
Voltage-Gated Sodium Channel NaV1.5 Controls NHE−1−Dependent Invasive Properties in Colon Cancer Cells

 , , , ,
, , , ,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
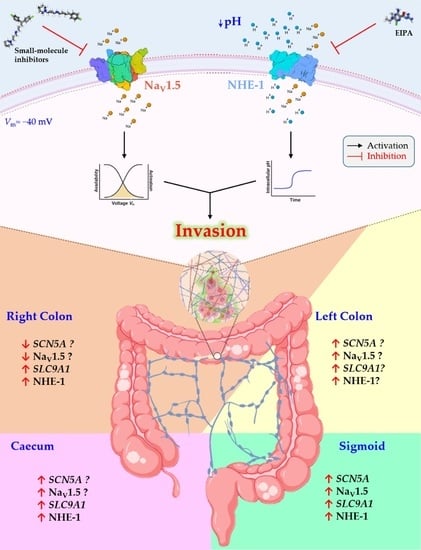
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Human Biopsies
2.3. RNA Extraction, Reverse Transcription and Real-Time PCR
2.4. Colorectal Cancer Gene Array and RNA Sequencing Datasets
2.5. Culture of Cell Lines
2.6. Transfection of Small-Interfering RNA
2.7. Preparation of Colon Tumor-Derived Primary Cell Cultures
2.8. Cellular Electrophysiology
2.9. Epifluorescence Microscopy
2.10. Measurement of Intracellular pH
2.11. Two-Dimension Cancer Cell Invasion Assays
2.12. Cell Viability
2.13. Chemical, Antibodies and Small-Molecule NaV1.5 Inhibitor Compounds
2.14. Statistical Analysis
3. Results
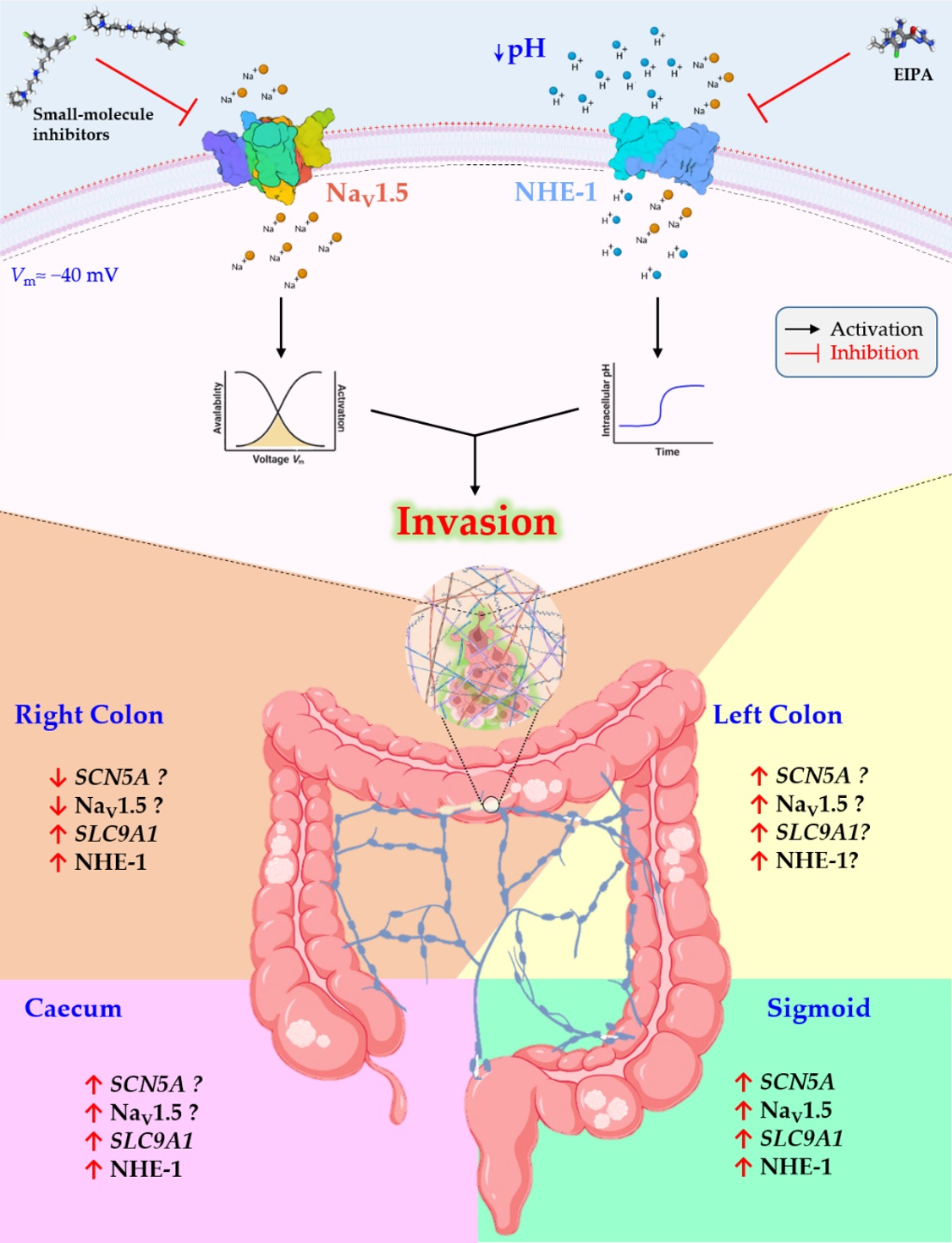
3.1. Stage- and Anatomy-Dependent Expression of SCN5A Gene in Colon Cancer
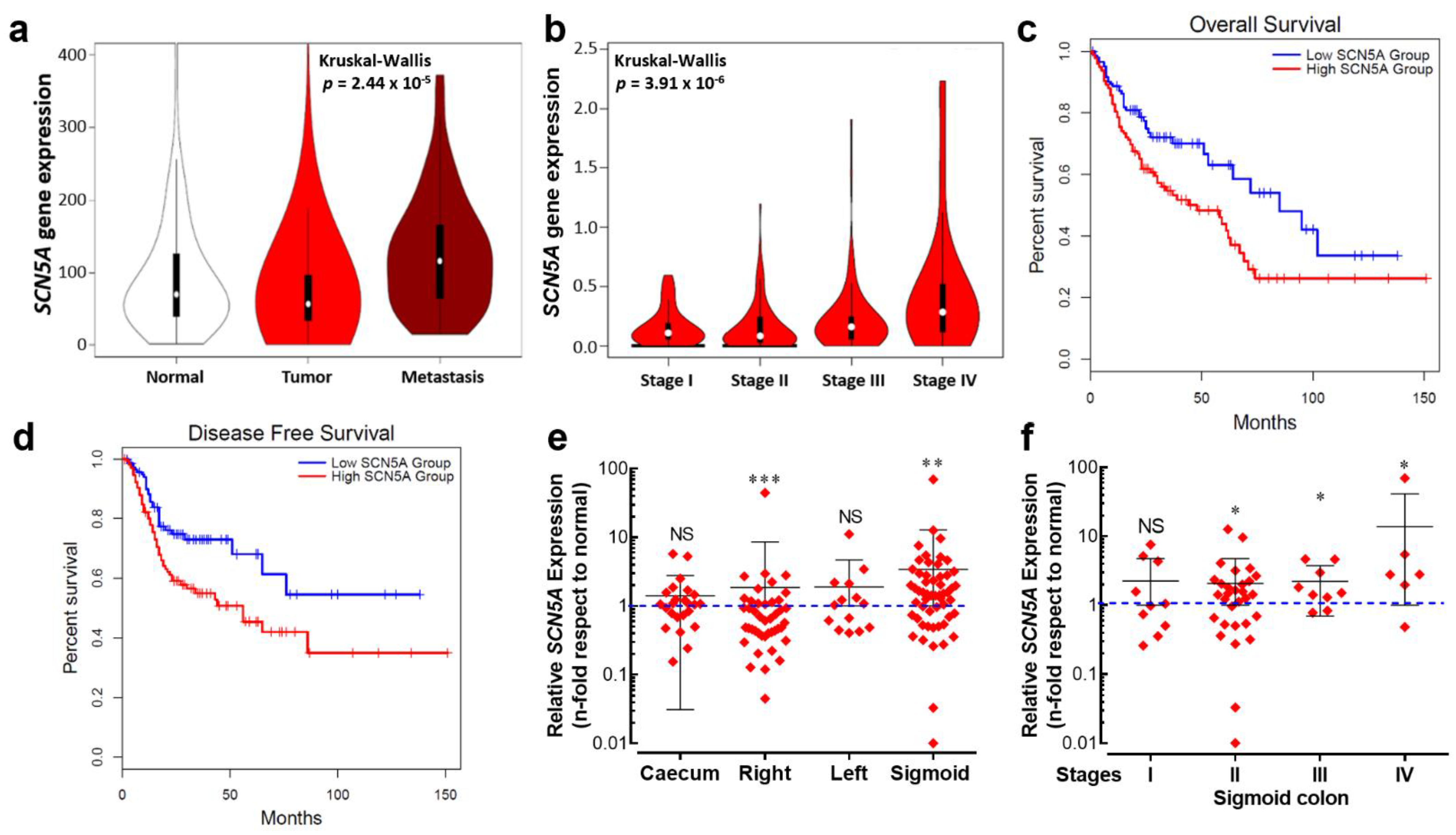
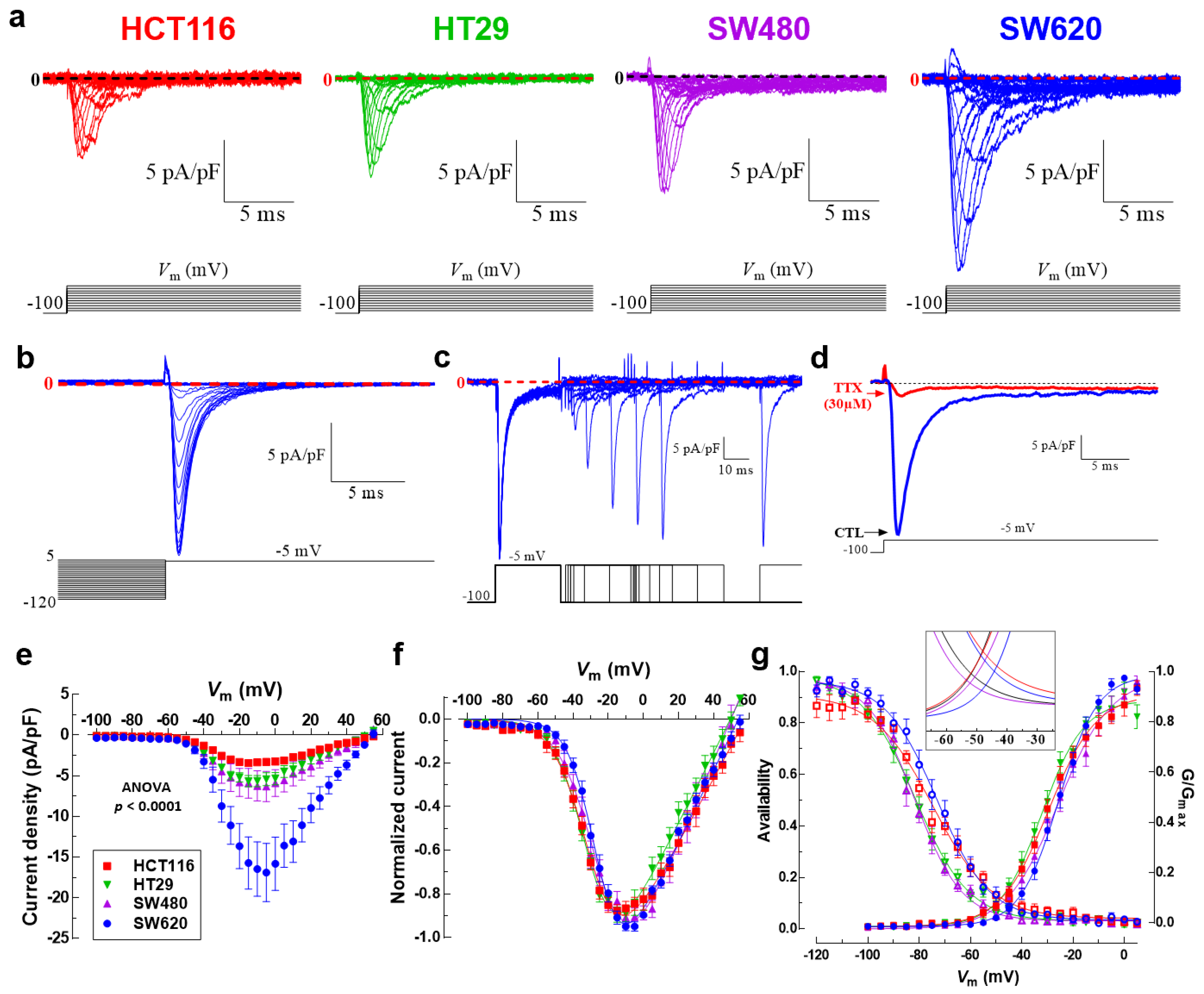
3.2. Nav1.5 Is the Main Pore-Forming Alpha-Subunit Isoform Expressed in Colon Cancer Cells
3.3. NaV1.5 Sodium Channels Are Responsible for the Generation of Macroscopic Na+ Currents in Colon Cancer Cells
3.4. Colon Tumor-Derived Primary Cells Express Functional NaV1.5 Sodium Channels
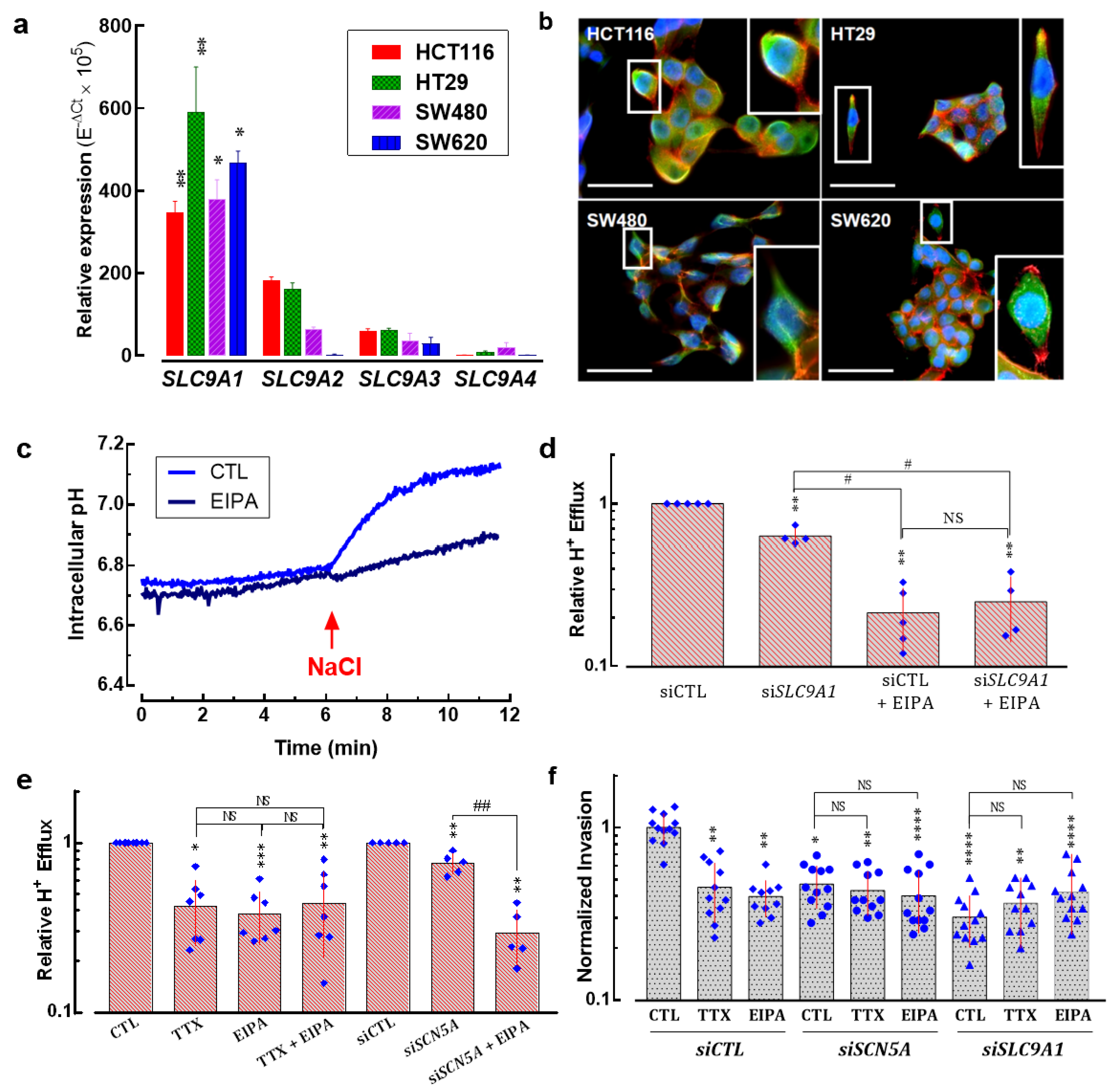
3.5. SLC9A1 Gene, Encoding for the Na+/H+ Exchanger Type 1, Is Differentially Expressed Regarding Anatomical Location of Colon Tumors
3.6. Na+/H+ Exchanger Type 1 Contributes to Colon Cancer Cell Invasiveness
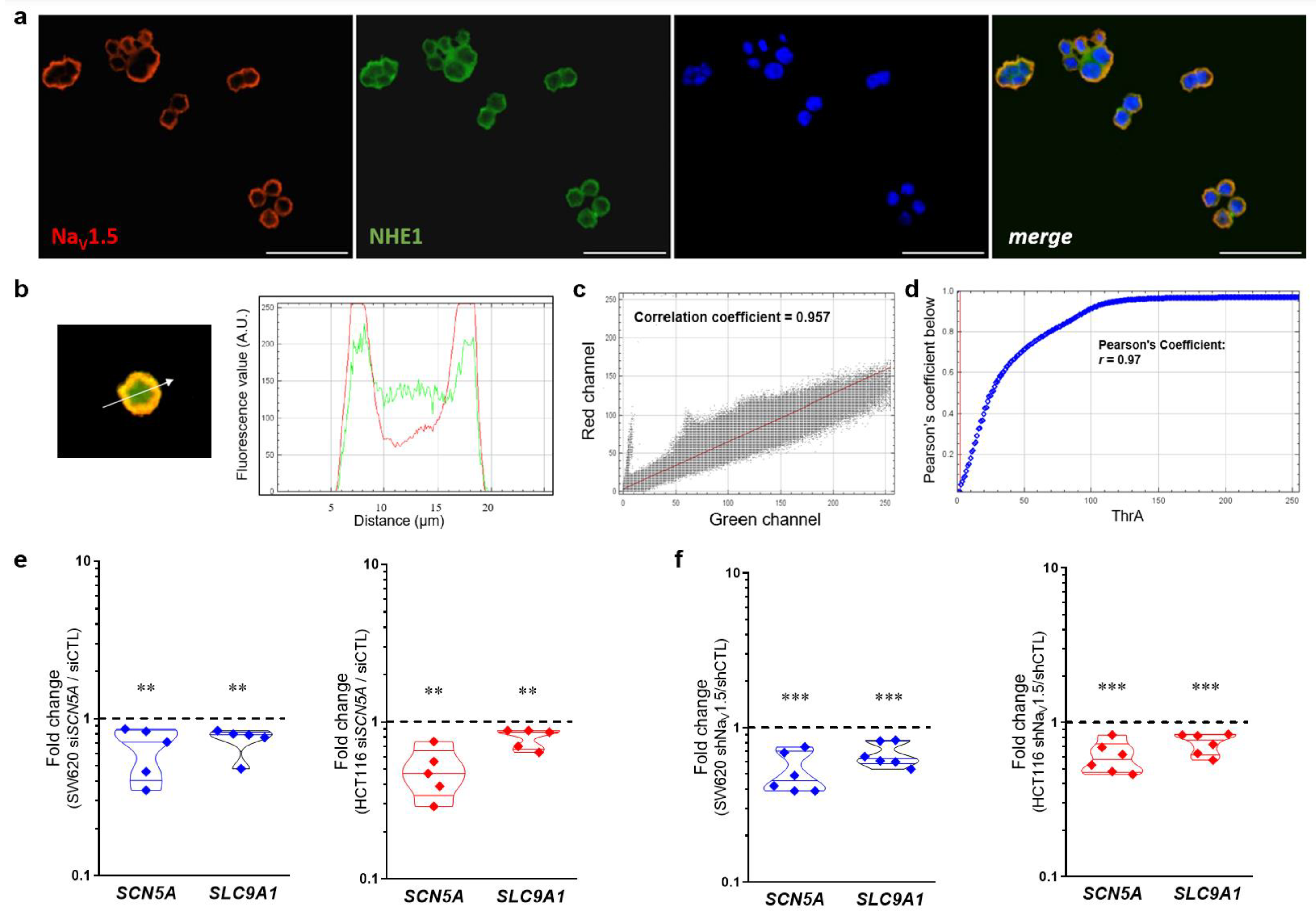
3.7. NaV1.5 Sodium Channels and NHE-1 Exchanger Proteins Colocalize in Colon Cancer Cells
3.8. New Small-Molecule NaV1.5 Inhibitors with Potential Anti-Metastatic Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Xi, Y.; Xu, P. Global Colorectal Cancer Burden in 2020 and Projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- van der Geest, L.G.M.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.G.; de Wilt, J.H.W. Nationwide Trends in Incidence, Treatment and Survival of Colorectal Cancer Patients with Synchronous Metastases. Clin. Exp. Metastasis 2015, 32, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [Green Version]
