa, D.C.F.; Silva, J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [Green Version]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.Y.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, M.Q.Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arner, E.S.J.; Wiman, K.G. APR-246/PRIMA-1(MET) inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.; Wiman, K.G.; Wilhelm, M.T. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalom-Feuerstein, R.; Serror, L.; Aberdam, E.; Muller, F.J.; van Bokhoven, H.; Wiman, K.G.; Zhou, H.; Aberdam, D.; Petit, I. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc. Natl. Acad. Sci. USA 2013, 110, 2152–2156. [Google Scholar] [CrossRef]
- Shen, J.; van den Bogaard, E.H.; Kouwenhoven, E.N.; Bykov, V.J.; Rinne, T.; Zhang, Q.; Tjabringa, G.S.; Gilissen, C.; van Heeringen, S.J.; Schalkwijk, J.; et al. APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 2157–2162. [Google Scholar] [CrossRef]
- Gourley, C.; Green, J.; Gabra, H.; Vergote, I.; Basu, B.; Brenton, J.D.; Bjorklund, U.; Smith, A.M.; Von Euler, M. PISARRO: A EUTROC phase Ib study of APR-246 in combination with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in platinum sensitive relapsed high grade serous ovarian cancer (HGSOC). J. Clin. Oncol. 2016, 34, 5571. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.S.; De Zern, A.E.; Sebert, M.; Garcia-Manero, G.; Rahme, R.; Steensma, D.P.; Che, J.L.; Roboz, G.J.; Madelaine, I.; et al. Long Term Follow-up and Combined Phase 2 Results of Eprenetapopt (APR-246) and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2021, 138, 246. [Google Scholar] [CrossRef]
- Loh, S.N. The missing zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef] [Green Version]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [Green Version]
- Maleki Vareki, S.; Salim, K.Y.; Danter, W.R.; Koropatnick, J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS ONE 2018, 13, e0191766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [Green Version]
- Bordeira-Carrico, R.; Pego, A.P.; Santos, M.; Oliveira, C. Cancer syndromes and therapy by stop-codon readthrough. Trends Mol. Med. 2012, 18, 667–678. [Google Scholar] [CrossRef]
- Martin, L.; Grigoryan, A.; Wang, D.; Wang, J.; Breda, L.; Rivella, S.; Cardozo, T.; Gardner, L.B. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014, 74, 3104–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Makarov, D.E. Effect of Mutation on an Aggregation-Prone Segment of p53: From Monomer to Dimer to Multimer. J. Phys. Chem. B 2016, 120, 11665–11673. [Google Scholar] [CrossRef]
