
Development and Challenges of Diclofenac-Based Novel Therapeutics: Targeting Cancer and Complex Diseases
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Diclofenac: Pharmacokinetics and Pharmacology
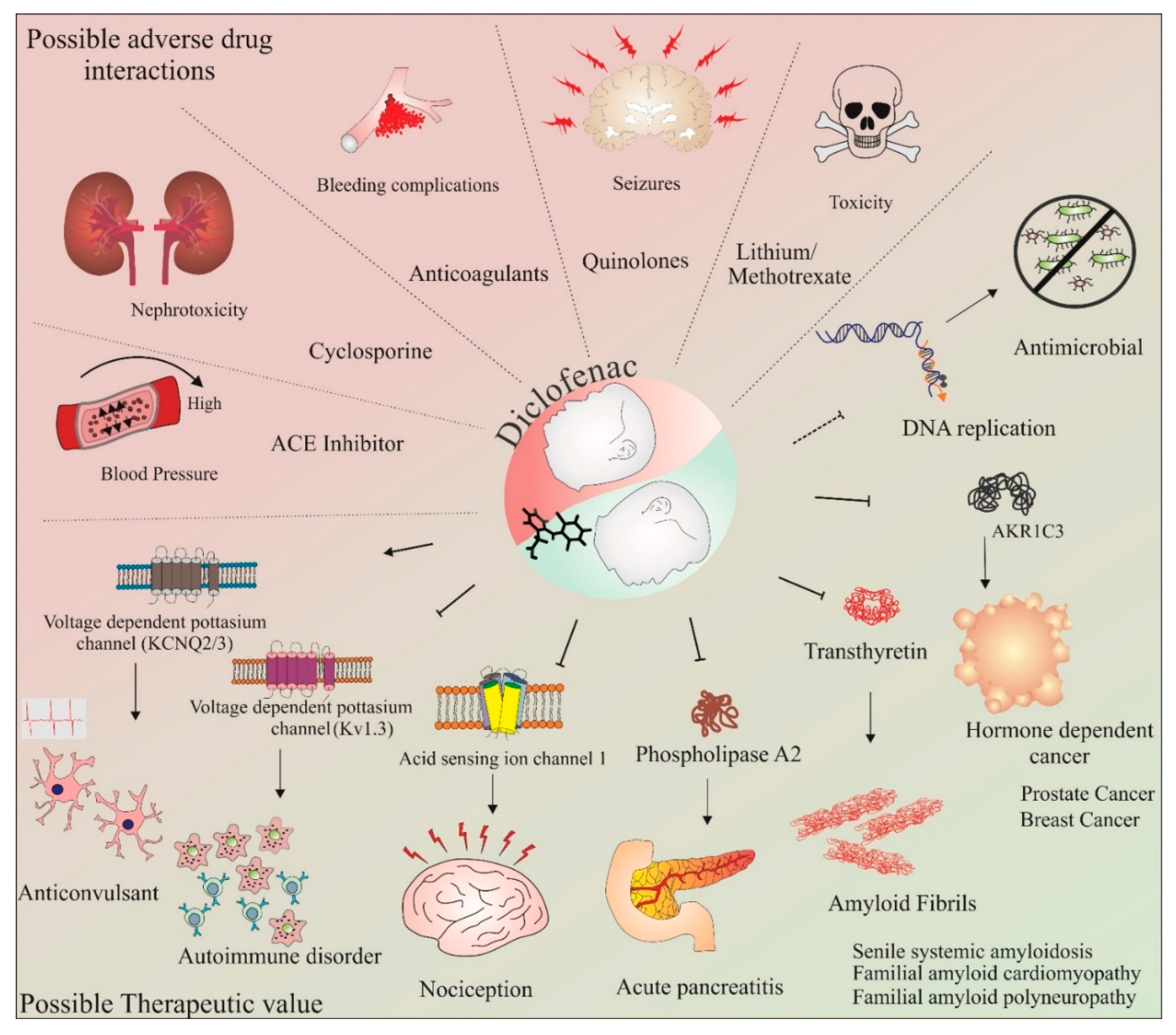
3. Diclofenac: Side Effects, Adverse Drug Interactions, and Therapeutic Potential beyond Anti-Inflammation
3.1. Side Effects
3.2. Adverse Drug Interactions
3.3. Therapeutic Potential beyond Anti-Inflammation
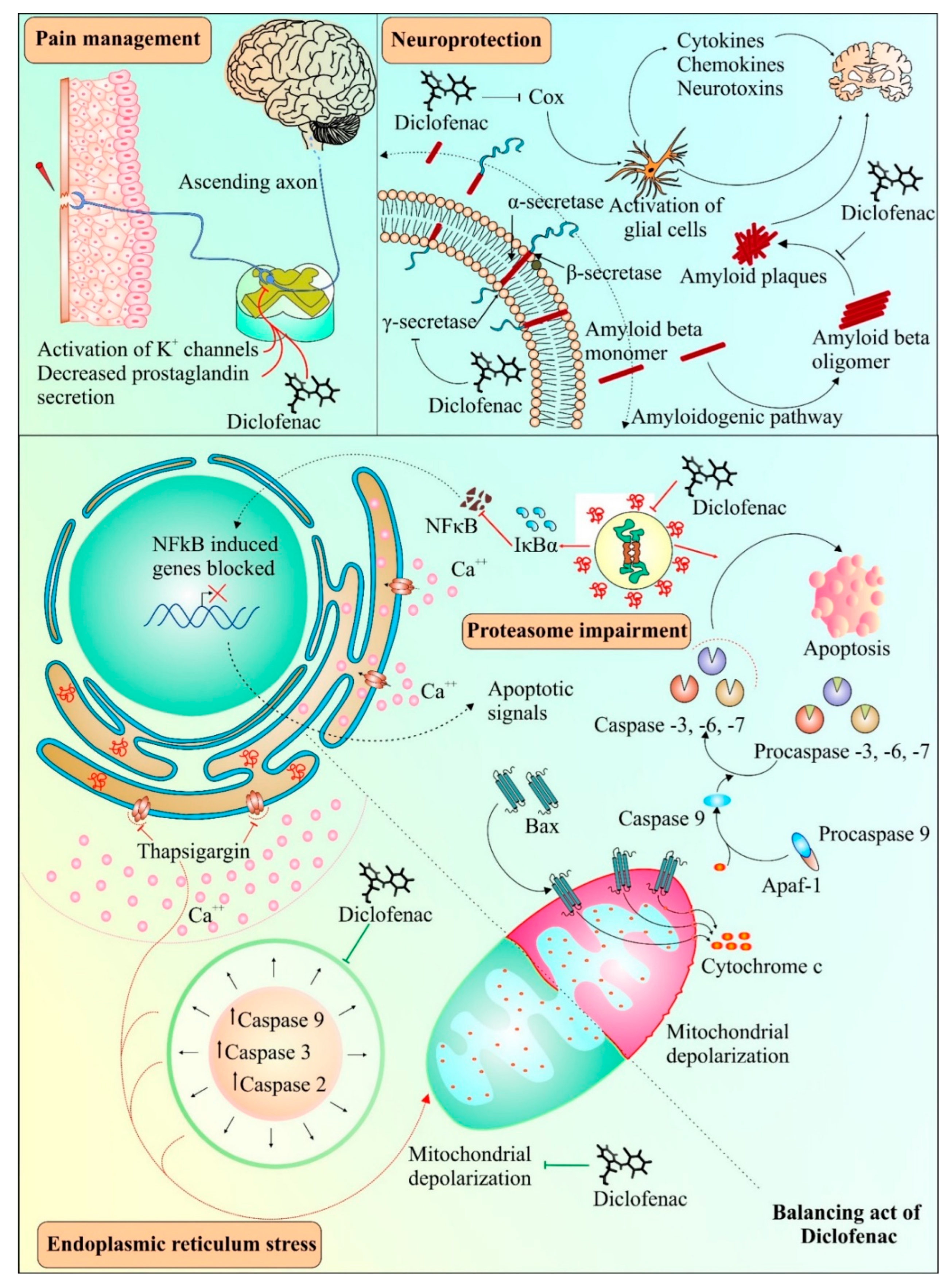
4. Neuroprotective Abilities of Diclofenac: A Unique Possibility
5. Diclofenac: Several Targets, Multiple Outcomes
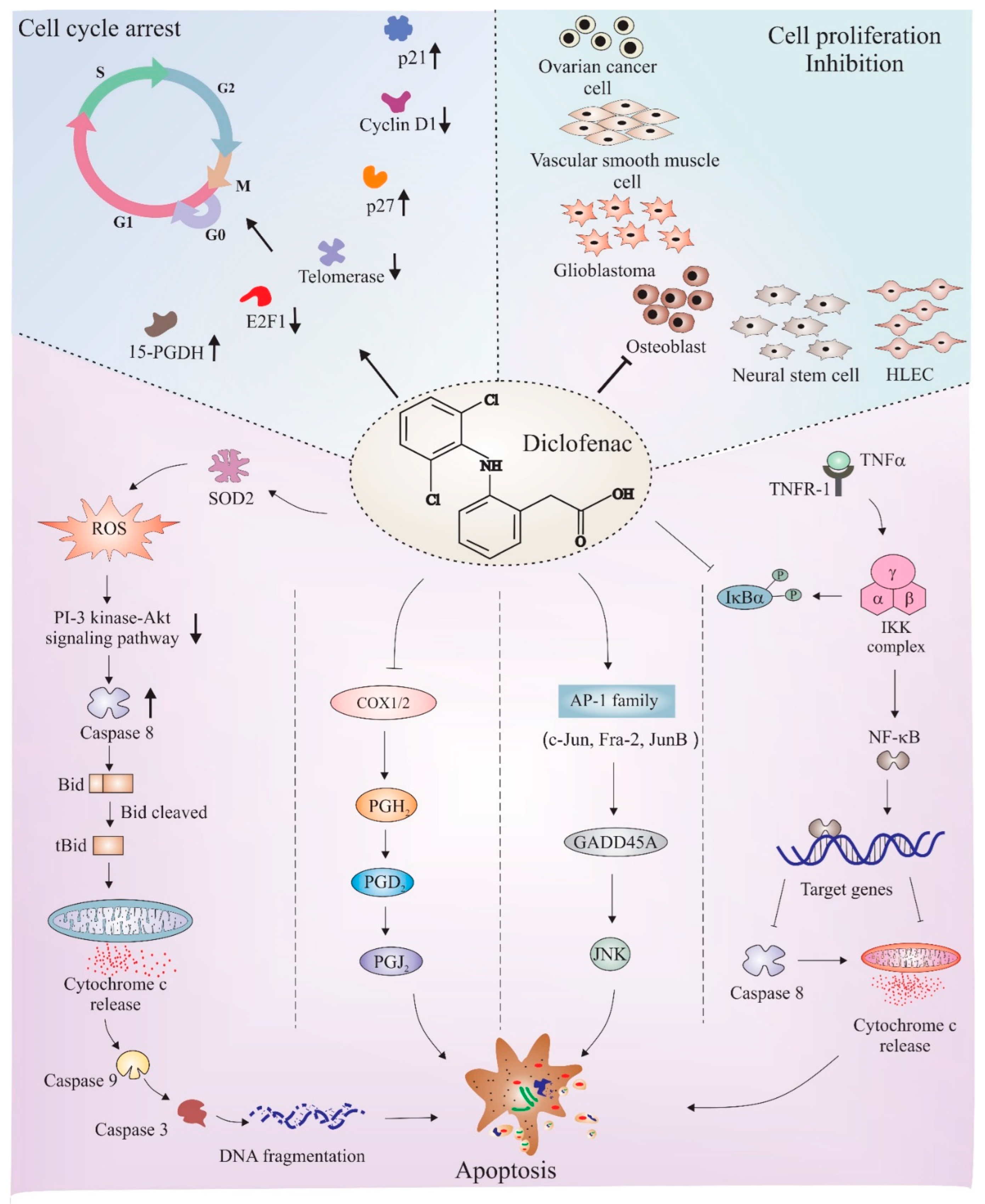
6. Diclofenac as an Anticancer Agent: Role of Cell Cycle Regulation and Apoptosis
6.1. Cell Cycle Regulation
6.2. Apoptosis
7. Discussion
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKR1C3 | Aldo-keto-reductase family 1 member C3 |
| Mrp-2 | Multidrug resistance associated protein-2 |
| NF-kB | Nuclear factor kappa light chain enhancer of activated B cells |
| NMDA | N-methyl D-aspartate receptor |
| PGI 2 | Prostaglandin-I (2) |
| ROS | Reactive oxygen species |
| Tnf-α | Tumor necrosis factor alpha |
| TxA 2 | Thromboxane-A (2) |
References
- Harizi, H.; Corcuff, J.B.; Gualde, N. Arachidonic-acid-derived eicosanoids: Roles in biology and immunopathology. Trends Mol. Med. 2008, 14, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Xue, D.; Zheng, Q.; Li, H.; Qian, S.; Zhang, B.; Pan, Z. Selective COX-2 inhibitor versus nonselective COX-1 and COX-2 inhibitor in the prevention of heterotopic ossification after total hip arthroplasty: A meta-analysis of randomised trials. Int. Orthop. 2011, 35, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.-J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. IJPR 2011, 10, 655–683. [Google Scholar]
- Harris, R.E.; Beebe, J.; Alshafie, G.A. Reduction in cancer risk by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. J. Exp. Pharmacol. 2012, 4, 91–96. [Google Scholar] [CrossRef]
- Khan, M.; Fraser, A. Cox-2 inhibitors and the risk of cardiovascular thrombotic events. Ir. Med. J. 2012, 105, 119–121. [Google Scholar]
- Nussmeier, N.A.; Whelton, A.A.; Brown, M.T.; Langford, R.M.; Hoeft, A.; Parlow, J.L.; Boyce, S.W.; Verburg, K.M. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N. Engl. J. Med. 2005, 352, 1081–1091. [Google Scholar] [CrossRef]
- Brueggemann, L.I.; Mani, B.K.; Mackie, A.R.; Cribbs, L.L.; Byron, K.L. Novel Actions of Nonsteroidal Anti-Inflammatory Drugs on Vascular Ion Channels: Accounting for Cardiovascular Side Effects and Identifying New Therapeutic Applications. Mol. Cell. Pharmacol. 2010, 2, 15–19. [Google Scholar]
- Lu, W.; Cheng, F.; Jiang, J.; Zhang, C.; Deng, X.; Xu, Z.; Zou, S.; Shen, X.; Tang, Y.; Huang, J. FXR antagonism of NSAIDs contributes to drug-induced liver injury identified by systems pharmacology approach. Sci. Rep. 2015, 5, 8114. [Google Scholar] [CrossRef]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Neuroprotective effects of nonsteroidal anti-inflammatory drugs on neurodegenerative diseases. Curr. Pharm. Des. 2004, 10, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Dell’Omo, G.; Crescenti, D.; Vantaggiato, C.; Parravicini, C.; Borroni, A.P.; Rizzi, N.; Garofalo, M.; Pinto, A.; Recordati, C.; Scanziani, E.; et al. Inhibition of SIRT1 deacetylase and p53 activation uncouples the anti-inflammatory and chemopreventive actions of NSAIDs. Br. J. Cancer 2019, 120, 537–546. [Google Scholar] [CrossRef]
