
Involvement of Epithelial–Mesenchymal Transition Genes in Small Cell Lung Cancer Phenotypic Plasticity
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
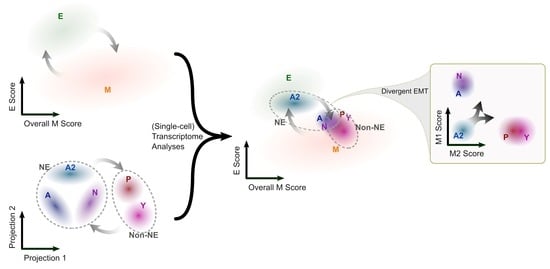
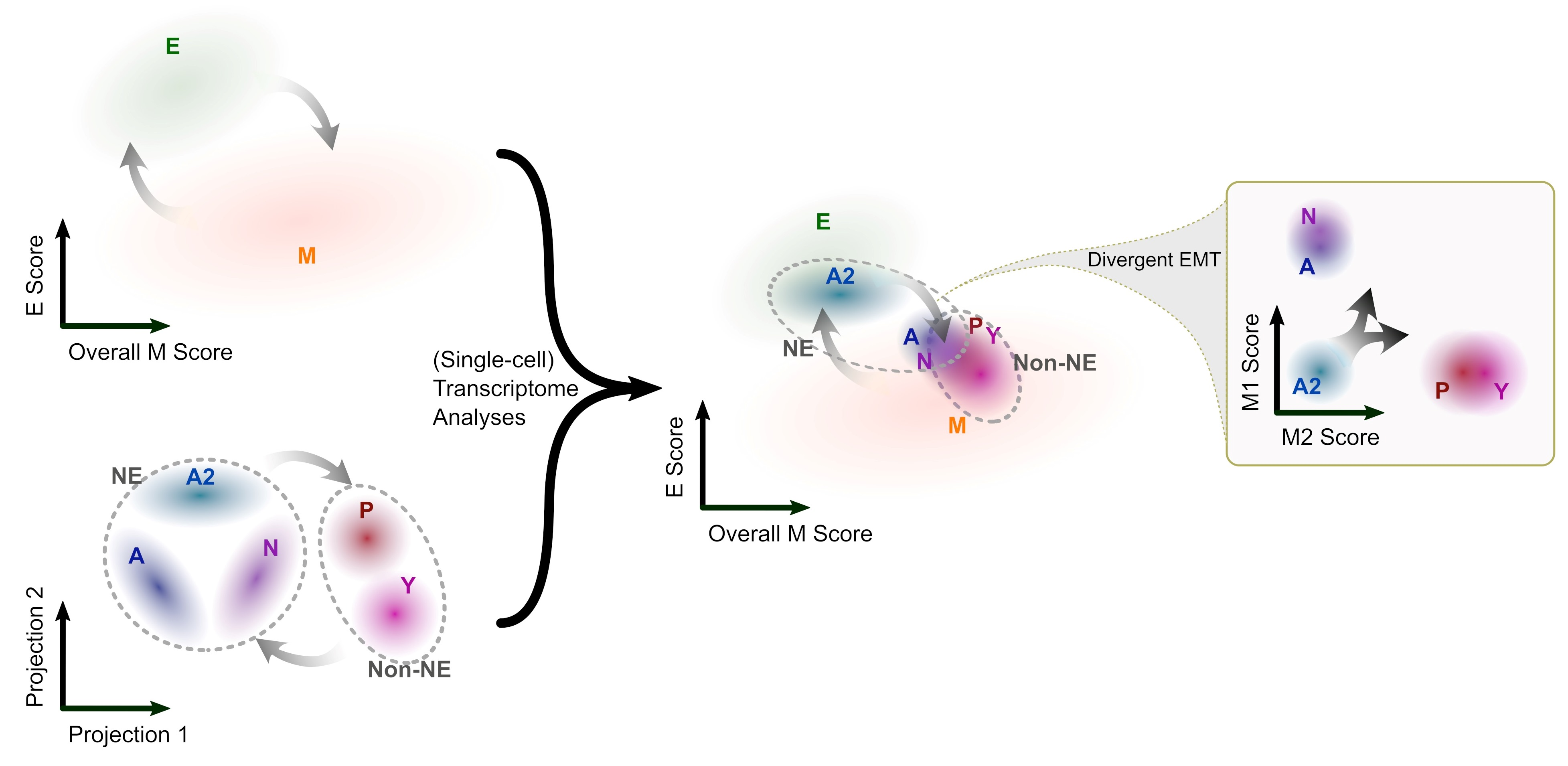{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. RNA-Sequencing Data
2.2. Subtyping of SCLC Cells
- (1)
- Find the mean bulk score x for a specialist, m.