- Ghosh, S.; Ghosh, D.; Ranganathan, S.; Anoop, A.; Kumar, P.S.; Jha, N.N.; Padinhateeri, R.; Maji, S.K. Investigating the intrinsic aggregation potential of evolutionarily conserved segments in p53. Biochemistry 2014, 53, 5995–6010. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Palanikumar, L.; Karpauskaite, L.; Al-Sayegh, M.; Chehade, I.; Alam, M.; Hassan, S.; Maity, D.; Ali, L.; Kalmouni, M.; Hunashal, Y.; et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 2021, 12, 3962. [Google Scholar] [CrossRef]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhou, L.; El-Deiry, W.S. Small-Molecule NSC59984 Induces Mutant p53 Degradation through a ROS-ERK2-MDM2 Axis in Cancer Cells. Mol. Cancer Res. 2022, 20, 622–636. [Google Scholar] [CrossRef]
- Song, H.; Wu, J.; Tang, Y.; Dai, Y.; Xiang, X.; Li, Y.; Wu, L.; Wu, J.; Liang, Y.; Xing, Y.; et al. Diverse rescue potencies of p53 mutations to ATO are predetermined by intrinsic mutational properties. Sci. Transl. Med. 2023, 15, eabn9155. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.L.; Liang, Y.; Tang, Y.G.; Song, H.X.; Wu, L.L.; Xing, Y.F.; Yan, N.; Li, Y.T.; Wang, Z.Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Tang, Y.; Song, H.; Wang, Z.; Xiao, S.; Xiang, X.; Zhan, H.; Wu, L.; Wu, J.; Xing, Y.; Tan, Y.; et al. Repurposing antiparasitic antimonials to noncovalently rescue temperature-sensitive p53 mutations. Cell Rep. 2022, 39, 110622. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Dumbrava, E.E.; Johnson, M.L.; Tolcher, A.W.; Shapiro, G.; Thompson, J.A.; El-Khoueiry, A.B.; Vandross, A.L.; Kummar, S.; Parikh, A.R.; Munster, P.N.; et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 2022, 40, 3003. [Google Scholar] [CrossRef]
- Guiley, K.Z.; Shokat, K.M. A Small Molecule Reacts with the p53 Somatic Mutant Y220C to Rescue Wild-type Thermal Stability. Cancer Discov. 2023, 13, 56–69. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Szekely, L.; Bao, W.; Selivanova, G. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res. 2010, 70, 3372–3381. [Google Scholar] [CrossRef] [Green Version]
- Weilbacher, A.; Gutekunst, M.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38. Cell Death Dis. 2014, 5, e1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmakin, M.; Shi, Y.; Hedstrom, E.; Kogner, P.; Selivanova, G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin. Cancer Res. 2013, 19, 5092–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, S.; Archer, T.K. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 2005, 15, 565–567. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Saini, N.; Parris, A.B.; Zhao, M.; Yang, X. Ganetespib induces G2/M cell cycle arrest and apoptosis in gastric cancer cells through targeting of receptor tyrosine kinase signaling. Int. J. Oncol. 2017, 51, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Badros, A.Z.; Jagannath, S.; Tarantolo, S.; Wolf, J.L.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin with bortezomib: Activity in relapsed/refractory patients with multiple myeloma. Br. J. Haematol. 2010, 150, 428–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Z.; Qian, X.G.; Sha, B.D. Heat Shock Protein 40: Structural Studies and Their Functional Implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef] [Green Version]