- Gan, T.J. Diclofenac: An update on its mechanism of action and safety profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef]
- Wehling, M. Non-steroidal anti-inflammatory drug use in chronic pain conditions with special emphasis on the elderly and patients with relevant comorbidities: Management and mitigation of risks and adverse effects. Eur. J. Clin. Pharmacol. 2014, 70, 1159–1172. [Google Scholar] [CrossRef]
- Alfaro, R.A.; Davis, D.D. Diclofenac. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sareddy, G.R.; Kesanakurti, D.; Kirti, P.B.; Babu, P.P. Nonsteroidal anti-inflammatory drugs diclofenac and celecoxib attenuates Wnt/beta-catenin/Tcf signaling pathway in human glioblastoma cells. Neurochem. Res. 2013, 38, 2313–2322. [Google Scholar] [CrossRef]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New aspects of an old drug--diclofenac targets MYC and glucose metabolism in tumor cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef]
- Mehndiratta, M.M.; Wadhai, S.A.; Tyagi, B.K.; Gulati, N.S.; Sinha, M. Drug repositioning. Int. J. Epilepsy 2016, 3, 91–94. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Massey, T.H.; Robertson, N.P. Repurposing drugs to treat neurological diseases. J. Neurol. 2018, 265, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, C.; Kalra, S. Bromocriptine in type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2011, 15, S17–S24. [Google Scholar] [CrossRef]
- Pushpakom, S. Chapter 1 Introduction and Historical Overview of Drug Repurposing Opportunities. In Drug Repurposing; The Royal Society of Chemistry: London, UK, 2022; pp. 1–13. [Google Scholar]
- Derry, P.; Derry, S.; Moore, R.A.; McQuay, H.J. Single dose oral diclofenac for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2009, CD004768. [Google Scholar] [CrossRef]
- Atzeni, F.; Masala, I.F.; Sarzi-Puttini, P. A Review of Chronic Musculoskeletal Pain: Central and Peripheral Effects of Diclofenac. Pain Ther. 2018, 7, 163–177. [Google Scholar] [CrossRef]
- Rodrigues, E.B.; Farah, M.E.; Bottós, J.M.; Aggio, F.B. CHAPTER 29—Nonsteroidal anti-inflammatory drugs (NSAIDs) in the treatment of retinal diseases. In Retinal Pharmacotherapy; Nguyen, Q.D., Rodrigues, E.B., Farah, M.E., Mieler, W.F., Eds.; W.B. Saunders: Edinburgh, Scotland, 2010; pp. 196–200. [Google Scholar]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID Development: Evolution of Diclofenac Products Using Pharmaceutical Technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef]
- Fini, A.; Bassini, G.; Monastero, A.; Cavallari, C. Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions. Pharmaceutics 2012, 4, 413–429. [Google Scholar] [CrossRef]