- (2)
- Choose a random sample of size na, where na is the number of specialists, with replacement from the remaining cells (i.e., cells that are not specialists, including generalists and other specialist cells). Find the mean bulk score for this sample. N.B. Because some time points have very few cells, we sample evenly from each time point to ensure adequate representation across the time points.
- (3)
- Repeat this random selection 1000 times.
- (4)
- Generate a p-value, which is equal to the percentage of means from this random distribution above m.
- (5)
- Using statsmodels.states.multitest, correct p-values for multiple tests. We used the Bonferroni–Holm method to control the family-wise error rate. Consider q < 0.1 significant.
2.3. Single Sample Gene Set Enrichment Analysis (ssGSEA)
2.4. Non-Negative Principal Component Analysis (nnPCA)
3. Results
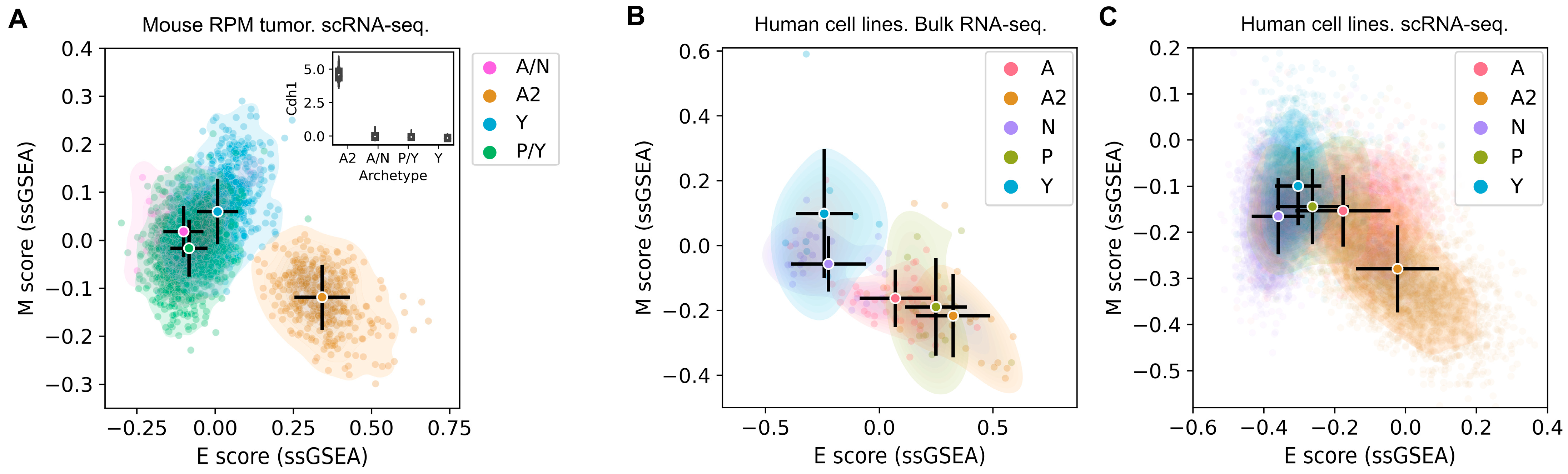
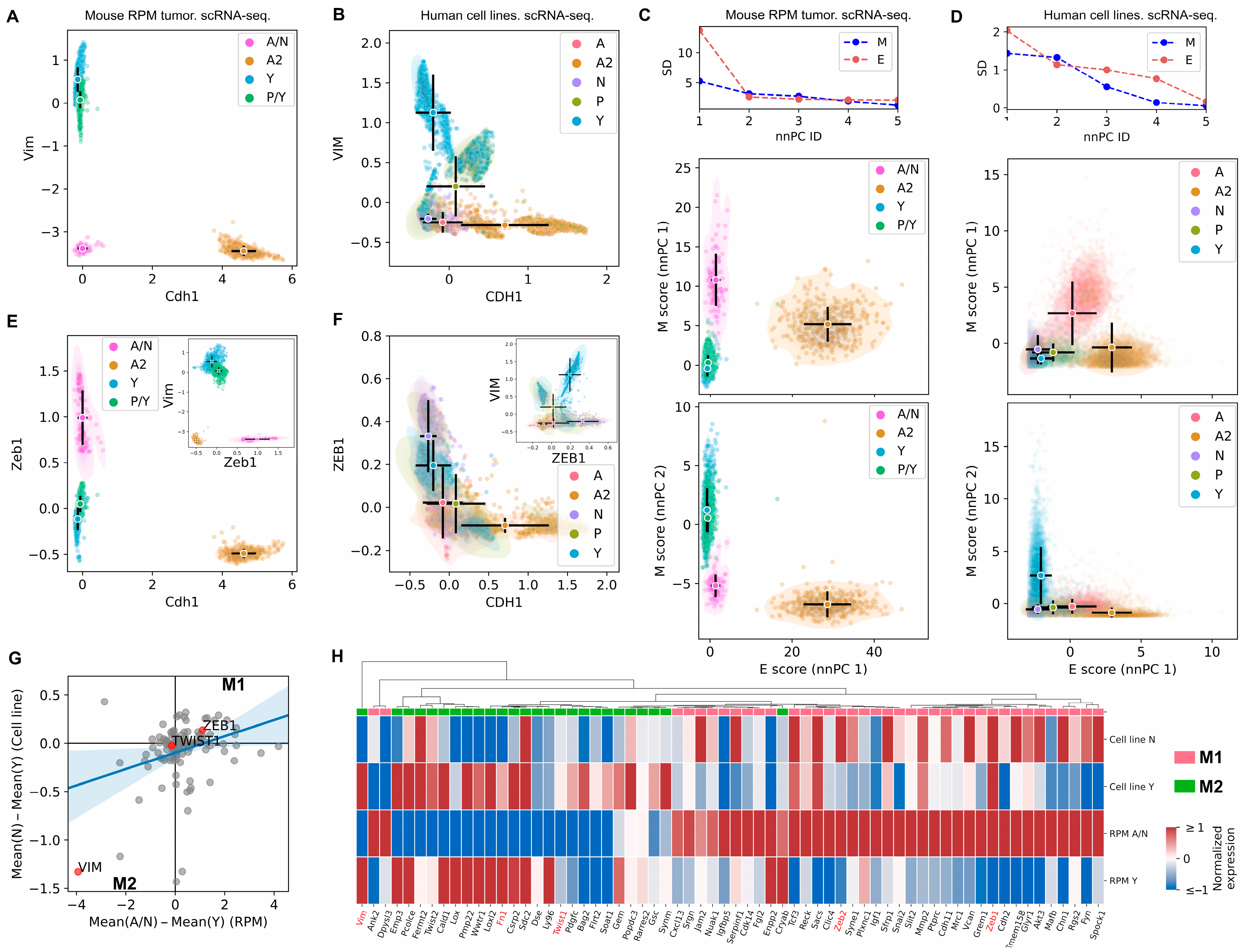
3.1. The SCLC-A2 Subtype Is Highly Enriched in Epithelial Gene Expression
3.2. Mesenchymal Scoring Is Diverse across Non-A2 Subtypes
3.3. The Highly Epithelial A2 Subtype and Diverse M Gene Expression Patterns Are Detectable in Human SCLC Tumor
3.4. Intratumor Heterogeneity of SCLC Indicates Strong A2–Epithelial Association at Single-Cell Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- Borges, M.; Linnoila, R.I.; Van De Velde, H.J.K.; Chen, H.; Nelkin, B.D.; Mabry, M.; Baylin, S.B.; Ball, D.W. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 1997, 386, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Yao, E.; Lin, C.; Gacayan, R.; Chen, M.-H.; Chuang, P.-T. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 17531–17536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calbo, J.; van Montfort, E.; Proost, N.; van Drunen, E.; Beverloo, H.B.; Meuwissen, R.; Berns, A. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011, 19, 244–256. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Shue, Y.T.; Lim, J.S.; Sage, J. Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl. Lung Cancer Res. 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, D.J.; Groves, S.M.; Tyson, D.R.; Liu, Q.; Lim, J.S.; Albert, R.; Lopez, C.F.; Sage, J.; Quaranta, V. Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. PLoS Comput. Biol. 2019, 15, e1007343. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360. [Google Scholar] [CrossRef]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell 2020, 38, 60–78. [Google Scholar] [CrossRef]