- Alalem, M.; Bhosale, M.; Ranjan, A.; Yamamoto, S.; Kaida, A.; Nishikawa, S.; Parrales, A.; Farooki, S.; Anant, S.; Padhye, S.; et al. Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin. Cancers 2022, 14, 4187. [Google Scholar] [CrossRef]
- Nishikawa, S.; Kaida, A.; Parrales, A.; Ranjan, A.; Alalem, M.; Ren, H.Y.; Schoenen, F.J.; Johnson, D.K.; Iwakuma, T. DNAJA1-and conformational mutant p53-dependent inhibition of cancer cell migration by a novel compound identified through a virtual screen. Cell Death Discov. 2022, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tahaney, W.M.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecularly targeted therapies for p53-mutant cancers. Cell Mol. Life Sci. 2017, 74, 4171–4187. [Google Scholar] [CrossRef]
- Osman, A.A.; Monroe, M.M.; Ortega Alves, M.V.; Patel, A.A.; Katsonis, P.; Fitzgerald, A.L.; Neskey, D.M.; Frederick, M.J.; Woo, S.H.; Caulin, C.; et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol. Cancer Ther. 2015, 14, 608–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; Weberpals, J.I.; Provencher, D.M.; Grischke, E.-M.; Hall, M.; Uyar, D.; Estevez-Diz, M.D.P.; Marme, F.; Kuzmin, A.; Rosenberg, P.; et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (P/C) for the treatment of women with platinum-sensitive, TP53-mutant ovarian cancer. J. Clin. Oncol. 2015, 33, 5506. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.-M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with chemotherapy (CT) in patients (pts) with platinum-resistant ovarian cancer (PPROC): An open label, four-arm, phase II study. J. Clin. Oncol. 2019, 37, 5513. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.Y.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
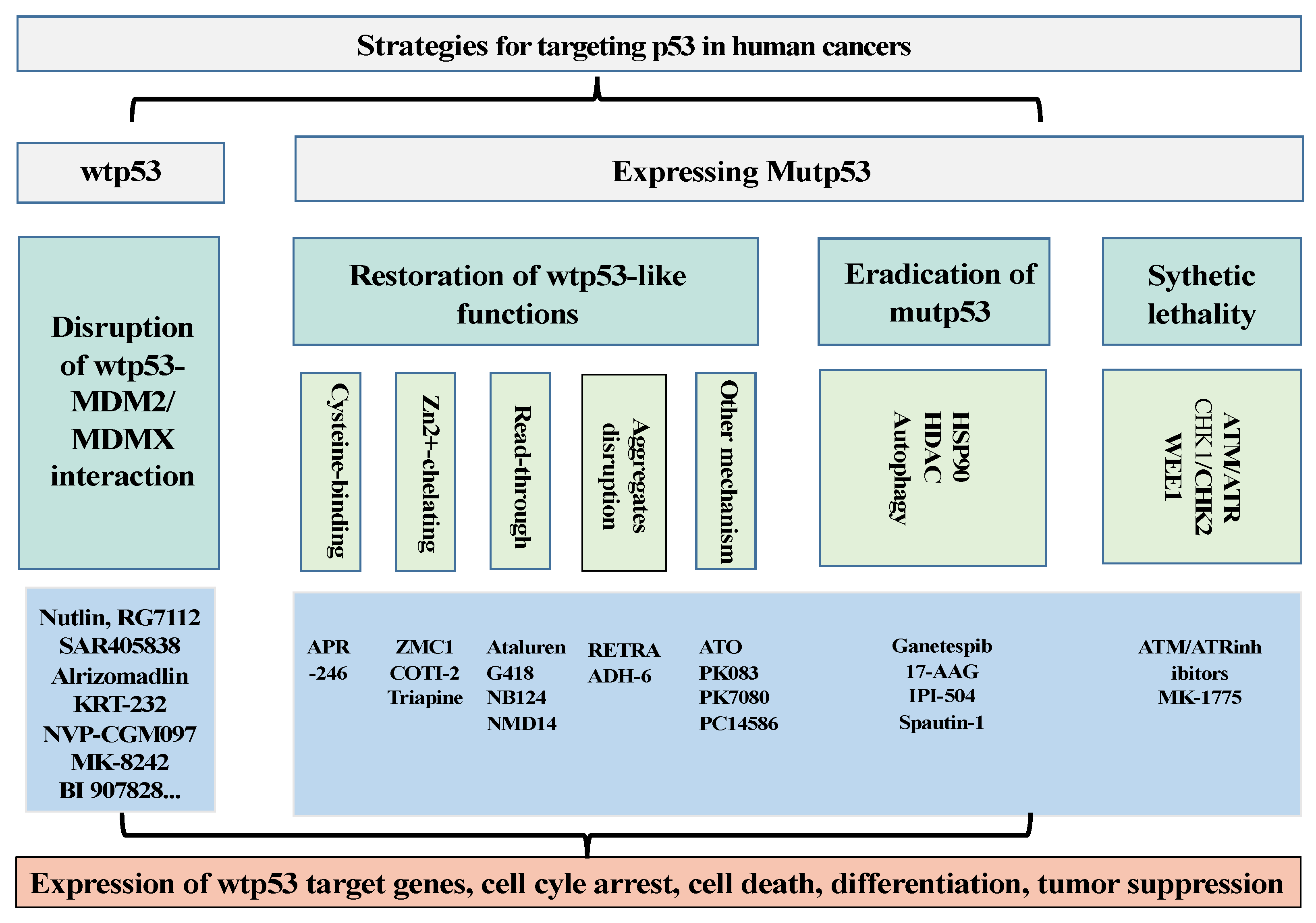
Figure 1.
Summary of the strategies to target wtp53 and mutp53 in human cancers. For activating wtp53, drug development has been focused on strategies to disrupt the interaction between p53 and its negative regulators MDM2/MDMX. For human cancers expressing mutant p53, drug development has been focused on strategies to restore wild-type p53 activities to mutp53. In addition, drugs to eradicate mutp53 and induce synthetic lethality of tumor cells expressing p53 mutants have also been explored.
Figure 1.
Summary of the strategies to target wtp53 and mutp53 in human cancers. For activating wtp53, drug development has been focused on strategies to disrupt the interaction between p53 and its negative regulators MDM2/MDMX. For human cancers expressing mutant p53, drug development has been focused on strategies to restore wild-type p53 activities to mutp53. In addition, drugs to eradicate mutp53 and induce synthetic lethality of tumor cells expressing p53 mutants have also been explored.