- Besson, P.; Driffort, V.; Bon, É.; Gradek, F.; Chevalier, S.; Roger, S. How Do Voltage-Gated Sodium Channels Enhance Migration and Invasiveness in Cancer Cells? Biochim. Et Biophys. Acta (BBA)—Biomembr. 2015, 1848, 2493–2501. [Google Scholar] [CrossRef] [Green Version]
- Roger, S.; Gillet, L.; Le Guennec, J.-Y.; Besson, P. Voltage-Gated Sodium Channels and Cancer: Is Excitability Their Primary Role? Front. Pharmacol. 2015, 6, 152. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Charcas, O.; Pukkanasut, P.; Velu, S.E.; Brackenbury, W.J.; Hales, T.G.; Besson, P.; Gomora, J.C.; Roger, S. Pharmacological and Nutritional Targeting of Voltage-Gated Sodium Channels in the Treatment of Cancers. iScience 2021, 24, 102270. [Google Scholar] [CrossRef]
- Driffort, V.; Gillet, L.; Bon, E.; Marionneau-Lambot, S.; Oullier, T.; Joulin, V.; Collin, C.; Pagès, J.-C.; Jourdan, M.-L.; Chevalier, S.; et al. Ranolazine Inhibits NaV1.5-Mediated Breast Cancer Cell Invasiveness and Lung Colonization. Mol. Cancer 2014, 13, 264. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The Sodium Channel-Blocking Antiepileptic Drug Phenytoin Inhibits Breast Tumour Growth and Metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef] [Green Version]
- Bon, E.; Driffort, V.; Gradek, F.; Martinez-Caceres, C.; Anchelin, M.; Pelegrin, P.; Cayuela, M.-L.; Marionneau-Lambot, S.; Oullier, T.; Guibon, R.; et al. SCN4B Acts as a Metastasis-Suppressor Gene Preventing Hyperactivation of Cell Migration in Breast Cancer. Nat. Commun. 2016, 7, 13648. [Google Scholar] [CrossRef]
- Doray, A.; Lemoine, R.; Severin, M.; Chadet, S.; Lopez-Charcas, O.; Héraud, A.; Baron, C.; Besson, P.; Monteil, A.; Pedersen, S.F.; et al. The Voltage-Gated Sodium Channel Beta4 Subunit Maintains Epithelial Phenotype in Mammary Cells. Cells 2021, 10, 1624. [Google Scholar] [CrossRef] [PubMed]