- Ko, J.; Winslow, M.M.; Sage, J. Mechanisms of small cell lung cancer metastasis. EMBO Mol. Med. 2021, 13, e13122. [Google Scholar] [CrossRef]
- Kwon, M.-c.; Proost, N.; Song, J.-Y.; Sutherland, K.D.; Zevenhoven, J.; Berns, A. Paracrine signaling between tumor subclones of mouse SCLC: A critical role of ETS transcription factor Pea3 in facilitating metastasis. Genes Dev. 2015, 29, 1587–1592. [Google Scholar] [CrossRef]
- Udyavar, A.R.; Wooten, D.J.; Hoeksema, M.; Bansal, M.; Califano, A.; Estrada, L.; Schnell, S.; Irish, J.M.; Massion, P.P.; Quaranta, V. Novel Hybrid Phenotype Revealed in Small Cell Lung Cancer by a Transcription Factor Network Model That Can Explain Tumor HeterogeneityHybrid Phenotype in SCLC. Cancer Res. 2017, 77, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Yazawa, T. Recent advances in histogenesis research of lung neuroendocrine cancers: Evidence obtained from functional analyses of primitive neural/neuroendocrine cell-specific transcription factors. Pathol. Int. 2015, 65, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Groves, S.M.; Ildefonso, G.V.; McAtee, C.O.; Ozawa, P.M.M.; Ireland, A.S.; Stauffer, P.E.; Wasdin, P.T.; Huang, X.; Qiao, Y.; Lim, J.S.; et al. Archetype tasks link intratumoral heterogeneity to plasticity and cancer hallmarks in small cell lung cancer. Cell Syst. 2022, 13, 690–710.e617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Girard, L.; Zhang, Y.-A.; Haruki, T.; Papari-Zareei, M.; Stastny, V.; Ghayee, H.K.; Pacak, K.; Oliver, T.G.; Minna, J.D. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl. Lung Cancer Res. 2018, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Guo, J.; Liu, Y.; Zheng, Q.; Li, X.; Wu, C.; Fang, D.; Chen, X.; Ma, L.; Xu, P. YAP drives fate conversion and chemoresistance of small cell lung cancer. Sci. Adv. 2021, 7, eabg1850. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Furukawa, K.T.; Morimoto, M. Pulmonary neuroendocrine cells: Physiology, tissue homeostasis and disease. Dis. Model. Mech. 2020, 13, dmm046920. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.a.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, K.D.; Ireland, A.S.; Oliver, T.G. Killing SCLC: Insights into how to target a shapeshifting tumor. Genes Dev. 2022, 36, 241–258. [Google Scholar] [CrossRef]
- Watanabe, K.; Panchy, N.; Noguchi, S.; Suzuki, H.; Hong, T. Combinatorial perturbation analysis reveals divergent regulations of mesenchymal genes during epithelial-to-mesenchymal transition. Npj Syst. Biol. Appl. 2019, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-β -induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7, ra91. [Google Scholar] [CrossRef]
