TNF Signaling Is Required for Castration-Induced Vascular Damage Preceding Prostate Cancer Regression


 and
and 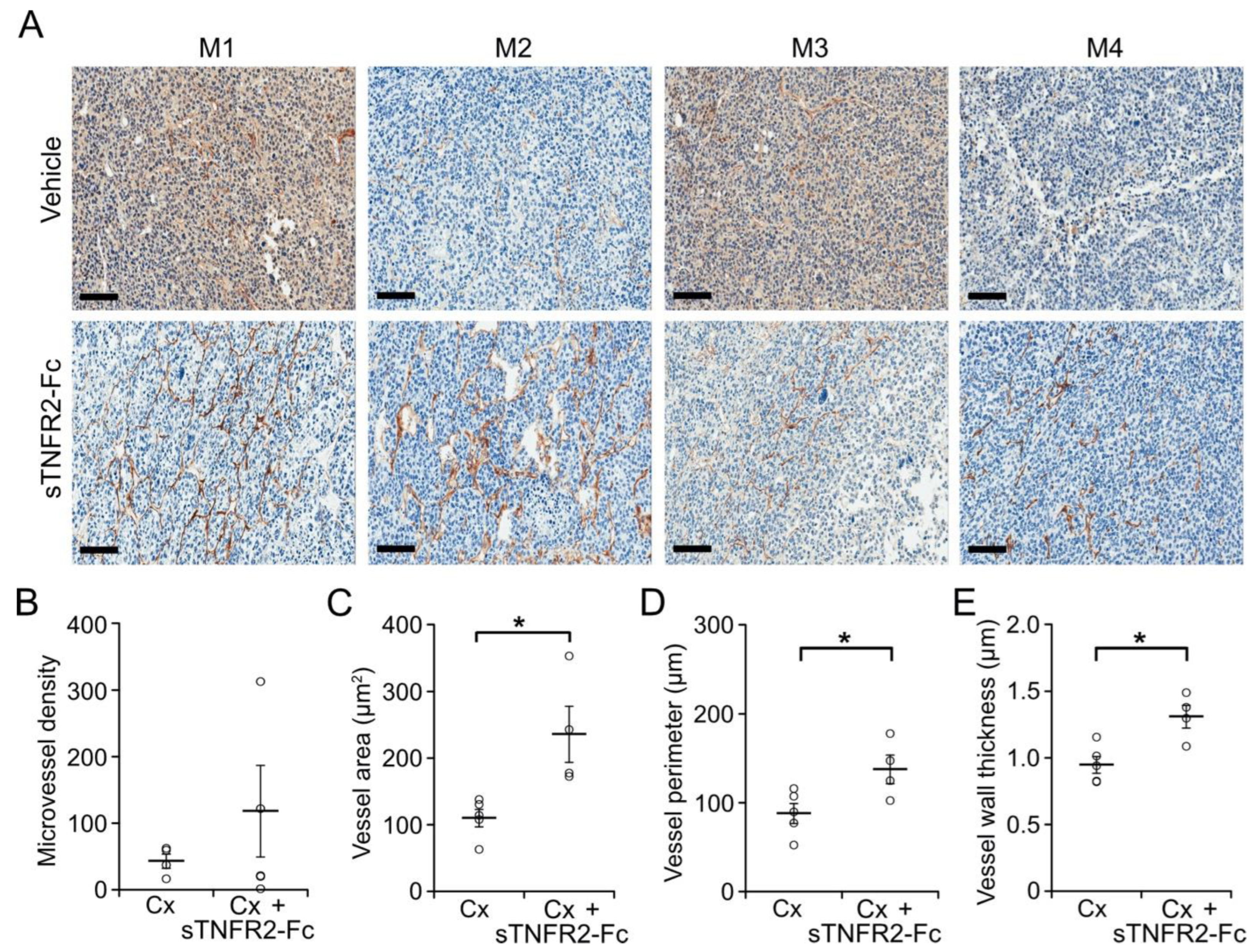{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animals
2.3. Anatomic and Functional Imaging
2.4. Immunohistochemistry
2.5. Statistical Analysis
2.6. Data and Materials Availability
3. Results
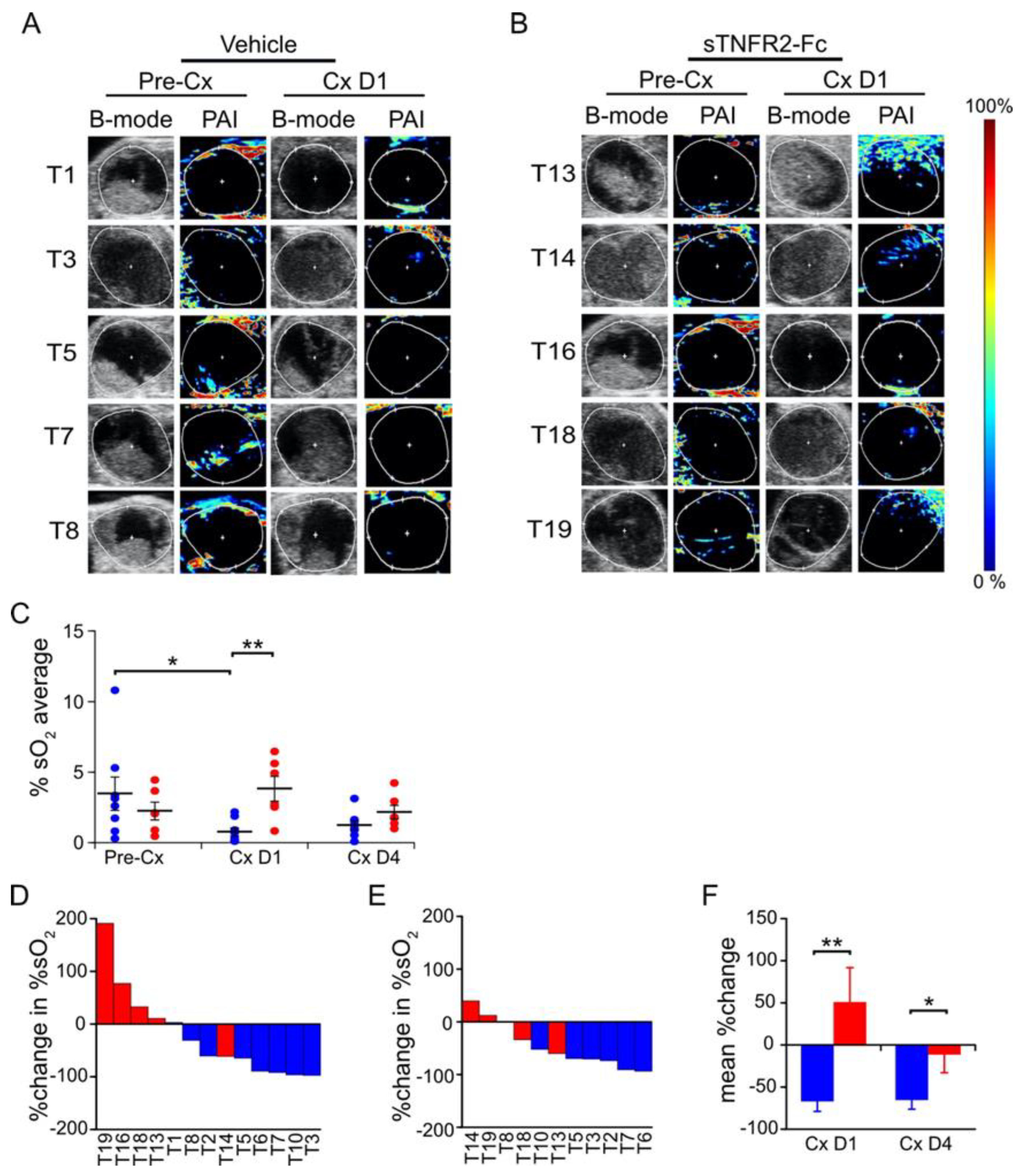
3.1. TNF Is Necessary for Castration-Induced Microvasculature Damage in Myc-CaP Allografts
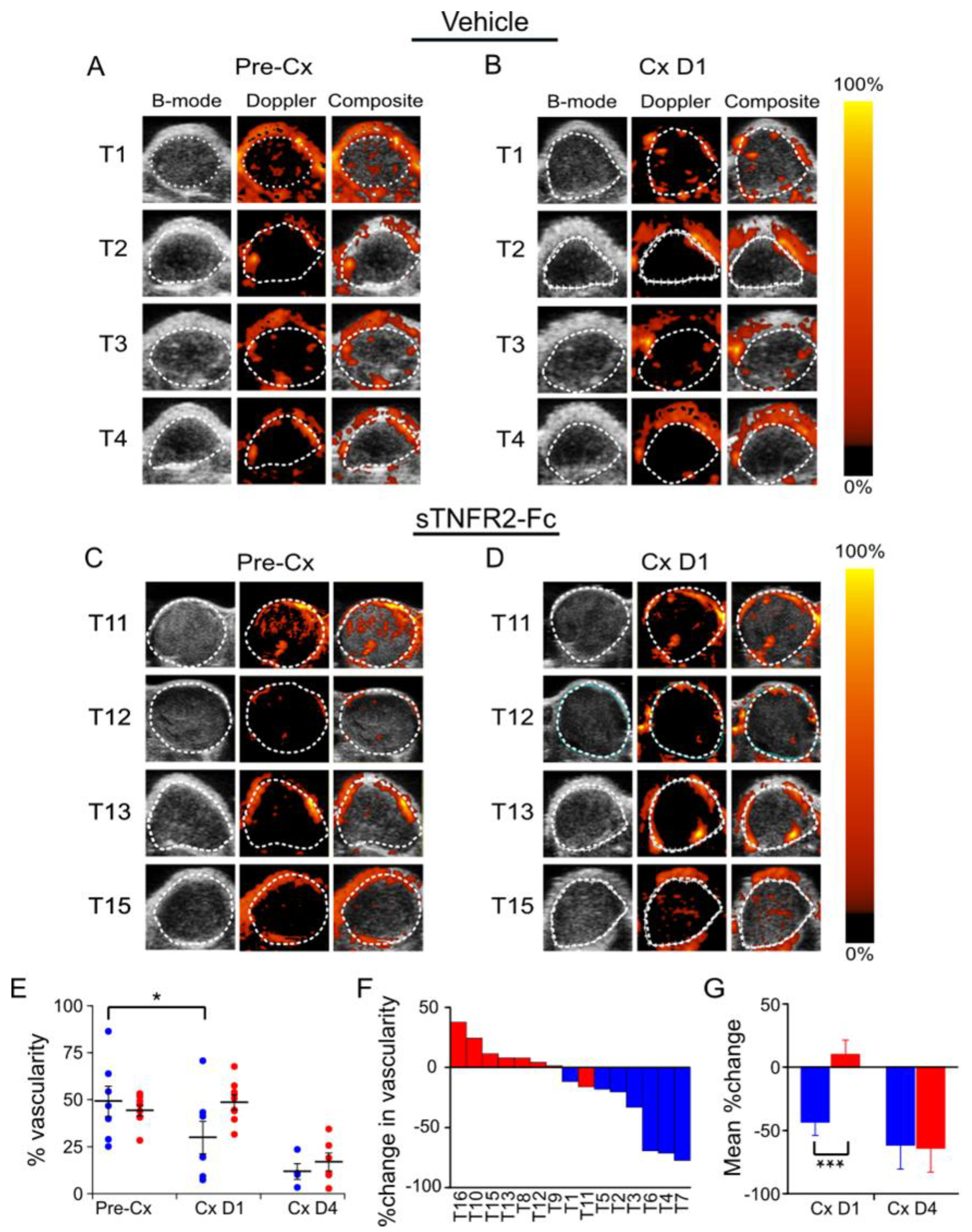
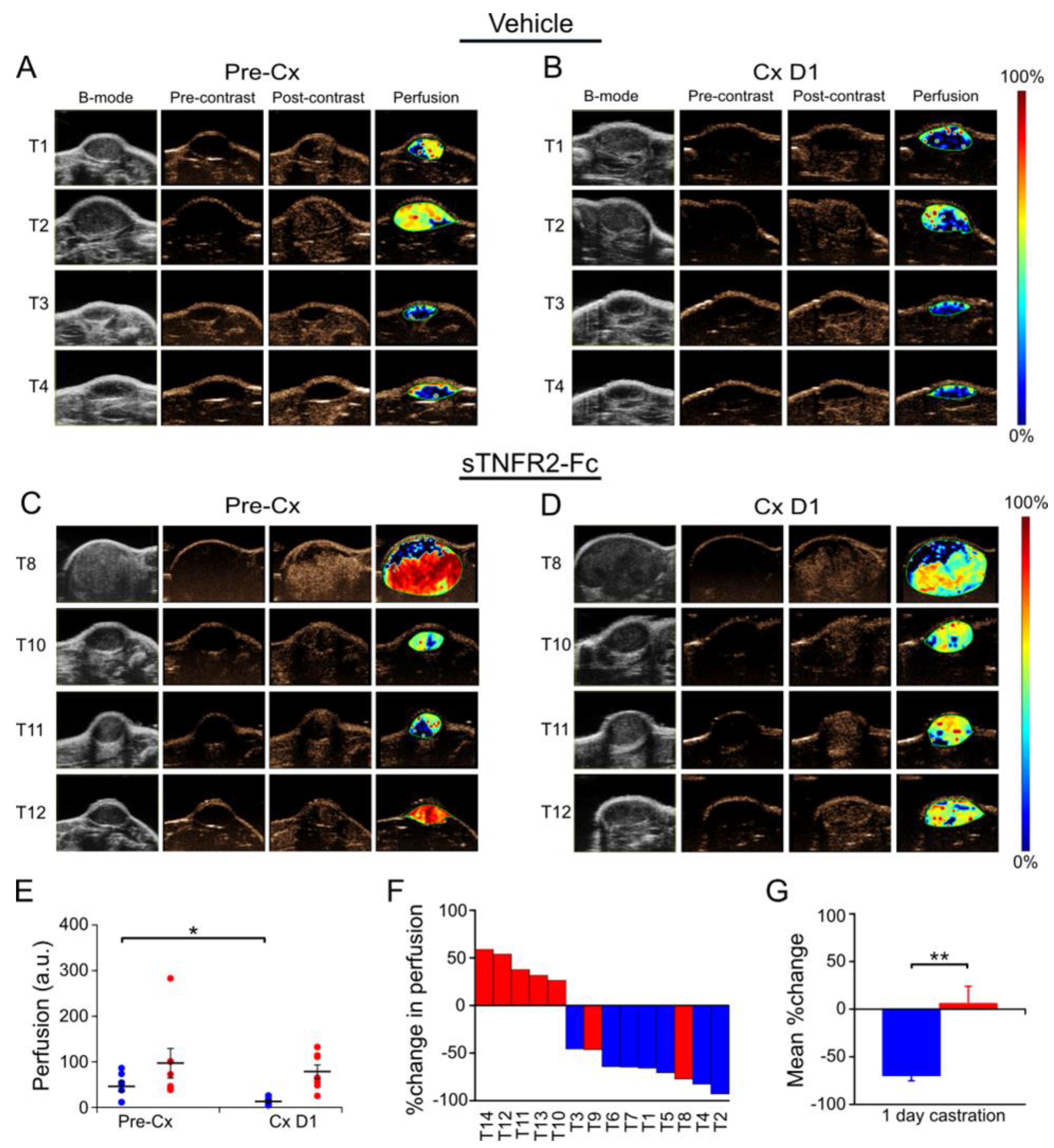
3.2. Castration-Induced Reduction in Blood Flow and Perfusion Is Prevented by Blocking TNF Signaling
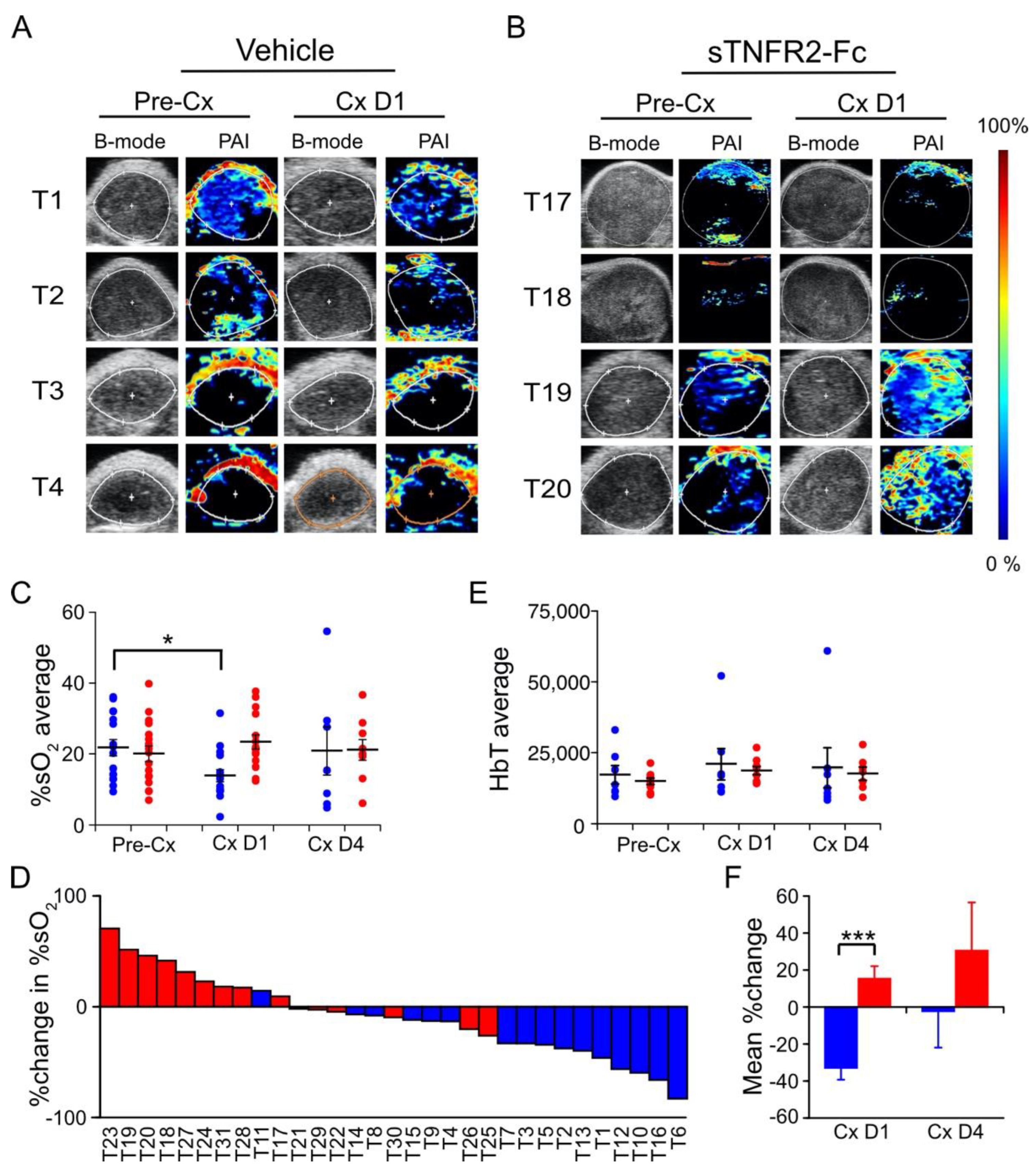
3.3. TNF Is Required for Castration-Induced Hypoxia in Myc-CaP Allografts
3.4. TNF Is Required for Castration-Induced Hypoxia in an Autochthonous Prostate Cancer Model
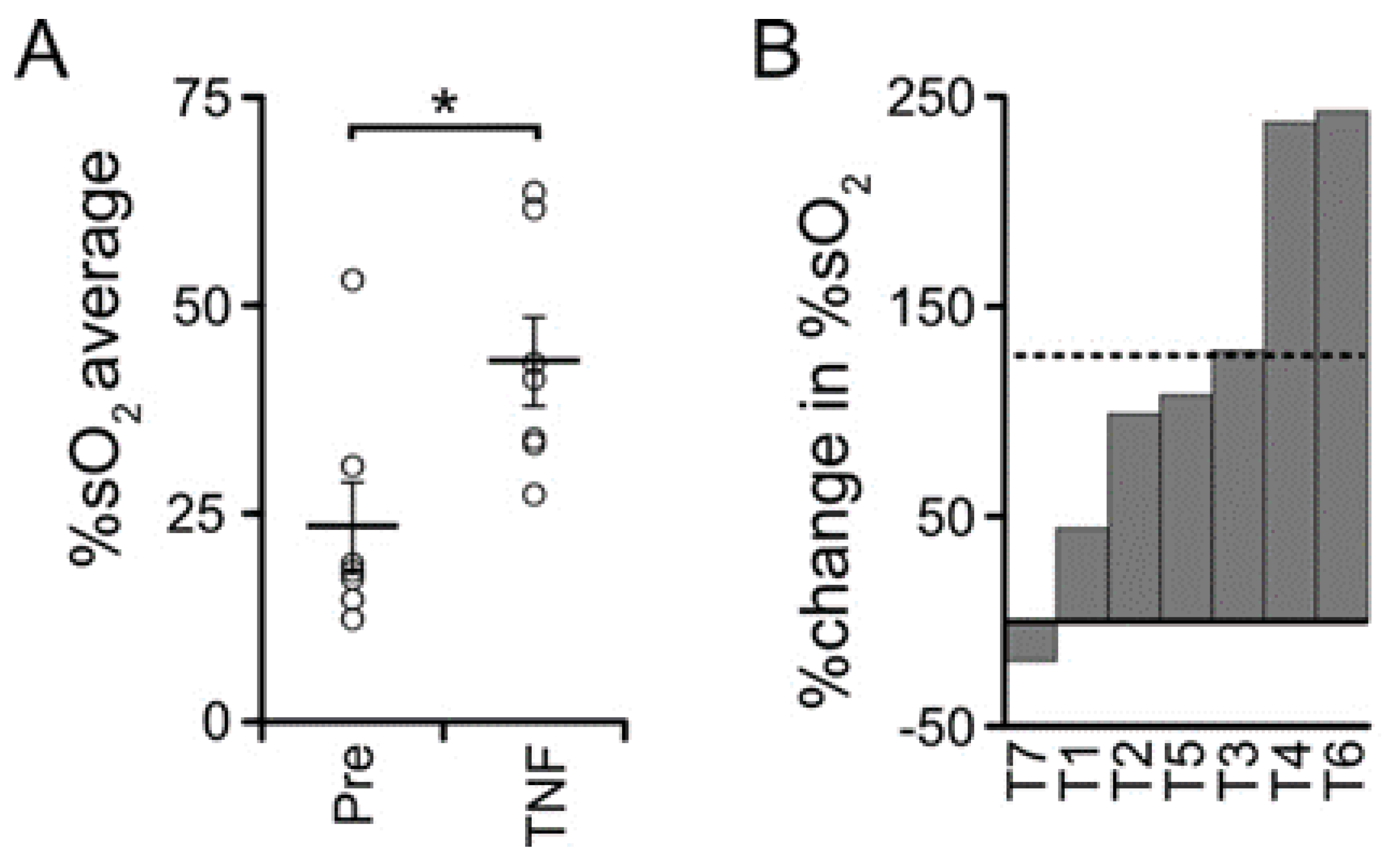
3.5. TNF Is Necessary but Not Sufficient for Castration-Induced Hypoxia in Myc-CaP Allografts
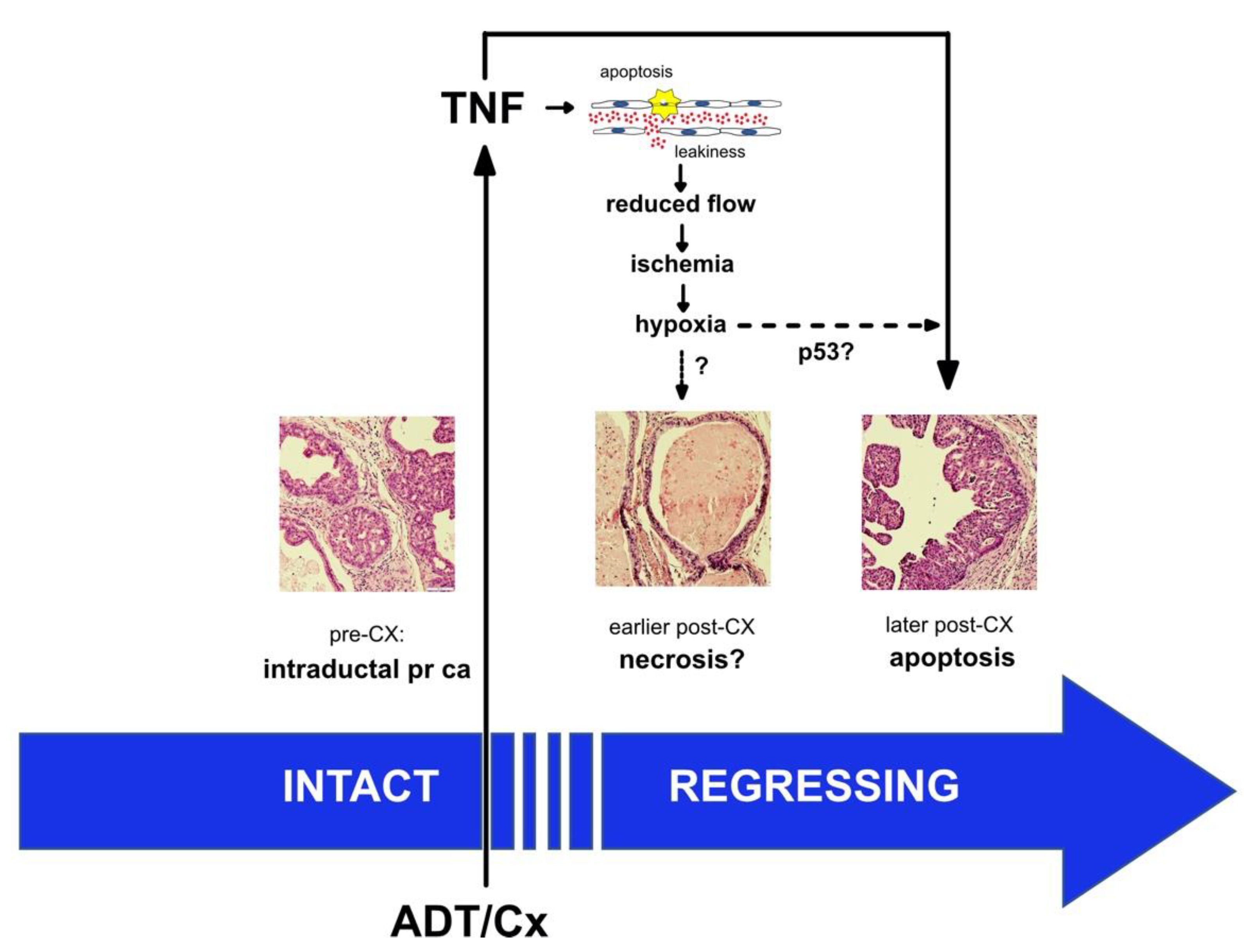
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Lin, X.S.; Isaacs, J.T. Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate 1996, 28, 251–265. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Kurita, T.; Wang, Y.Z.; Donjacour, A.A.; Zhao, C.; Lydon, J.P.; O’Malley, B.W.; Isaacs, J.T.; Dahiya, R.; Cunha, G.R. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ. 2001, 8, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.S.; Nastiuk, K.L.; Krolewski, J.J. TNF is necessary for castration-induced prostate regression, whereas TRAIL and FasL are dispensable. Mol. Endocrinol. 2011, 25, 611–620. [Google Scholar] [CrossRef]
