
Growth and Migration Blocking Effect of Nanaomycin K, a Compound Produced by Streptomyces sp., on Prostate Cancer Cell Lines In Vitro and In Vivo
 , ,
, ,  , , , and
, , , and 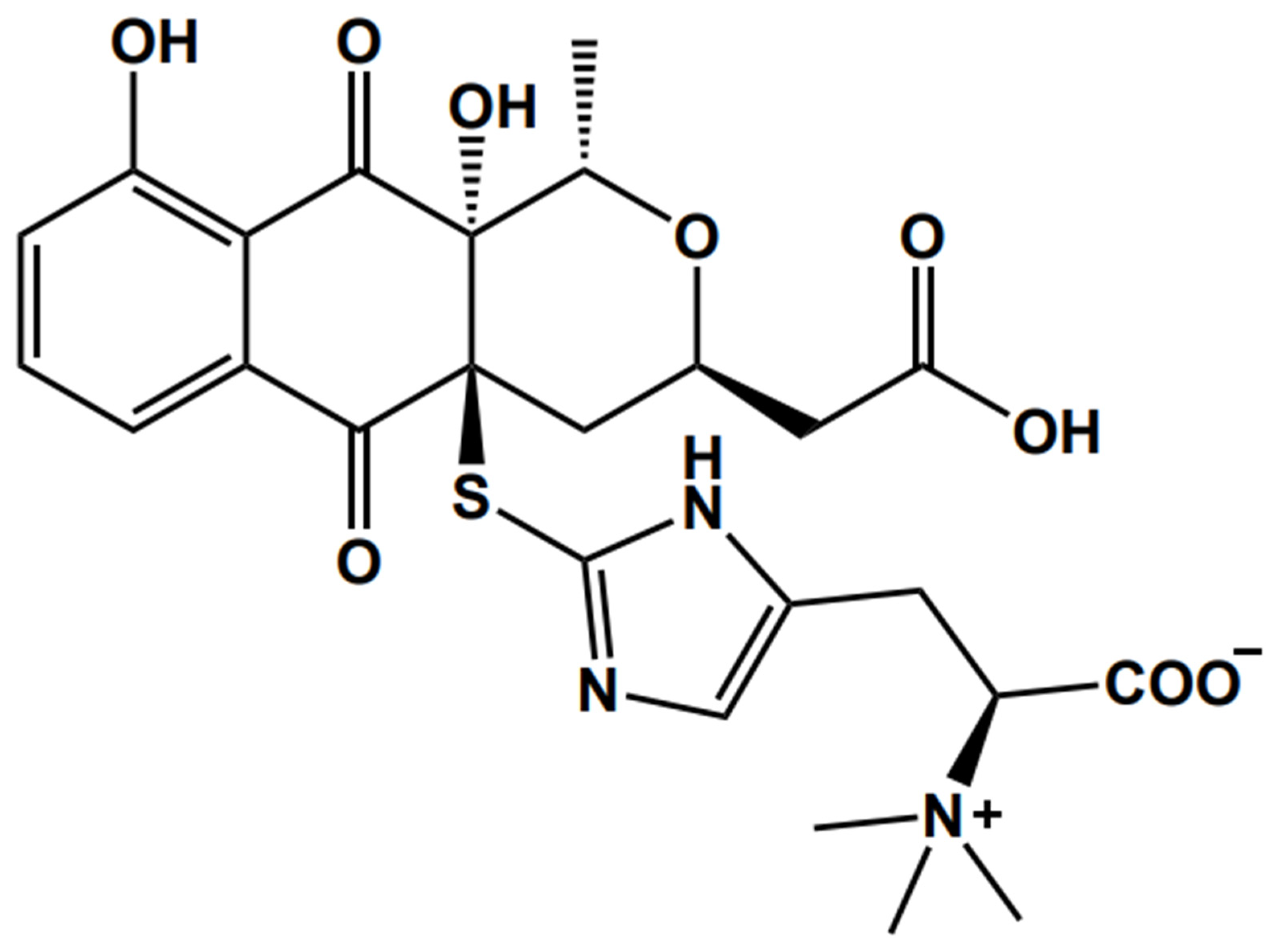{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Cell Proliferation Assays
2.3. Wound Healing Assays
2.4. Western Blotting
2.5. Animal Experiments
2.6. Immunohistochemical Staining
2.7. Immunohistochemical Analysis
2.8. Ethical Approval
2.9. Statistical Analysis
3. Results
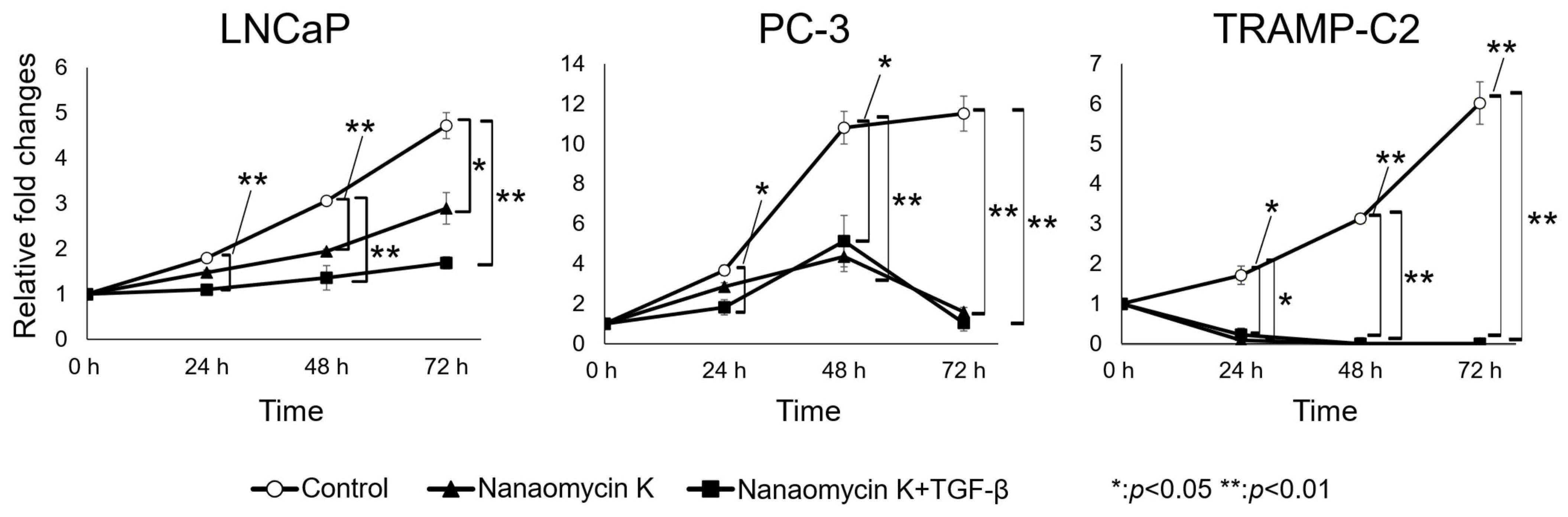
3.1. Nanaomycin K Inhibited the Growth of Prostate Cancer Cells
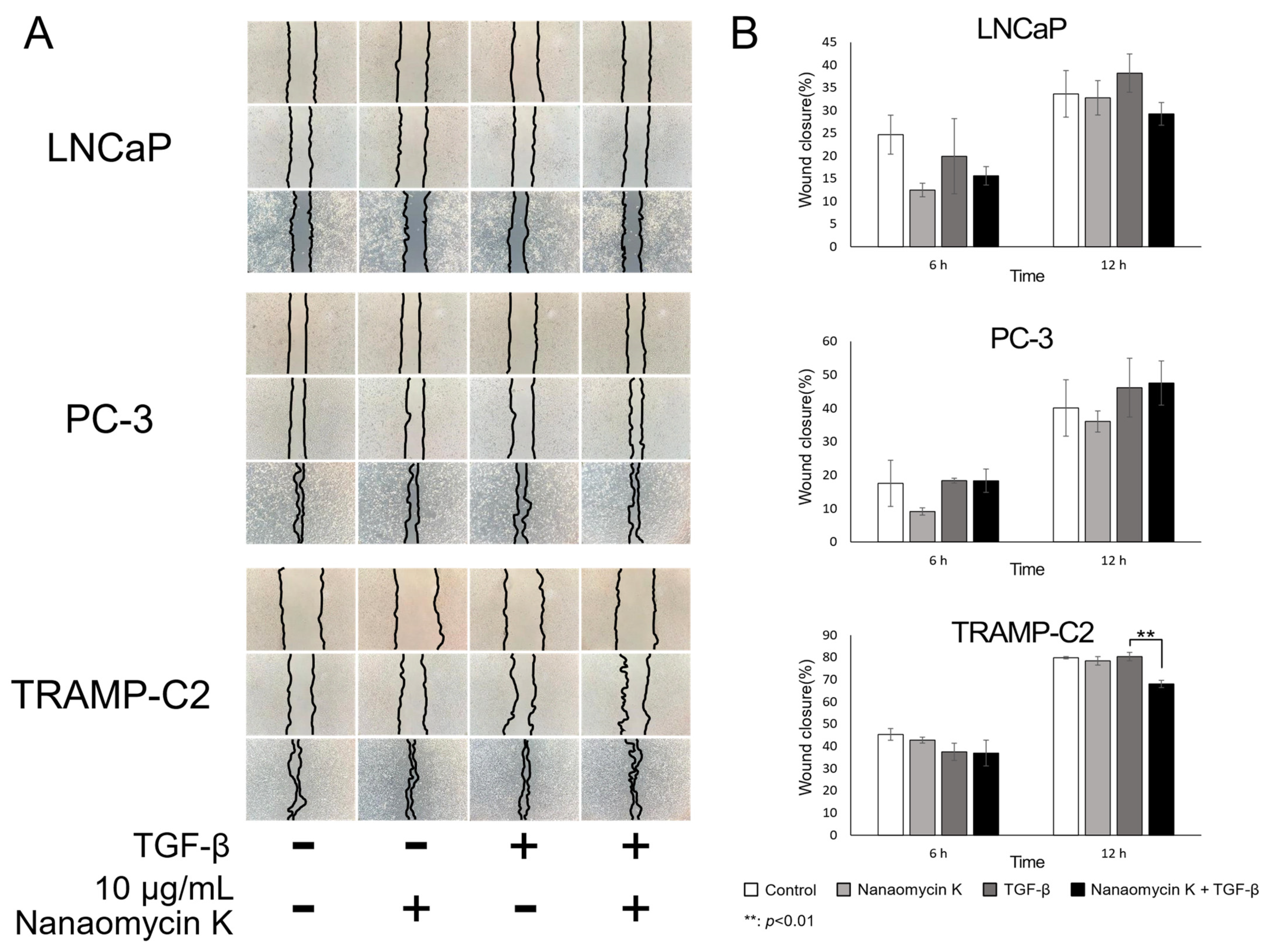
3.2. Migration-Inhibitory Effect of Nanaomycin K
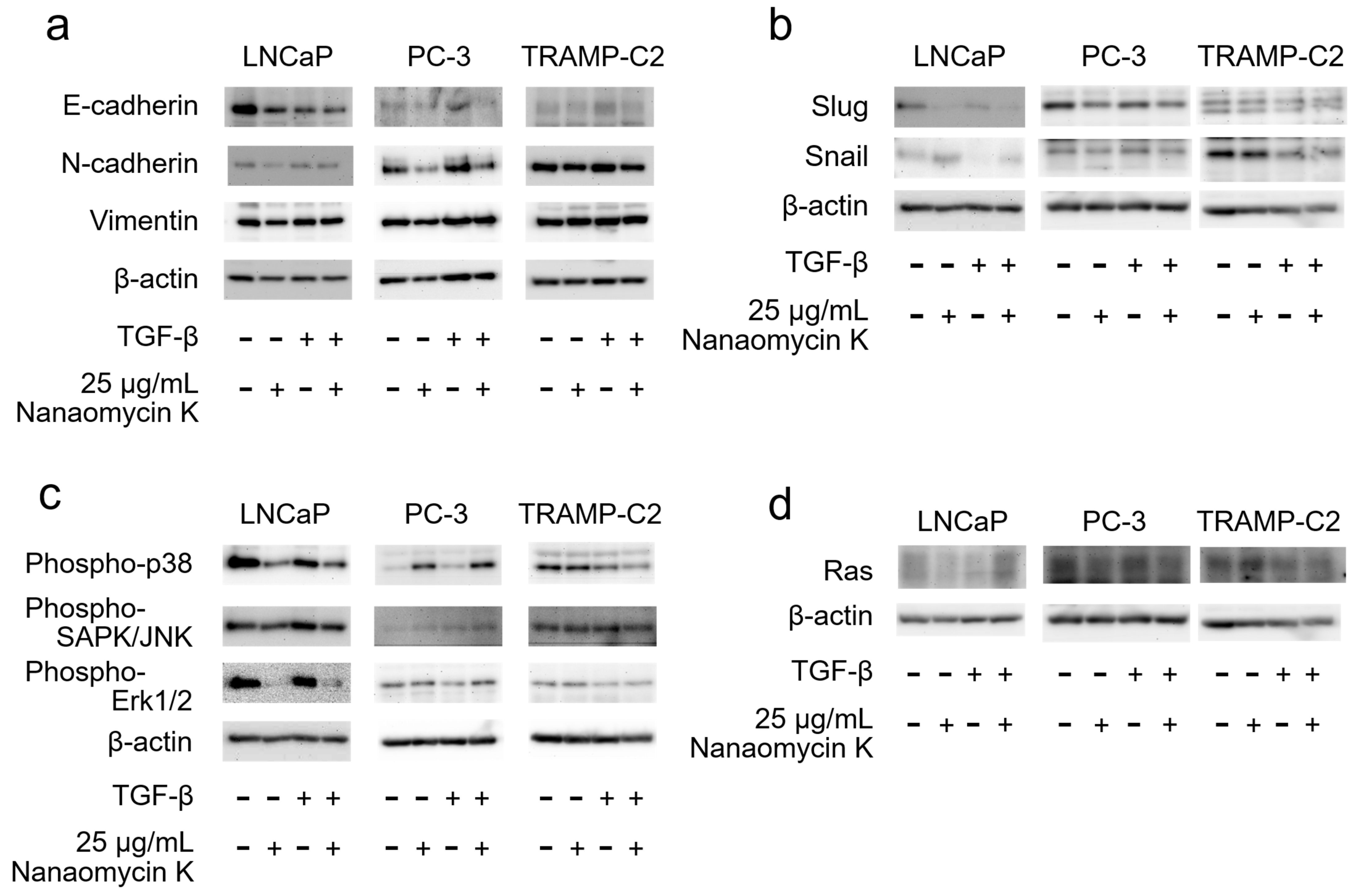
3.3. Expression of EMT-Related Protein and MAPK Signaling after Culture with Nanaomycin K
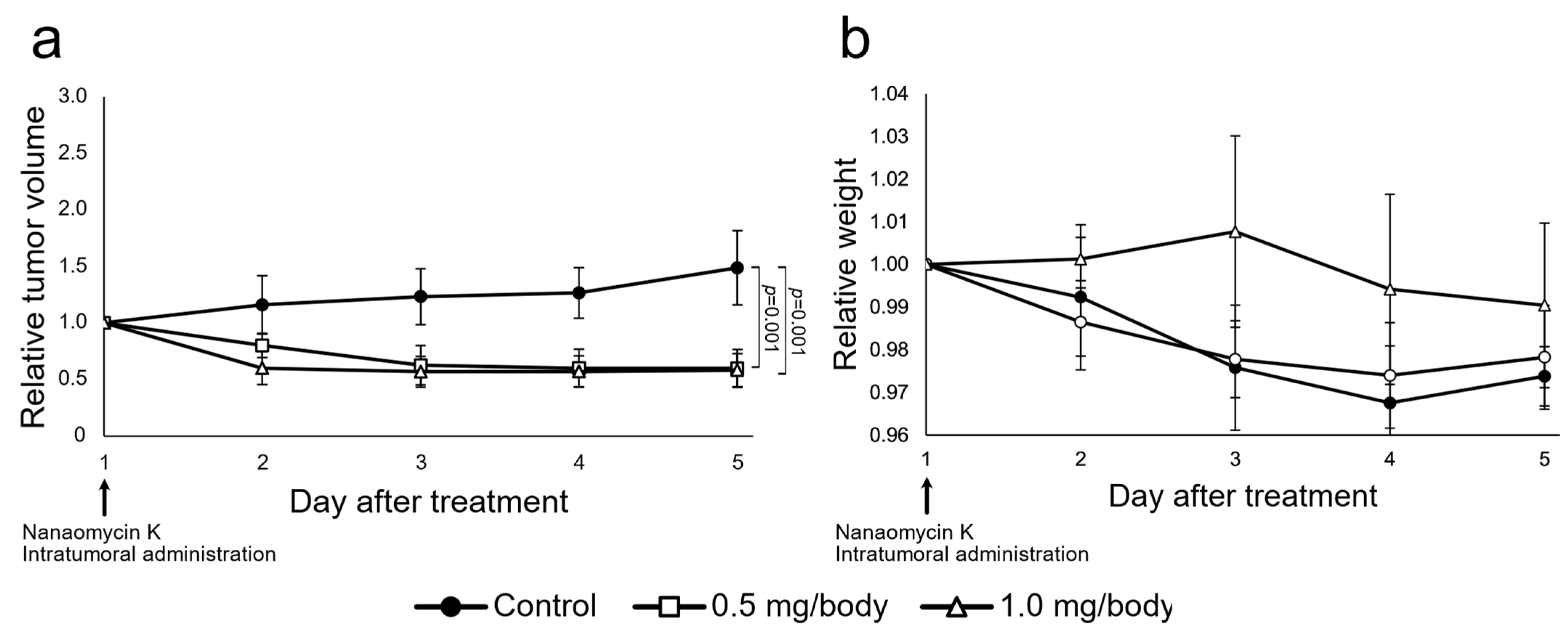
3.4. Nanaomycin K Inhibited Tumor Growth In Vivo
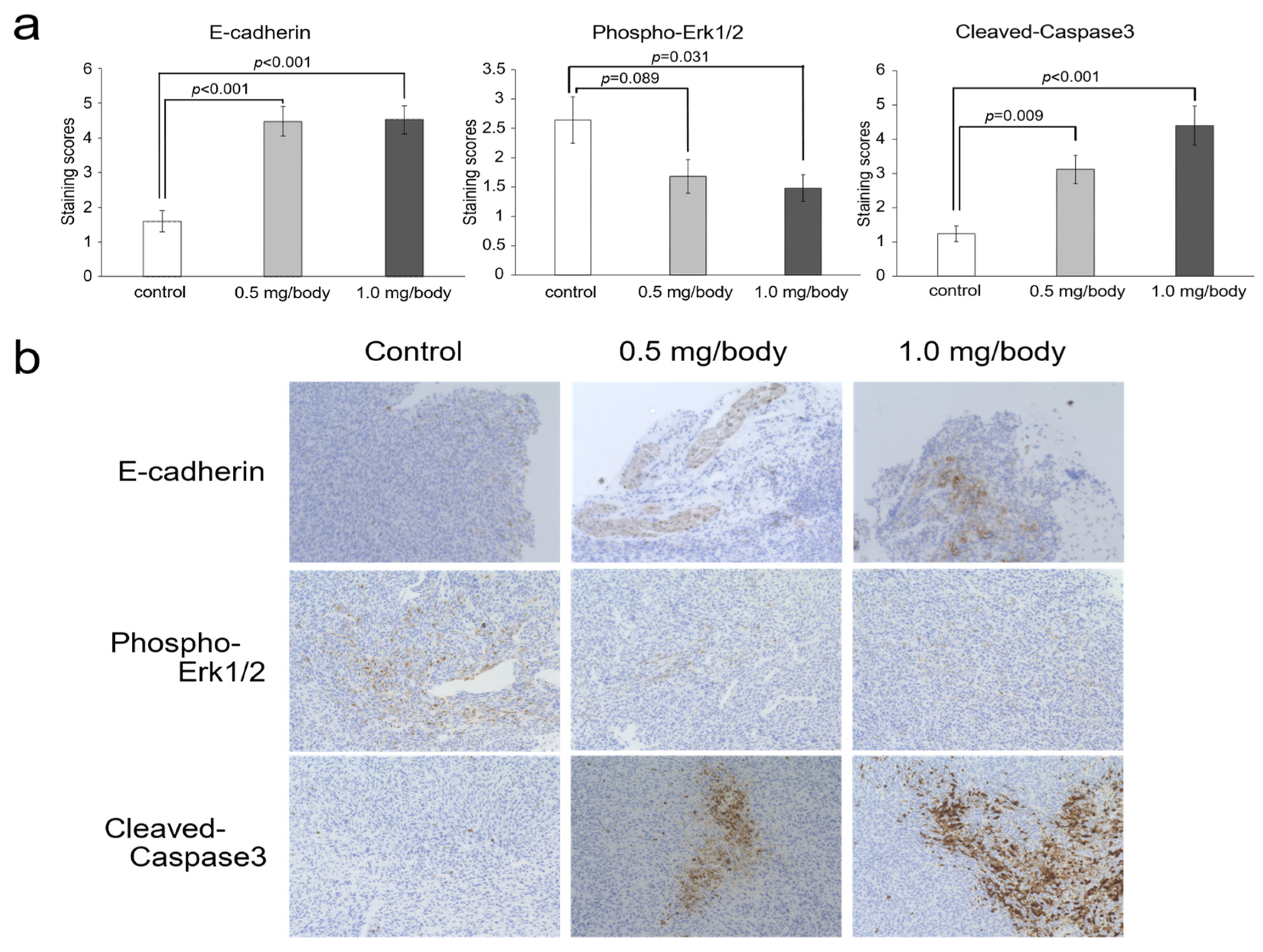
3.5. Changes of E-Cadherin, Phosho-ERK1/2 and Cleaved-Caspase-3 in Tumor Tissues after Treatment with Nanaomycin K
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.A.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 904. [Google Scholar] [CrossRef] [PubMed]