{kind=link}
Table 1.
Summary of clinical trials of drugs to activate wild-type activities of p53 in human cancers.
Table 1.
Summary of clinical trials of drugs to activate wild-type activities of p53 in human cancers.
| Drug Name | Combination with Other Therapy | Type of Malignancies | Clinical Trials | Clinical Trial ID |
|---|---|---|---|---|
| RG7112 | Hematologic | 1 | NCT00623870 | |
| Idasanutlin | Cytarabine | AML | 3 | NCT02545283 |
| Chemotherapy/ Venetoclax | AML Solid tumors | 2 | NCT04029688 | |
| Solid tumors | 2 | NCT04589845 | ||
| Glioblastoma | 2 | NCT03158389 | ||
| SAR405838 | Malignant neoplasm | 1 | NCT01636479 | |
| Malignant neoplasm | 1 | NCT01985191 | ||
| Milademetan | Advanced solid tumor, Lymphoma | 1 | NCT01877382 | |
| Solid tumor | 2 | NCT05012397 | ||
| Alrizomadlin | Pembrolizumab | Metastatic melanomas Advanced solid tumors | 2 | NCT03611868 |
| KRT-232 | MCC | 2 | NCT03787602 | |
| NVP-CGM097 | Solid tumors | 1 | NCT01760525 | |
| MK-8242 | Cytarabine | AML | 1 | NCT01451437 |
| Solid tumors | 1 | NCT01463696 | ||
| BI 907828 | Solid tumors | 1 | NCT03449381 | |
| Siremadlin | Advanced solid tumors Hematological neoplasm | 1 | NCT02143635 | |
| LEE011 | Solid tumors | 1 | NCT02343172 | |
| ALRN-6924 | Advanced solid tumors | 1 | NCT05622058 | |
| APR-246 | Refractory hematologic tumor Prostate cancer | 1 | NCT00900614 | |
| Carboplatin+ Pegylated Liposomal Doxorubicin Hydrochloride (PLD) | Platinum sensitive recurrent high-grade serous ovarian Cancer with p53 Mutations | 2 | NCT02098343 | |
| Myeloid Neoplasms with p53 mutations | 2 | NCT03072043 | ||
| Azacitidine | MDS with p53 mutations | 3 | NCT03745716 | |
| COTI-2 | Cisplatin | Advanced solid tumors with p53 mutations | 1 | NCT02433626 |
| Tanespimycin | Bortezomib | Multiple myeloma with p53 mutations | 3 | NCT00514371 |
| Atorvastatin Calcium | Colorectal Carcinoma Ulcerative Colitis with p53 mutations | 2 | NCT04767984 | |
| Atorvastatin | Advanced solid tumors with p53 mutations | 1 | NCT03560882 | |
| MK-1775 | Paclitaxel+ Carboplatin | Platinum-sensitive Ovarian Tumors with p53 Mutations | 2 | NCT01357161 |
| Carboplatin | Epithelial ovarian cancer with p53 Mutation | 2 | NCT01164995 | |
| Paclitaxel/Carboplatin/Gemcitabine/PLD | Ovarian, Fallopian Tube, Peritoneal cancer with p53 mutations | 2 | NCT02272790 |
AML: Acute Myeloid Leukemia; MDS, myelodysplastic syndrome.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lu, Y.; Wu, M.; Xu, Y.; Yu, L. The Development of p53-Targeted Therapies for Human Cancers. Cancers 2023, 15, 3560. https://doi.org/10.3390/cancers15143560
AMA Style
Lu Y, Wu M, Xu Y, Yu L. The Development of p53-Targeted Therapies for Human Cancers. Cancers. 2023; 15(14):3560. https://doi.org/10.3390/cancers15143560
Chicago/Turabian StyleLu, Yier, Meng Wu, Yang Xu, and Lili Yu. 2023. "The Development of p53-Targeted Therapies for Human Cancers" Cancers 15, no. 14: 3560. https://doi.org/10.3390/cancers15143560
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.