- Buttyan, R.; Ghafar, M.A.; Shabsigh, A. The effects of androgen deprivation on the prostate gland: Cell death mediated by vascular regression. Curr. Opin. Urol. 2000, 10, 415–420. [Google Scholar] [CrossRef]
- Godoy, A.; Montecinos, V.P.; Gray, D.R.; Sotomayor, P.; Yau, J.M.; Vethanayagam, R.R.; Singh, S.; Mohler, J.L.; Smith, G.J. Androgen deprivation induces rapid involution and recovery of human prostate vasculature. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E263–E275. [Google Scholar] [CrossRef] [Green Version]
- Shabisgh, A.; Tanji, N.; D’Agati, V.; Burchardt, M.; Rubin, M.; Goluboff, E.T.; Heitjan, D.; Kiss, A.; Buttyan, R. Early effects of castration on the vascular system of the rat ventral prostate gland. Endocrinology 1999, 140, 1920–1926. [Google Scholar] [CrossRef]
- Shabsigh, A.; Chang, D.T.; Heitjan, D.F.; Kiss, A.; Olsson, C.A.; Puchner, P.J.; Buttyan, R. Rapid reduction in blood flow to the rat ventral prostate gland after castration: Preliminary evidence that androgens influence prostate size by regulating blood flow to the prostate gland and prostatic endothelial cell survival. Prostate 1998, 36, 201–206. [Google Scholar] [CrossRef]
- Lekas, E.; Johansson, M.; Widmark, A.; Bergh, A.; Damber, J.E. Decrement of blood flow precedes the involution of the ventral prostate in the rat after castration. Urol. Res. 1997, 25, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kalmuk, J.; Folaron, M.; Buchinger, J.; Pili, R.; Seshadri, M. Multimodal imaging guided preclinical trials of vascular targeting in prostate cancer. Oncotarget 2015, 6, 24376–24392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, K.; Mikalsen, L.T.; van der Kogel, A.J.; Bussink, J.; Lyng, H.; Ree, A.H.; Marignol, L.; Olsen, D.R. Vascular responses to radiotherapy and androgen-deprivation therapy in experimental prostate cancer. Radiat. Oncol. 