- Goetz, H.; Melendez-Alvarez, J.R.; Chen, L.; Tian, X.-J. A plausible accelerating function of intermediate states in cancer metastasis. PLoS Comput. Biol. 2020, 16, e1007682. [Google Scholar] [CrossRef]
- Tian, X.-J.; Zhang, H.; Xing, J. Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Nordick, B.; Park, M.C.-Y.; Quaranta, V.; Hong, T. Cooperative RNA degradation stabilizes intermediate epithelial-mesenchymal states and supports phenotypic continuum. iScience 2022, 25, 105224. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.M.; Quintanal-Villalonga, Á.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021, 39, 1479–1496. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D. Recovering gene interactions from single-cell data using data diffusion. Cell 2018, 174, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Hart, Y.; Sheftel, H.; Hausser, J.; Szekely, P.; Ben-Moshe, N.B.; Korem, Y.; Tendler, A.; Mayo, A.E.; Alon, U. Inferring biological tasks using Pareto analysis of high-dimensional data. Nat. Methods 2015, 12, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef] [PubMed]
- Krug, K.; Mertins, P.; Zhang, B.; Hornbeck, P.; Raju, R.; Ahmad, R.; Szucs, M.; Mundt, F.; Forestier, D.; Jane-Valbuena, J. A Curated Resource for Phosphosite-specific Signature Analysis. Mol. Cell. Proteom. 2019, 18, 576–593. [Google Scholar] [CrossRef] [Green Version]
- Panchy, N.; Watanabe, K.; Takahashi, M.; Willems, A.; Hong, T. Comparative single-cell transcriptomes of dose and time dependent epithelial-mesenchymal spectrums. NAR Genom. Bioinform. 2022, 4, lqac072. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Panchy, N.; Watanabe, K.; Hong, T. Interpretable, Scalable, and Transferrable Functional Projection of Large-Scale Transcriptome Data Using Constrained Matrix Decomposition. Front. Genet. 2021, 12, 1555. [Google Scholar] [CrossRef] [PubMed]
- Sigg, C.D.; Buhmann, J.M. Expectation-maximization for sparse and non-negative PCA. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008; pp. 960–967. [Google Scholar]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Zur Hausen, A. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.-I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- Altschuler, S.J.; Wu, L.F. Cellular heterogeneity: Do differences make a difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáez-Ayala, M.; Montenegro, M.F.; Sánchez-del-Campo, L.; Fernández-Pérez, M.P.; Chazarra, S.; Freter, R.; Middleton, M.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Goding, C.R. Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell 2013, 24, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431. [Google Scholar] [CrossRef] [Green Version]