- Leslie, T.K.; James, A.D.; Zaccagna, F.; Grist, J.T.; Deen, S.; Kennerley, A.; Riemer, F.; Kaggie, J.D.; Gallagher, F.A.; Gilbert, F.J.; et al. Sodium Homeostasis in the Tumour Microenvironment. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2019, 1872, 188304. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Brackenbury, W.J. Membrane Potential and Cancer Progression. Front. Physiol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.-Y. Voltage-Gated Sodium Channel Activity Promotes Cysteine Cathepsin-Dependent Invasiveness and Colony Growth of Human Cancer Cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef] [Green Version]
- Brisson, L.; Gillet, L.; Calaghan, S.; Besson, P.; Le Guennec, J.-Y.; Roger, S.; Gore, J. NaV1.5 Enhances Breast Cancer Cell Invasiveness by Increasing NHE1-Dependent H+ Efflux in Caveolae. Oncogene 2011, 30, 2070–2076. [Google Scholar] [CrossRef] [Green Version]
- Brisson, L.; Driffort, V.; Benoist, L.; Poet, M.; Counillon, L.; Antelmi, E.; Rubino, R.; Besson, P.; Labbal, F.; Chevalier, S.; et al. NaV1.5 Sodium Channels Allosterically Regulate the NHE-1 Exchanger and Promote Breast Cancer Cell Invadopodial Activity. J. Cell Sci. 2013, 126, 4835–4842. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Charcas, O.; Espinosa, A.M.; Alfaro, A.; Herrera-Carrillo, Z.; Ramirez-Cordero, B.E.; Cortes-Reynosa, P.; Perez Salazar, E.; Berumen, J.; Gomora, J.C. The Invasiveness of Human Cervical Cancer Associated to the Function of NaV1.6 Channels Is Mediated by MMP-2 Activity. Sci. Rep. 2018, 8, 12995. [Google Scholar] [CrossRef] [Green Version]
- House, C.D.; Vaske, C.J.; Schwartz, A.M.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G.; et al. Voltage-Gated Na+ Channel SCN5A Is a Key Regulator of a Gene Transcriptional Network That Controls Colon Cancer Invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef] [Green Version]