2012, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.; Rudolfsson, S.H.; Kilter, S.; Bergh, A. Targeting castration-induced tumour hypoxia enhances the acute effects of castration therapy in a rat prostate cancer model. BJU Int. 2011, 107, 1818–1824. [Google Scholar] [CrossRef]
- Franck-Lissbrant, I.; Haggstrom, S.; Damber, J.E.; Bergh, A. Testosterone stimulates angiogenesis and vascular regrowth in the ventral prostate in castrated adult rats. Endocrinology 1998, 139, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Shabsigh, A.; Ghafar, M.A.; de la Taille, A.; Burchardt, M.; Kaplan, S.A.; Anastasiadis, A.G.; Buttyan, R. Biomarker analysis demonstrates a hypoxic environment in the castrated rat ventral prostate gland. J. Cell Biochem. 2001, 81, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F.J.; Lienard, D.; Matter, M.; Ruegg, C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. 2006, 6, 6. [Google Scholar]
- Petrache, I.; Verin, A.D.; Crow, M.T.; Birukova, A.; Liu, F.; Garcia, J.G. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1168–L1178. [Google Scholar] [CrossRef]
- Watson, P.A.; Ellwood-Yen, K.; King, J.C.; Wongvipat, J.; Lebeau, M.M.; Sawyers, C.L. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005, 65, 11565–11571. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.; Lehet, K.; Ramakrishnan, S.; Adelaiye, R.; Miles, K.M.; Wang, D.; Liu, S.; Atadja, P.; Carducci, M.A.; Pili, R. Concurrent HDAC and mTORC1 inhibition attenuate androgen receptor and hypoxia signaling associated with alterations in microRNA expression. PLoS ONE 2011, 6, e27178. [Google