- Skoutakis, V.A.; Carter, C.A.; Mickle, T.R.; Smith, V.H.; Arkin, C.R.; Alissandratos, J.; Petty, D.E. Review of diclofenac and evaluation of its place in therapy as a nonsteroidal antiinflammatory agent. Drug Intell. Clin. Pharm. 1988, 22, 850–859. [Google Scholar] [CrossRef]
- Davies, N.M.; Anderson, K.E. Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef]
- Reiner, V.; Reiner, A.; Reiner, G.; Conti, M. Increased absorption rate of diclofenac from fast acting formulations containing its potassium salt. Arzneim. Forsch. 2001, 51, 885–890. [Google Scholar] [CrossRef]
- Macia, M.A.; Frias, J.; Carcas, A.J.; Guerra, P.; Valiente, R.; Lucero, M.L. Comparative bioavailability of a dispersible formulation of diclofenac and finding of double plasma peaks. Int. J. Clin. Pharmacol. Ther. 1995, 33, 333–339. [Google Scholar] [PubMed]
- Garbacz, G.; Weitschies, W. Investigation of dissolution behavior of diclofenac sodium extended release formulations under standard and biorelevant test conditions. Drug Dev. Ind. Pharm. 2010, 36, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, B.; Buvanendran, A. 40—Nonsteroidal Anti-inflammatory Drugs, Acetaminophen, and COX-2 Inhibitors. In Practical Management of Pain, 5th ed.; Benzon, H.T., Rathmell, J.P., Wu, C.L., Turk, D.C., Argoff, C.E., Hurley, R.W., Eds.; Mosby: Philadelphia, PA, USA, 2014; pp. 553–568.e555. [Google Scholar]
- John, V.A. The pharmacokinetics and metabolism of diclofenac sodium (Voltarol) in animals and man. Rheumatol. Rehabil. 1979, 2, 22–37. [Google Scholar] [PubMed]
- Lagas, J.S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Hepatic clearance of reactive glucuronide metabolites of diclofenac in the mouse is dependent on multiple ATP-binding cassette efflux transporters. Mol. Pharmacol. 2010, 77, 687–694. [Google Scholar] [CrossRef]
- King, C.; Tang, W.; Ngui, J.; Tephly, T.; Braun, M. Characterization of Rat and Human UDP-Glucuronosyltransferases Responsible for the in Vitro Glucuronidation of Diclofenac. Toxicol. Sci. 2001, 61, 49–53. [Google Scholar] [CrossRef]
- Tang, W.; Stearns, R.A.; Bandiera, S.M.; Zhang, Y.; Raab, C.; Braun, M.P.; Dean, D.C.; Pang, J.; Leung, K.H.; Doss, G.A.; et al. Studies on cytochrome P-450-mediated bioactivation of diclofenac in rats and in human hepatocytes: Identification of glutathione conjugated metabolites. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 365–372. [Google Scholar]
- Poon, G.K.; Chen, Q.; Teffera, Y.; Ngui, J.S.; Griffin, P.R.; Braun, M.P.; Doss, G.A.; Freeden, C.; Stearns, R.A.; Evans, D.C.; et al. Bioactivation of diclofenac via benzoquinone imine intermediates-identification of urinary mercapturic acid derivatives in rats and humans. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 1608–1613. [Google Scholar]
- Marel, C.D.; Anderson, B.J.; Rømsing, J.; Jacqz-Aigrain, E.; Tibboel, D. Diclofenac and metabolite pharmacokinetics in children. Pediatric Anesth. 2004, 14, 443–451. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Meineke, I.; Steinbach, N.; Meisel, C.; Roots, I.; Brockmöller, J. Pharmacokinetics of diclofenac and inhibition of cyclooxygenases 1 and 2: No relationship to the CYP2C9 genetic polymorphism in humans. Br. J. Clin. Pharmacol. 2003, 55, 51–61. [Google Scholar] [CrossRef]