- Stovold, R.; Meredith, S.L.; Bryant, J.L.; Babur, M.; Williams, K.J.; Dean, E.J.; Dive, C.; Blackhall, F.H.; White, A. Neuroendocrine and epithelial phenotypes in small-cell lung cancer: Implications for metastasis and survival in patients. Br. J. Cancer 2013, 108, 1704–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kudoh, S.; Ichimura, T.; Fujino, K.; Hassan, W.A.M.A.; Udaka, N. Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: Significance of inactive Notch signaling and expression of achaete-scute complex homologue 1. Hum. Cell 2017, 30, 1–10. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Bastian, C.; Liu, D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef] [Green Version]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Villarreal-Ponce, A.; Gutierrez Jr, G.; Nguyen, Q.; Sun, P.; Wu, T.; Sui, B.; Berx, G.; Brabletz, T.; Kessenbrock, K. Coordinate control of basal epithelial cell fate and stem cell maintenance by core EMT transcription factor Zeb1. Cell Rep. 2022, 38, 110240. [Google Scholar] [CrossRef]
- Cursons, J.; Pillman, K.A.; Scheer, K.G.; Gregory, P.A.; Foroutan, M.; Hediyeh-Zadeh, S.; Toubia, J.; Crampin, E.J.; Goodall, G.J.; Bracken, C.P.; et al. Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst. 2018, 7, 77–91.e77. [Google Scholar] [CrossRef]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordick, B.; Yu, P.Y.; Liao, G.; Hong, T. Nonmodular oscillator and switch based on RNA decay drive regeneration of multimodal gene expression. Nucleic Acids Res. 2022, 50, 3693. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groves, S.M.; Panchy, N.; Tyson, D.R.; Harris, L.A.; Quaranta, V.; Hong, T. Involvement of Epithelial–Mesenchymal Transition Genes in Small Cell Lung Cancer Phenotypic Plasticity. Cancers 2023, 15, 1477. https://doi.org/10.3390/cancers15051477
Groves SM, Panchy N, Tyson DR, Harris LA, Quaranta V, Hong T. Involvement of Epithelial–Mesenchymal Transition Genes in Small Cell Lung Cancer Phenotypic Plasticity. Cancers. 2023; 15(5):1477. https://doi.org/10.3390/cancers15051477
Chicago/Turabian StyleGroves, Sarah M., Nicholas Panchy, Darren R. Tyson, Leonard A. Harris, Vito Quaranta, and Tian Hong. 2023. "Involvement of Epithelial–Mesenchymal Transition Genes in Small Cell Lung Cancer Phenotypic Plasticity" Cancers 15, no. 5: 1477. https://doi.org/10.3390/cancers15051477