- Baptista-Hon, D.T.; Robertson, F.M.; Robertson, G.B.; Owen, S.J.; Rogers, G.W.; Lydon, E.L.; Lee, N.H.; Hales, T.G. Potent Inhibition by Ropivacaine of Metastatic Colon Cancer SW620 Cell Invasion and Na V 1.5 Channel Function. Br. J. Anaesth. 2014, 113, i39–i48. [Google Scholar] [CrossRef] [Green Version]
- House, C.D.; Wang, B.-D.; Ceniccola, K.; Williams, R.; Simaan, M.; Olender, J.; Patel, V.; Baptista-Hon, D.T.; Annunziata, C.M.; Silvio Gutkind, J.; et al. Voltage-Gated Na+ Channel Activity Increases Colon Cancer Transcriptional Activity and Invasion Via Persistent MAPK Signaling. Sci. Rep. 2015, 5, 11541. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. TNMplot.Com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. IJMS 2021, 22, 2622. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, P.; Fexova, S.; George, N.; Manning, J.R.; Miao, Z.; Mohammed, S.; Muñoz-Pomer, A.; Fullgrabe, A.; Bi, Y.; Bush, N.; et al. Expression Atlas Update: Gene and Protein Expression in Multiple Species. Nucleic Acids Res. 2022, 50, D129–D140. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.-L.; Sripadhan, P.; Chen, C.; et al. Conditional Reprogramming and Long-Term Expansion of Normal and Tumor Cells from Human Biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef]
- Poisson, L.; Lopez-Charcas, O.; Chadet, S.; Bon, E.; Lemoine, R.; Brisson, L.; Ouaissi, M.; Baron, C.; Besson, P.; Roger, S.; et al. Rock Inhibition Promotes NaV1.5 Sodium Channel-Dependent SW620 Colon Cancer Cell Invasiveness. Sci. Rep. 2020, 10, 13350. [Google Scholar] [CrossRef] [PubMed]
- Gradek, F.; Lopez-Charcas, O.; Chadet, S.; Poisson, L.; Ouldamer, L.; Goupille, C.; Jourdan, M.-L.; Chevalier, S.; Moussata, D.; Besson, P.; et al. Sodium Channel Nav1.5 Controls Epithelial-to-Mesenchymal Transition and Invasiveness in Breast Cancer Cells Through Its Regulation by the Salt-Inducible Kinase-1. Sci. Rep. 2019, 9, 18652. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Lopez Charcas, O.; Tanner, S.; Gradek, F.; Driffort, V.; Roger, S.; Selander, K.; Velu, S.E.; Brouillette, W. Discovery and Evaluation of NNav1.5 Sodium Channel Blockers with Potent Cell Invasion Inhibitory Activity in Breast Cancer Cells. Bioorganic Med. Chem. 2018, 26, 2428–2436. [Google Scholar] [CrossRef]