- Dorado, P.; Berecz, R.; Cáceres, M.C.; Llerena, A. Analysis of diclofenac and its metabolites by high-performance liquid chromatography: Relevance of CYP2C9 genotypes in diclofenac urinary metabolic ratios. J. Chromatogr. B 2003, 789, 437–442. [Google Scholar] [CrossRef]
- Ngui, J.S.; Tang, W.; Stearns, R.A.; Shou, M.; Miller, R.R.; Zhang, Y.; Lin, J.H.; Baillie, T.A. Cytochrome P450 3A4-mediated interaction of diclofenac and quinidine. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 1043–1050. [Google Scholar] [PubMed]
- Brogden, R.N.; Heel, R.C.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Diclofenac Sodium: A Review of its Pharmacological Properties and Therapeutic Use in Rheumatic Diseases and Pain of Varying Origin. Drugs 1980, 20, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Suhail, M.; Wu, P.-C.; Minhas, M.U. Development and characterization of pH-sensitive chondroitin sulfate-co-poly(acrylic acid) hydrogels for controlled release of diclofenac sodium. J. Saudi Chem. Soc. 2021, 25, 101212. [Google Scholar] [CrossRef]
- Lyons, J.L.; Tovar-y-Romo, L.B.; Thakur, K.T.; McArthur, J.C.; Haughey, N.J. Chapter 28—Pathobiology of CNS Human Immunodeficiency Virus Infection. In Neurobiology of Brain Disorders; Zigmond, M.J., Rowland, L.P., Coyle, J.T., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 444–466. [Google Scholar]
- Litalien, C.; Beaulieu, P. Chapter 117—Molecular Mechanisms of Drug Actions: From Receptors to Effectors. In Pediatric Critical Care, 4th ed.; Fuhrman, B.P., Zimmerman, J.J., Eds.; Mosby: Saint Louis, Missouri, 2011; pp. 1553–1568. [Google Scholar]
- Al-Nimer, M.S.M.; Hameed, H.G.; Mahmood, M.M. Antiproliferative effects of aspirin and diclofenac against the growth of cancer and fibroblast cells: In vitro comparative study. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2015, 23, 483–486. [Google Scholar] [CrossRef]
- Power, I.; Chambers, W.A.; Greer, I.A.; Ramage, D.; Simon, E. Platelet function after intramuscular diclofenac. Anaesthesia 2007, 45, 916–919. [Google Scholar] [CrossRef]
- Niu, X.; de Graaf, I.A.; van de Vegte, D.; Langelaar-Makkinje, M.; Sekine, S.; Groothuis, G.M. Consequences of Mrp2 deficiency for diclofenac toxicity in the rat intestine ex vivo. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2015, 29, 168–175. [Google Scholar] [CrossRef]
- Seitz, S.; Kretz-Rommel, A.; Oude Elferink, R.P.; Boelsterli, U.A. Selective protein adduct formation of diclofenac glucuronide is critically dependent on the rat canalicular conjugate export pump (Mrp2). Chem. Res. Toxicol. 1998, 11, 513–519. [Google Scholar] [CrossRef]
- Dong, X.D.; Svensson, P.; Cairns, B.E. The analgesic action of topical diclofenac may be mediated through peripheral NMDA receptor antagonism. Pain 2009, 147, 36–45. [Google Scholar] [CrossRef]
- Russell, R.I. Non-steroidal anti-inflammatory drugs and gastrointestinal damage—Problems and solutions. Postgrad. Med. J. 2001, 77, 82. [Google Scholar] [CrossRef]