Scholar] [CrossRef]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Singh, S.; Pan, C.; Wood, R.; Yeh, C.R.; Yeh, S.; Sha, K.; Krolewski, J.J.; Nastiuk, K.L. Quantitative volumetric imaging of normal, neoplastic and hyperplastic mouse prostate using ultrasound. BMC Urol. 2015, 15, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.; Singh, S.; Sahasrabudhe, D.M.; Chakkalakal, J.V.; Krolewski, J.J.; Nastiuk, K.L. TGFbeta Superfamily Members Mediate Androgen Deprivation Therapy-Induced Obese Frailty in Male Mice. Endocrinology 2016, 157, 4461–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, L.J.; Seshadri, M. Photoacoustic monitoring of tumor and normal tissue response to radiation. Sci. Rep. 2016, 6, 21237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, L.J.; Seshadri, M. Photoacoustic imaging of vascular hemodynamics: Validation with blood oxygenation level-dependent MR imaging. Radiology 2015, 275, 110–118. [Google Scholar] [CrossRef]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Maolake, A.; Zhang, R.; Sha, K.; Singh, S.; Pan, C.; Xu, B.; Chatta, G.; Mastri, M.; Eng, K.H.; Krolewski, J.J.; et al. Glucocorticoid signaling delays castration-induced regression in murine models of prostate cancer. BioRxiv 2022. [Google Scholar] [CrossRef]
- Goertz, D.E.; Yu, J.L.; Kerbel, R.S.; Burns, P.N.; Foster, F.S. High-frequency Doppler ultrasound monitors the effects of antivascular therapy on tumor blood flow. Cancer Res. 2002, 62, 6371–6375. [Google Scholar]
- Hamper, U.M.; DeJong, M.R.; Caskey, C.I.; Sheth, S. Power Doppler imaging: Clinical experience and correlation with color Doppler US and other imaging modalities. Radiographics 1997, 17, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.I.; Kim, T.K.; Han, J.K.; Chung, J.W.; Park, J.H.; Han, M.C. Power versus conventional