- Guzel, R.M.; Ogmen, K.; Ilieva, K.M.; Fraser, S.P.; Djamgoz, M.B.A. Colorectal Cancer Invasiveness in Vitro: Predominant Contribution of Neonatal Nav1.5 under Normoxia and Hypoxia. J. Cell. Physiol. 2019, 234, 6582–6593. [Google Scholar] [CrossRef]
- Diaz, D.; Delgadillo, D.M.; Hernández-Gallegos, E.; Ramírez-Domínguez, M.E.; Hinojosa, L.M.; Ortiz, C.S.; Berumen, J.; Camacho, J.; Gomora, J.C. Functional Expression of Voltage-Gated Sodium Channels in Primary Cultures of Human Cervical Cancer. J. Cell. Physiol. 2007, 210, 469–478. [Google Scholar] [CrossRef]
- Beltrán, A.R.; Ramírez, M.A.; Carraro-Lacroix, L.R.; Hiraki, Y.; Rebouças, N.A.; Malnic, G. NHE1, NHE2, and NHE4 Contribute to Regulation of Cell PH in T84 Colon Cancer Cells. Pflug. Arch.—Eur. J. Physiol. 2008, 455, 799–810. [Google Scholar] [CrossRef]
- Yugandhar, K.; Gromiha, M.M. Protein–Protein Binding Affinity Prediction from Amino Acid Sequence. Bioinformatics 2014, 30, 3583–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein–Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Hamada, H.; Tsuruo, T.; Ogata, E. Enhancement of Voltage-Gated Na+ Channel Current Associated with Multidrug Resistance in Human Leukemia Cells. Cancer Res. 1987, 47, 3736–3741. [Google Scholar] [PubMed]
- Grimes, J.A.; Fraser, S.P.; Stephens, G.J.; Downing, J.E.G.; Laniado, M.E.; Foster, C.S.; Abel, P.D.; Djamgoz, M.B.A. Differential Expression of Voltage-Activated Na+ Currents in Two Prostatic Tumour Cell Lines: Contribution to Invasiveness in Vitro. FEBS Lett. 1995, 369, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Roger, S.; Besson, P.; Le Guennec, J.-Y. Involvement of a Novel Fast Inward Sodium Current in the Invasion Capacity of a Breast Cancer Cell Line. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2003, 1616, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Roger, S.; Rollin, J.; Barascu, A.; Besson, P.; Raynal, P.-I.; Iochmann, S.; Lei, M.; Bougnoux, P.; Gruel, Y.; Le Guennec, J.-Y. Voltage-Gated Sodium Channels Potentiate the Invasive Capacities of Human Non-Small-Cell Lung Cancer Cell Lines. Int. J. Biochem. Cell Biol. 2007, 39, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Ou, Q.; Wu, X.; Zhang, R.; Zhao, Q.; Jiang, W.; Lu, Z.; Wan, D.; Pan, Z.; Fang, Y. Expression of Voltage-Gated Sodium Channel Nav1.5 in Non-Metastatic Colon Cancer and Its Associations with Estrogen Receptor (ER)-β Expression and Clinical Outcomes. Chin. J. Cancer 2017, 36, 89. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic Potential for Phenytoin: Targeting Nav1.5 Sodium Channels to Reduce Migration and Invasion in Metastatic Breast Cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Fraser, S.P.; Diss, J.K.J.; Chioni, A.-M.; Mycielska, M.E.; Pan, H.; Yamaci, R.F.; Pani, F.; Siwy, Z.; Krasowska, M.; Grzywna, Z.; et al. Voltage-Gated Sodium Channel Expression and Potentiation of Human Breast Cancer Metastasis. Clin. Cancer Res. 2005, 11, 5381–5389. [Google Scholar] [CrossRef] [Green Version]
- Onkal, R.; Mattis, J.H.; Fraser, S.P.; Diss, J.K.J.; Shao, D.; Okuse, K.; Djamgoz, M.B.A. Alternative Splicing of Nav1.5: An Electrophysiological Comparison of ‘Neonatal’ and ‘Adult’ Isoforms and Critical Involvement of a Lysine Residue. J. Cell. Physiol. 2008, 216, 716–726. [Google Scholar] [CrossRef]
- Fraser, S.P.; Diss, J.K.J.; Lloyd, L.J.; Pani, F.; Chioni, A.-M.; George, A.J.T.; Djamgoz, M.B.A. T-Lymphocyte Invasiveness: Control by Voltage-Gated Na+ Channel Activity. FEBS Lett. 2004, 569, 191–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Huang, N.; Huang, H.; Sun, L.; Dong, S.; Su, J.; Zhang, J.; Wang, L.; Lin, L.; Shi, M.; et al. Voltage-Gated Sodium Channel Na v 1.7 Promotes Gastric Cancer Progression through MACC1-Mediated Upregulation of NHE1: NaV1.7 Promotes Gastric Cancer Progression. Int. J. Cancer 2016, 139, 2553–2569. [Google Scholar] [CrossRef] [PubMed]