- McGettigan, P.; Henry, D. Use of non-steroidal anti-inflammatory drugs that elevate cardiovascular risk: An examination of sales and essential medicines lists in low-, middle-, and high-income countries. PLoS Med. 2013, 10, e1001388. [Google Scholar] [CrossRef]
- Boelsterli, U.A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef]
- Dhanvijay, P.; Misra, A.K.; Varma, S.K. Diclofenac induced acute renal failure in a decompensated elderly patient. J. Pharmacol. Pharmacother. 2013, 4, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Struthmann, L.; Hellwig, N.; Pircher, J.; Sohn, H.Y.; Buerkle, M.A.; Klauss, V.; Mannell, H.; Pohl, U.; Krotz, F. Prothrombotic effects of diclofenac on arteriolar platelet activation and thrombosis in vivo. J. Thromb. Haemost. JTH 2009, 7, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Moore, N. Coronary Risks Associated with Diclofenac and Other NSAIDs: An Update. Drug Saf. 2020, 43, 301–318. [Google Scholar] [CrossRef]
- Schmidt, M.; Sørensen, H.T.; Pedersen, L. Diclofenac use and cardiovascular risks: Series of nationwide cohort studies. BMJ 2018, 362, k3426. [Google Scholar] [CrossRef] [Green Version]
- Shainhouse, J.Z.; Grierson, L.M.; Naseer, Z. A long-term, open-label study to confirm the safety of topical diclofenac solution containing dimethyl sulfoxide in the treatment of the osteoarthritic knee. Am. J. Ther. 2010, 17, 566–576. [Google Scholar] [CrossRef]
- Nair, B.; Taylor-Gjevre, R. A Review of Topical Diclofenac Use in Musculoskeletal Disease. Pharmaceuticals 2010, 3, 1892–1908. [Google Scholar] [CrossRef]
- Jha, A.A.; Bohra, V.; Behera, V. Severe anaphylactic reaction to diclofenac. Med. J. Armed Forces India 2015, 71, S279–S281. [Google Scholar] [CrossRef]
- Yarishkin, O.V.; Hwang, E.M.; Kim, D.; Yoo, J.C.; Kang, S.S.; Kim, D.R.; Shin, J.-H.-J.; Chung, H.-J.; Jeong, H.-S.; Kang, D.; et al. Diclofenac, a Non-steroidal Anti-inflammatory Drug, Inhibits L-type Ca2+ Channels in Neonatal Rat Ventricular Cardiomyocytes. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2009, 13, 437–442. [Google Scholar] [CrossRef]
- Cantoni, L.; Valaperta, R.; Ponsoda, X.; Castell, J.V.; Barelli, D.; Rizzardini, M.; Mangolini, A.; Hauri, L.; Villa, P. Induction of hepatic heme oxygenase-1 by diclofenac in rodents: Role of oxidative stress and cytochrome P-450 activity. J. Hepatol. 2003, 38, 776–783. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Ponsoda, X.; O’Connor, E.; Donato, T.; Castell, J.V.; Jover, R. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem. Pharmacol. 2003, 66, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, W.J.; Olayaki, L.A. Diclofenac—Induced hepatotoxicity: Low dose of omega-3 fatty acids have more protective effects. Toxicol. Rep. 2018, 5, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Lee, W.; Park, S.-H.; Lee, K.-Y.; Choi, Y.-J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.-S.; Hong, S.-S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, T.; Qureshi, G.S.; Perveen, S. Adverse effects of diclofenac sodium on renal parenchyma of adult albino rats. J. Pak. Med. Assoc. JPMA 2007, 57, 349–351. [Google Scholar]