color Doppler sonography: Comparison in the depiction of vasculature in liver tumors. Radiology 1996, 200, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, M.R.; Gonzalez, I.Q.; O’Connor, J.P.; Abeyakoon, O.; Parker, G.J.; Williams, K.J.; Gilbert, F.J.; Bohndiek, S.E. Oxygen Enhanced Optoacoustic Tomography (OE-OT) Reveals Vascular Dynamics in Murine Models of Prostate Cancer. Theranostics 2017, 7, 2900–2913. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.M.; Heinrich, M.C.; Lim, J.; Nelson, D.; Beadling, C.; Warrick, A.; Neff, T.; Higano, C.S.; Garzotto, M.; Qian, D.; et al. Genetic profiling to determine risk of relapse-free survival in high-risk localized prostate cancer. Clin. Cancer Res. 2014, 20, 1306–1312. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Azad, A.K.; Karanika, S.; Basourakos, S.P.; Zuo, X.; Wang, J.; Yang, L.; Yang, G.; Korentzelos, D.; Yin, J.; et al. Enzalutamide and CXCR7 inhibitor combination treatment suppresses cell growth and angiogenic signaling in castration-resistant prostate cancer models. Int. J. Cancer 2018, 142, 2163–2174. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Mitsunari, K.; Asai, A.; Takehara, K.; Mochizuki, Y.; Sakai, H. Pathological significance and prognostic role of microvessel density, evaluated using CD31, CD34, and CD105 in prostate cancer patients after radical prostatectomy with neoadjuvant therapy. Prostate 2015, 75, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Naito, H.; Iba, T.; Wakabayashi, T.; Tai-Nagara, I.; Suehiro, J.I.; Jia, W.; Eino, D.; Sakimoto, S.; Muramatsu, F.; Kidoya, H.; et al. TAK1 Prevents Endothelial Apoptosis and Maintains Vascular Integrity. Dev. Cell 2019, 48, 151–166.e7. [Google Scholar] [CrossRef] [Green Version]