- Putnam, R.W.; Roos, A. Intracellular pH. In Comprehensive Physiology; Terjung, R., Ed.; Wiley: Hoboken, NJ, USA, 1997; pp. 389–440. ISBN 978-0-470-65071-4. [Google Scholar]
- Cameron, I.L.; Smith, N.K.; Pool, T.B.; Sparks, R.L. Intracellular Concentration of Sodium and Other Elements as Related to Mitogenesis and Oncogenesis in Vivo. Cancer Res. 1980, 40, 1493–1500. [Google Scholar]
- Hürter, T.; Bröcker, W.; Bosma, H.J. Investigations on Vasogenic and Cytotoxic Brain Edema, Comparing Results from X-Ray Microanalysis and Flame Photometry. Microsc. Acta 1982, 85, 285–293. [Google Scholar] [PubMed]
- Long, Z.; Chen, B.; Liu, Q.; Zhao, J.; Yang, Z.; Dong, X.; Xia, L.; Huang, S.; Hu, X.; Song, B.; et al. The Reverse-Mode NCX1 Activity Inhibitor KB-R7943 Promotes Prostate Cancer Cell Death by Activating the JNK Pathway and Blocking Autophagic Flux. Oncotarget 2016, 7, 42059–42070. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, B.; Liskova, V.; Babula, P.; Krizanova, O. Role of Sodium/Calcium Exchangers in Tumors. Biomolecules 2020, 10, 1257. [Google Scholar] [CrossRef]
- Sennoune, S.R.; Santos, J.M.; Hussain, F.; Martínez-Zaguilán, R. Sodium Calcium Exchanger Operates in the Reverse Mode in Metastatic Human Melanoma Cells. Cell Mol. Biol. 2015, 61, 40–49. [Google Scholar]
- Li, X.; Prins, D.; Michalak, M.; Fliegel, L. Calmodulin-Dependent Binding to the NHE1 Cytosolic Tail Mediates Activation of the Na+ /H+ Exchanger by Ca 2+ and Endothelin. Am. J. Physiol.-Cell Physiol. 2013, 305, C1161–C1169. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biophysical Parameter | HCT116 | HT29 | SW480 | SW620 | Primary Cells |
|---|---|---|---|---|---|
| INa+ density (pA/pF) | −3.3 ± 0.4 | −5.4 ± 0.8 | −6.3 ± 0.4 | −16.8 ± 3.5 | −3.1 ± 1.0 |
| V50 activation (mV) | −29.6 ± 0.7 | −32.1 ± 0.7 | −25.7 ± 1.1 | −25.6 ± 0.5 | −14.3 ± 0.6 |