- Ghosh, R.; Goswami, S.K.; Feitoza, L.F.; Hammock, B.; Gomes, A.V. Diclofenac induces proteasome and mitochondrial dysfunction in murine cardiomyocytes and hearts. Int. J. Cardiol. 2016, 223, 923–935. [Google Scholar] [CrossRef]
- Amanullah, A.; Upadhyay, A.; Chhangani, D.; Joshi, V.; Mishra, R.; Yamanaka, K.; Mishra, A. Proteasomal Dysfunction Induced By Diclofenac Engenders Apoptosis Through Mitochondrial Pathway. J. Cell. Biochem. 2017, 118, 1014–1027. [Google Scholar] [CrossRef]
- Izhar, M.; Alausa, T.; Folker, A.; Hung, E.; Bakris, G.L. Effects of COX inhibition on blood pressure and kidney function in ACE inhibitor-treated blacks and hispanics. Hypertension 2004, 43, 573–577. [Google Scholar] [CrossRef]
- Colombo, M.D.; Perego, R.; Bellia, G. Cyclosporine-Associated Nephrotoxicity. Open J. Nephrol. 2013, 3, 13. [Google Scholar] [CrossRef]
- Willis, J.V.; Kendall, M.J.; Jack, D.B. A study of the effect of aspirin on the pharmacokinetics of oral and intravenous diclofenac sodium. Eur. J. Clin. Pharmacol. 1980, 18, 415–418. [Google Scholar] [CrossRef]
- Knijff-Dutmer, E.A.J.; Schut, G.A.; van de Laar, M.A.F.J. Concomitant Coumarin–NSAID Therapy and Risk for Bleeding. Ann. Pharmacother. 2003, 37, 12–16. [Google Scholar] [CrossRef]
- Shrivastava, M.P.; Makde, S.D.; Paranjpe, B.D. Interaction of ciprofloxacin with diclofenac and paracetamol in relation to it’s epileptogenic effect. Indian J. Physiol. Pharmacol. 1997, 41, 164–166. [Google Scholar] [PubMed]
- Grissinger, M. Severe Harm and Death Associated With Errors and Drug Interactions Involving Low-Dose Methotrexate. Pharm. Ther. 2018, 43, 191–248. [Google Scholar]
- Kawase, A.; Yamamoto, T.; Egashira, S.; Iwaki, M. Stereoselective Inhibition of Methotrexate Excretion by Glucuronides of Nonsteroidal Anti-inflammatory Drugs via Multidrug Resistance Proteins 2 and 4. J. Pharmacol. Exp. Ther. 2016, 356, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.D.; Sorathia, Z.H. Tramadol/Diclofenac Fixed-Dose Combination: A Review of Its Use in Severe Acute Pain. Pain Ther. 2020, 9, 113–128. [Google Scholar] [CrossRef]
- Peretz, A.; Degani, N.; Nachman, R.; Uziyel, Y.; Gibor, G.; Shabat, D.; Attali, B. Meclofenamic Acid and Diclofenac, Novel Templates of KCNQ2/Q3 Potassium Channel Openers, Depress Cortical Neuron Activity and Exhibit Anticonvulsant Properties. Mol. Pharmacol. 2005, 67, 1053. [Google Scholar] [CrossRef]
- Ihara, Y.; Tomonoh, Y.; Deshimaru, M.; Zhang, B.; Uchida, T.; Ishii, A.; Hirose, S. Retigabine, a Kv7.2/Kv7.3-Channel Opener, Attenuates Drug-Induced Seizures in Knock-In Mice Harboring Kcnq2 Mutations. PLoS ONE 2016, 11, e0150095. [Google Scholar] [CrossRef]
- Gwanyanya, A.; Macianskiene, R.; Mubagwa, K. Insights into the effects of diclofenac and other non-steroidal anti-inflammatory agents on ion channels. J. Pharm. Pharmacol. 2012, 64, 1359–1375. [Google Scholar] [CrossRef]
- Lam, J.; Wulff, H. The Lymphocyte Potassium Channels Kv1.3 and KCa3.1 as Targets for Immunosuppression. Drug Dev. Res. 2011, 72, 573–584. [Google Scholar] [CrossRef]