- Kaszak, I.; Witkowska-Piłaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Ngosa Toka, F.; Jurka, P. Role of Cadherins in Cancer—A Review. Int. J. Mol. Sci. 2020, 21, 7624. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting Epithelial–Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug Resistance in Metastatic Castration-Resistant Prostate Cancer. Nat. Rev. Clin. Oncol. 2011, 8, 12–23. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Sreekumar, S.; Zhou, D.; Mpoy, C.; Schenk, E.; Scott, J.; Arbeit, J.M.; Xu, J.; Rogers, B.E. Preclinical Efficacy of a PARP-1 Targeted Auger-Emitting Radionuclide in Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 3083. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523. [Google Scholar] [CrossRef]
- Omura, S.; Tanaka, H.; Koyama, Y.; Oiwa, R.; Katagiri, M.; Awaya, J.; Nagai, T.; Hata, T. Nanaomycins A and B*, New Antibiotics Produced by a Strain of Streptomyces. J. Antibiot. 1974, 27, 363–365. [Google Scholar] [CrossRef]
- Tanaka, H.; Marumo, H.; Nagai, T.; Okada, M.; Taniguchi, K. Nanaomycins, New Antibiotics Produced by a Strain of Streptomyces. III. A New Component, Nanaomycin C, and Biological Activities of Nanaomycin Derivatives. J. Antibiot. 1975, 28, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Kasai, M.; Shirahata, K.; Ishii, S.; Mineura, K.; Marumo, H.; Tanaka, H.; Omura, S. Structure of Nanaomycin E, a New Nanaomycin. J. Antibiot. 1979, 32, 442–445. [Google Scholar] [CrossRef]