- Clauss, M.; Sunderkotter, C.; Sveinbjornsson, B.; Hippenstiel, S.; Willuweit, A.; Marino, M.; Haas, E.; Seljelid, R.; Scheurich, P.; Suttorp, N.; et al. A permissive role for tumor necrosis factor in vascular endothelial growth factor-induced vascular permeability. Blood 2001, 97, 1321–1329. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Lee, P.; Wang, H.; Gerald, W.; Adler, M.; Zhang, L.; Wang, Y.F.; Wang, Z. The androgen receptor directly targets the cellular Fas/FasL-associated death domain protein-like inhibitory protein gene to promote the androgen-independent growth of prostate cancer cells. Mol. Endocrinol. 2005, 19, 1792–1802. [Google Scholar] [CrossRef] [Green Version]
- Nastiuk, K.L.; Kim, J.W.; Mann, M.; Krolewski, J.J. Androgen regulation of FLICE-like inhibitory protein gene expression in the rat prostate. J. Cell Physiol. 2003, 196, 386–393. [Google Scholar] [CrossRef]
- Nastiuk, K.L.; Krolewski, J.J. FLIP-ping out: Death receptor signaling in the prostate. Cancer Biol. Ther. 2008, 7, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Prins, G.S.; Birch, L.; Greene, G.L. Androgen receptor localization in different cell types of the adult rat prostate. Endocrinology 1991, 129, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hadoke, P.W.; Takov, K.; Korczak, A.; Denvir, M.A.; Smith, L.B. Influence of Androgen Receptor in Vascular Cells on Reperfusion following Hindlimb Ischaemia. PLoS ONE 2016, 11, e0154987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graeber, T.G.; Peterson, J.F.; Tsai, M.; Monica, K.; Fornace, A.J., Jr.; Giaccia, A.J. Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol. Cell Biol. 1994, 14, 6264–6277. [Google Scholar]