- Martínez-Mármol, R.; Styrczewska, K.; Pérez-Verdaguer, M.; Vallejo-Gracia, A.; Comes, N.; Sorkin, A.; Felipe, A. Ubiquitination mediates Kv1.3 endocytosis as a mechanism for protein kinase C-dependent modulation. Sci. Rep. 2017, 7, 42395. [Google Scholar] [CrossRef]
- Villalonga, N.; David, M.; Bielanska, J.; Gonzalez, T.; Parra, D.; Soler, C.; Comes, N.; Valenzuela, C.; Felipe, A. Immunomodulatory effects of diclofenac in leukocytes through the targeting of Kv1.3 voltage-dependent potassium channels. Biochem. Pharmacol. 2010, 80, 858–866. [Google Scholar] [CrossRef]
- Voilley, N.; de Weille, J.; Mamet, J.; Lazdunski, M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 8026–8033. [Google Scholar] [CrossRef]
- Murray, B.; Carter, R.; Imrie, C.; Evans, S.; O’Suilleabhain, C. Diclofenac reduces the incidence of acute pancreatitis after endoscopic retrograde cholangiopancreatography. Gastroenterology 2003, 124, 1786–1791. [Google Scholar] [CrossRef]
- Makela, A.; Kuusi, T.; Schroder, T. Inhibition of serum phospholipase-A2 in acute pancreatitis by pharmacological agents in vitro. Scand. J. Clin. Lab. Investig. 1997, 57, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Oza, V.B.; Smith, C.; Raman, P.; Koepf, E.K.; Lashuel, H.A.; Petrassi, H.M.; Chiang, K.P.; Powers, E.T.; Sachettinni, J.; Kelly, J.W. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J. Med. Chem. 2002, 45, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Gobec, S.; Brozic, P.; Rizner, T.L. Nonsteroidal anti-inflammatory drugs and their analogues as inhibitors of aldo-keto reductase AKR1C3: New lead compounds for the development of anticancer agents. Bioorganic Med. Chem. Lett. 2005, 15, 5170–5175. [Google Scholar] [CrossRef]
- Mazumdar, K.; Dastidar, S.G.; Park, J.H.; Dutta, N.K. The anti-inflammatory non-antibiotic helper compound diclofenac: An antibacterial drug target. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2009, 28, 881–891. [Google Scholar] [CrossRef]
- Ortiz, M.I.; Torres-Lopez, J.E.; Castaneda-Hernandez, G.; Rosas, R.; Vidal-Cantu, G.C.; Granados-Soto, V. Pharmacological evidence for the activation of K(+) channels by diclofenac. Eur. J. Pharmacol. 2002, 438, 85–91. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, H.I.; Shin, Y.K.; Lee, C.S.; Park, M.; Song, J.H. Diclofenac inhibition of sodium currents in rat dorsal root ganglion neurons. Brain Res. 2003, 992, 120–127. [Google Scholar] [CrossRef]
- Dorofeeva, N.A.; Barygin, O.I.; Staruschenko, A.; Bolshakov, K.V.; Magazanik, L.G. Mechanisms of non-steroid anti-inflammatory drugs action on ASICs expressed in hippocampal interneurons. J. Neurochem. 2008, 106, 429–441. [Google Scholar] [CrossRef]
- Benesic, A.; Rahm, N.L.; Ernst, S.; Gerbes, A.L. Human monocyte-derived cells with individual hepatocyte characteristics: A novel tool for personalized in vitro studies. Lab. Investig. 2012, 92, 926–936. [Google Scholar] [CrossRef]