- Matsuo, H.; Nakanishi, J.; Noguchi, Y.; Kitagawa, K.; Shigemura, K.; Sunazuka, T.; Takahashi, Y.; Ōmura, S.; Nakashima, T. Nanaomycin K, a New Epithelial–Mesenchymal Transition Inhibitor Produced by the Actinomycete “Streptomyces Rosa Subsp. Notoensis” OS-3966. J. Biosci. Bioeng. 2020, 129, 291–295. [Google Scholar] [CrossRef]
- Kitagawa, K.; Shigemura, K.; Ishii, A.; Nakashima, T.; Matsuo, H.; Takahashi, Y.; Omura, S.; Nakanishi, J.; Fujisawa, M. Nanaomycin K Inhibited Epithelial Mesenchymal Transition and Tumor Growth in Bladder Cancer Cells in Vitro and in Vivo. Sci. Rep. 2021, 11, 9217. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Shigemura, K.; Sung, S.-Y.; Chen, K.-C.; Huang, C.-C.; Chiang, Y.-T.; Liu, M.-C.; Huang, T.-W.; Yamamichi, F.; Shirakawa, T.; et al. Possible Correlation of Sonic Hedgehog Signaling with Epithelial–Mesenchymal Transition in Muscle-Invasive Bladder Cancer Progression. J. Cancer Res. Clin. Oncol. 2019, 145, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Kawata, M.; Koinuma, D.; Ogami, T.; Umezawa, K.; Iwata, C.; Watabe, T.; Miyazono, K. TGF-β-Induced Epithelial-Mesenchymal Transition of A549 Lung Adenocarcinoma Cells Is Enhanced by pro-Inflammatory Cytokines Derived from RAW 264.7 Macrophage Cells. J. Biochem. 