- Colombel, M.; Radvanyi, F.; Blanche, M.; Abbou, C.; Buttyan, R.; Donehower, L.A.; Chopin, D.; Thiery, J.P. Androgen suppressed apoptosis is modified in p53 deficient mice. Oncogene 1997, 10, 1269–1274. [Google Scholar]
- Berges, R.R.; Furuya, Y.; Remington, L.; English, H.F.; Jacks, T.; Isaacs, J.T. Cell proliferation, DNA repair, and p53 function are not required for programmed death of prostatic glandular cells induced by androgen ablation. Proc. Natl. Acad. Sci. USA 1993, 90, 8910–8914. [Google Scholar] [CrossRef] [Green Version]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folli, S.; Pelegrin, A.; Chalandon, Y.; Yao, X.; Buchegger, F.; Lienard, D.; Lejeune, F.; Mach, J.P. Tumor-necrosis factor can enhance radio-antibody uptake in human colon carcinoma xenografts by increasing vascular permeability. Int. J. Cancer 1993, 53, 829–836. [Google Scholar] [CrossRef]
- Longo, D.L.; Stefania, R.; Callari, C.; De Rose, F.; Rolle, R.; Conti, L.; Consolino, L.; Arena, F.; Aime, S. Water Soluble Melanin Derivatives for Dynamic Contrast Enhanced Photoacoustic Imaging of Tumor Vasculature and Response to Antiangiogenic Therapy. Adv. Healthc. Mater. 2017, 18, 1719. [Google Scholar] [CrossRef] [Green Version]
- Royall, J.A.; Berkow, R.L.; Beckman, J.S.; Cunningham, M.K.; Matalon, S.; Freeman, B.A. Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am. J. Physiol. 1989, 257 Pt 1, L399–L410. [Google Scholar] [CrossRef] [PubMed]
- Kallinowski, F.; Schaefer, C.; Tyler, G.; Vaupel, P. In vivo targets of recombinant human tumour necrosis factor-alpha: Blood flow, oxygen consumption and growth of isotransplanted rat tumours. Br. J. Cancer 1989, 60, 555–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Naylor, M.S.; Malik, S. Tumour necrosis factor as an anticancer agent. Eur. J. Cancer 1990, 26, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Curnis, F.; Sacchi, A.; Borgna, L.; Magni, F.; Gasparri, A.; Corti, A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat. Biotechnol. 