| k activation (ms) | 9.4 ± 0.6 | 8.3 ± 0.6 | 10.2 ± 0.9 | 7.6 ± 0.4 | 8.4 ± 0.5 |
| Vrev (mV) | 58.2 ± 1.3 | 49.5 ± 1.0 | 49.8 ± 1.2 | 54.1 ± 0.9 | 63.6 ± 5.2 |
| V50 inactivation (mV) | −74.6 ± 0.9 | −81.5 ± 0.8 | −81.7 ± 0.6 | −72.9 ± 0.8 | −49.5 ± 3.7 |
| kinactivation (ms) | 11.6 ± 0.8 | 10.5 ± 0.6 | 9.2 ± 0.6 | 10.2 ± 0.7 | 15.2 ± 3.1 |
| Em (mV) | −46.2 ± 4.2 | −38.5 ± 3.3 | −42.4 ± 3.8 | −27.7 ± 6.4 | −32.4 ± 8.3 |
| INa+ at —10 mV (pA/pF) | −3.4 ± 0.35 | −5.65 ± 0.68 | −6.24 ± 0.95 | −16.5 ± 3.34 | −2.05 ± 0.63 |
| Biophysical Parameter | CTL | Compound 1 | Compound 4 |
|---|---|---|---|
| INa+ density (pA/pF) | −9.7 ± 1.4 | −4.1 ± 1.3 | −3.6 ± 1.3 |
| V50 activation (mV) | −25.9 ± 0.4 | −34.2 ± 0.9 | −30.9 ± 0.7 |
| k activation (ms) | 7.8 ± 0.4 | 6.9 ± 0.8 | 6.4 ± 0.6 |
| Vrev (mV) | 51.1 ± 2.2 | 44.4 ± 3.9 | 43.4 ± 2.3 |
| V50 inactivation (mV) | −66.6 ± 1.2 | −82.9 ± 1.6 | −78.9 ± 1.7 |
| kinactivation (ms) | 11.8 ± 1.0 | 11.3 ± 1.3 | 14.5 ± 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Charcas, O.; Poisson, L.; Benouna, O.; Lemoine, R.; Chadet, S.; Pétereau, A.; Lahlou, W.; Guyétant, S.; Ouaissi, M.; Pukkanasut, P.; et al. Voltage-Gated Sodium Channel NaV1.5 Controls NHE−1−Dependent Invasive Properties in Colon Cancer Cells. Cancers 2023, 15, 46. https://doi.org/10.3390/cancers15010046
Lopez-Charcas O, Poisson L, Benouna O, Lemoine R, Chadet S, Pétereau A, Lahlou W, Guyétant S, Ouaissi M, Pukkanasut P, et al. Voltage-Gated Sodium Channel NaV1.5 Controls NHE−1−Dependent Invasive Properties in Colon Cancer Cells. Cancers. 2023; 15(1):46. https://doi.org/10.3390/cancers15010046
Chicago/Turabian StyleLopez-Charcas, Osbaldo, Lucile Poisson, Oumnia Benouna, Roxane Lemoine, Stéphanie Chadet, Adrien Pétereau, Widad Lahlou, Serge Guyétant, Mehdi Ouaissi, Piyasuda Pukkanasut, and et al. 2023. "Voltage-Gated Sodium Channel NaV1.5 Controls NHE−1−Dependent Invasive Properties in Colon Cancer Cells" Cancers 15, no. 1: 46. https://doi.org/10.3390/cancers15010046