2012, 151, 205–216. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Sun, D.-Y.; Wu, J.-Q.; He, Z.-H.; He, M.-F.; Sun, H.-B. Cancer-Associated Fibroblast Regulate Proliferation and Migration of Prostate Cancer Cells through TGF-β Signaling Pathway. Life Sci. 2019, 235, 116791. [Google Scholar] [CrossRef]
- Gilbert, S.; Péant, B.; Mes-Masson, A.-M.; Saad, F. IKKε Inhibitor Amlexanox Promotes Olaparib Sensitivity through the C/EBP-β-Mediated Transcription of Rad51 in Castrate-Resistant Prostate Cancer. Cancers 2022, 14, 3684. [Google Scholar] [CrossRef]
- Takahashi, K.; Akatsu, Y.; Podyma-Inoue, K.A.; Matsumoto, T.; Takahashi, H.; Yoshimatsu, Y.; Koinuma, D.; Shirouzu, M.; Miyazono, K.; Watabe, T. Targeting All Transforming Growth Factor-β Isoforms with an Fc Chimeric Receptor Impairs Tumor Growth and Angiogenesis of Oral Squamous Cell Cancer. J. Biol. Chem. 2020, 295, 12559–12572. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Osmulski, P.A.; Bouamar, H.; Mahalingam, D.; Lin, C.-L.; Liss, M.A.; Kumar, A.P.; Chen, C.-L.; Thompson, I.M.; Sun, L.