2000, 18, 1185–1190. [Google Scholar] [CrossRef]
- Bertilaccio, M.T.; Grioni, M.; Sutherland, B.W.; Degl’Innocenti, E.; Freschi, M.; Jachetti, E.; Greenberg, N.M.; Corti, A.; Bellone, M. Vasculature-targeted tumor necrosis factor-alpha increases the therapeutic index of doxorubicin against prostate cancer. Prostate 2008, 68, 1105–1115. [Google Scholar] [CrossRef]
- Probst, P.; Stringhini, M.; Ritz, D.; Fugmann, T.; Neri, D. Antibody-based Delivery of TNF to the Tumor Neovasculature Potentiates the Therapeutic Activity of a Peptide Anticancer Vaccine. Clin. Cancer Res. 2019, 25, 698–709. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krolewski, J.J.; Singh, S.; Sha, K.; Jaiswal, N.; Turowski, S.G.; Pan, C.; Rich, L.J.; Seshadri, M.; Nastiuk, K.L. TNF Signaling Is Required for Castration-Induced Vascular Damage Preceding Prostate Cancer Regression. Cancers 2022, 14, 6020. https://doi.org/10.3390/cancers14246020
Krolewski JJ, Singh S, Sha K, Jaiswal N, Turowski SG, Pan C, Rich LJ, Seshadri M, Nastiuk KL. TNF Signaling Is Required for Castration-Induced Vascular Damage Preceding Prostate Cancer Regression. Cancers. 2022; 14(24):6020. https://doi.org/10.3390/cancers14246020
Chicago/Turabian StyleKrolewski, John J., Shalini Singh, Kai Sha, Neha Jaiswal, Steven G. Turowski, Chunliu Pan, Laurie J. Rich, Mukund Seshadri, and Kent L. Nastiuk. 2022. "TNF Signaling Is Required for Castration-Induced Vascular Damage Preceding Prostate Cancer Regression" Cancers 14, no. 24: 6020. https://doi.org/10.3390/cancers14246020