-Z.; et al. TGF-β Signal Rewiring Sustains Epithelial-Mesenchymal Transition of Circulating Tumor Cells in Prostate Cancer Xenograft Hosts. Oncotarget 2016, 7, 77124–77137. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ren, D.; Guo, W.; Huang, S.; Wang, Z.; Li, Q.; Du, H.; Song, L.; Peng, X. N-Cadherin Promotes Epithelial-Mesenchymal Transition and Cancer Stem Cell-like Traits via ErbB Signaling in Prostate Cancer Cells. Int. J. Oncol. 2016, 48, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin as a Potential Molecular Target in Cancer Therapy Or Vimentin, an Overview and Its Potential as a Molecular Target for Cancer Therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Abou-Kheir, W.; Yin, J.J.; Fang, L.; Hynes, P.; Casey, O.; Hu, D.; Wan, Y.; Seng, V.; Sheppard-Tillman, H.; et al. Critical and Reciprocal Regulation of KLF4 and SLUG in Transforming Growth Factor β-Initiated Prostate Cancer Epithelial-Mesenchymal Transition. Mol. Cell. Biol. 2012, 32, 941–953. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Dijke, P. ten Signaling Interplay between Transforming Growth Factor-β Receptor and PI3K/AKT Pathways in Cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-MTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, G.; Getzenberg, R.H.; Veltri, R.W. The Upregulation of PI3K/Akt and MAP Kinase Pathways Is Associated with Resistance of Microtubule-Targeting Drugs in Prostate Cancer. J. Cell. Biochem. 2015, 116, 1341–1349. [Google Scholar] [CrossRef]
- Park, S.; Kwon, W.; Park, J.-K.; Baek, S.-M.; Lee, S.-W.; Cho, G.-J.; Ha, Y.-S.; Lee, J.N.; Kwon, T.G.; Kim, M.O.; et al. Suppression of Cathepsin a Inhibits Growth, Migration, and Invasion by Inhibiting the P38 MAPK Signaling Pathway in Prostate Cancer. Arch. Biochem. Biophys. 2020, 688, 108407. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, B.G.; Jerde, T.J.; Hollenhorst, P.C. Ras/ERK and PI3K/AKT Signaling Differentially Regulate Oncogenic ERG Mediated Transcription in Prostate Cells. PLoS Genet. 2021, 17, e1009708. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-R.; Mokgautsi, N.; Liu, Y.-N. Ras and Wnt Interaction Contribute in Prostate Cancer Bone Metastasis. Molecules 2020, 25, 2380. [Google Scholar] [CrossRef]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK Signaling Is a Therapeutic Target in Metastatic Castration Resistant Prostate Cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef]
- Ismy, J.; Sugandi, S.; Rachmadi, D.; Hardjowijoto, S.; Mustafa, A. The Effect of Exogenous Superoxide Dismutase (SOD) on Caspase-3 Activation and Apoptosis Induction in Pc-3 Prostate Cancer Cells. Res. Rep. Urol. 2020, 12, 503–508. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Boran, S.A.; O’Keane, C.; Coffey, R.N.T.; Hegarty, N.J.; Hegarty, P.; Gaffney, E.F.; Fitzpatrick, J.M.; Watson, R.W.G. Caspase 3 Expression in Benign Prostatic Hyperplasia and Prostate Carcinoma. Prostate 2001, 47, 183–188. [Google Scholar] [CrossRef]
- Rizzo, M. Mechanisms of Docetaxel Resistance in Prostate Cancer: The Key Role Played by MiRNAs. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2021, 1875, 188481. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, Cancer Stem Cells and Drug Resistance: An Emerging Axis of Evil in the War on Cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The Role of Epithelial–Mesenchymal Transition Drivers ZEB1 and ZEB2 in Mediating Docetaxel-resistant Prostate Cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef]
- Ren, J.; Chen, Y.; Song, H.; Chen, L.; Wang, R. Inhibition of ZEB1 Reverses EMT and Chemoresistance in Docetaxel-Resistant Human Lung Adenocarcinoma Cell Line. J. Cell. Biochem. 2013, 114, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Kunimura, N.; Kitagawa, K.; Fukui, Y.; Saito, H.; Narikiyo, K.; Ishiko, M.; Otsuki, N.; Nibu, K.; Fujisawa, M.; et al. Overexpression of SOCS3 Mediated by Adenovirus Vector in Mouse and Human Castration-Resistant Prostate Cancer Cells Increases the Sensitivity to NK Cells in Vitro and in Vivo. Cancer Gene 2019, 26, 388–399. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirata, Y.; Shigemura, K.; Moriwaki, M.; Iwatsuki, M.; Kan, Y.; Ooya, T.; Maeda, K.; Yang, Y.; Nakashima, T.; Matsuo, H.; et al. Growth and Migration Blocking Effect of Nanaomycin K, a Compound Produced by Streptomyces sp., on Prostate Cancer Cell Lines In Vitro and In Vivo. Cancers 2023, 15, 2684. https://doi.org/10.3390/cancers15102684
Hirata Y, Shigemura K, Moriwaki M, Iwatsuki M, Kan Y, Ooya T, Maeda K, Yang Y, Nakashima T, Matsuo H, et al. Growth and Migration Blocking Effect of Nanaomycin K, a Compound Produced by Streptomyces sp., on Prostate Cancer Cell Lines In Vitro and In Vivo. Cancers. 2023; 15(10):2684. https://doi.org/10.3390/cancers15102684
Chicago/Turabian StyleHirata, Yuto, Katsumi Shigemura, Michika Moriwaki, Masato Iwatsuki, Yuki Kan, Tooru Ooya, Koki Maeda, Youngmin Yang, Takuji Nakashima, Hirotaka Matsuo, and et al. 2023. "Growth and Migration Blocking Effect of Nanaomycin K, a Compound Produced by Streptomyces sp., on Prostate Cancer Cell Lines In Vitro and In Vivo" Cancers 15, no. 10: 2684. https://doi.org/10.3390/cancers15102684