- Benesic, A.; Jalal, K.; Gerbes, A.L. Drug-Drug Combinations Can Enhance Toxicity as Shown by Monocyte-Derived Hepatocyte-like Cells From Patients With Idiosyncratic Drug-Induced Liver Injury. Toxicol. Sci. 2019, 171, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Naeem, S.; Najam, R.; Khan, S.S.; Mirza, T.; Sikandar, B. Neuroprotective effect of diclofenac on chlorpromazine induced catalepsy in rats. Metab. Brain Dis. 2019, 34, 1191–1199. [Google Scholar] [CrossRef]
- Parmar, H.S.; Assaiya, A.; Agrawal, R.; Tiwari, S.; Mufti, I.; Jain, N.; Manivannan, E.; Banerjee, T.; Kumar, A. Inhibition of Aβ(1-42)Oligomerization, Fibrillization and Acetylcholinesterase Activity by Some Anti-Inflammatory Drugs: An in vitro Study. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2017, 15, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Galant, N.J.; Bugyei-Twum, A.; Rakhit, R.; Walsh, P.; Sharpe, S.; Arslan, P.E.; Westermark, P.; Higaki, J.N.; Torres, R.; Tapia, J.; et al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: A potential diagnostic and therapeutic for TTR amyloidoses. Sci. Rep. 2016, 6, 25080. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T. Amyloidosis of pancreatic islets in primary amyloidosis (AL type). Pathol. Int. 2005, 55, 223–227. [Google Scholar] [CrossRef]
- Fortin, J.S.; Benoit-Biancamano, M.O. Inhibition of islet amyloid polypeptide aggregation and associated cytotoxicity by nonsteroidal anti-inflammatory drugs. Can. J. Physiol. Pharmacol. 2016, 94, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef]
- Tzeng, S.F.; Hsiao, H.Y.; Mak, O.T. Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 335–340. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-Steroidal Anti-Inflammatory Drugs and Brain Inflammation: Effects on Microglial Functions. Pharmaceuticals 2010, 3, 1949–1964. [Google Scholar] [CrossRef]
- Dehlaghi Jadid, K.; Davidsson, J.; Lidin, E.; Hånell, A.; Angéria, M.; Mathiesen, T.; Risling, M.; Günther, M. COX-2 Inhibition by Diclofenac Is Associated with Decreased Apoptosis and Lesion Area After Experimental Focal Penetrating Traumatic Brain Injury in Rats. Front. Neurol. 2019, 10, 811. [Google Scholar] [CrossRef]
- Hassan, H.; Varney, M.; Dave, B.J.; Singh, R.K. Diclofenac Induces Apoptosis and Suppresses Diffuse Large B-Cell Lymphoma Proliferation Independent of P53 Status. Blood 2014, 124, 5485. [Google Scholar] [CrossRef]
- Ashton, M.; Hanson, P.J. Disparate effects of non-steroidal anti-inflammatory drugs on apoptosis in guinea-pig gastric mucous cells: Inhibition of basal apoptosis by diclofenac. Br. J. Pharmacol. 2002, 135, 407–416. [Google Scholar] [CrossRef]
- Yamazaki, T.; Muramoto, M.; Oe, T.; Morikawa, N.; Okitsu, O.; Nagashima, T.; Nishimura, S.; Katayama, Y.; Kita, Y. Diclofenac, a non-steroidal anti-inflammatory drug, suppresses apoptosis induced by endoplasmic reticulum stresses by inhibiting caspase signaling. Neuropharmacology 2006, 50, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Muranaka, S.; Fujita, H.; Kanno, T.; Tamai, H.; Utsumi, K. Molecular mechanism of diclofenac-induced apoptosis of promyelocytic leukemia: Dependency on reactive oxygen species, Akt, Bid, cytochrome and caspase pathway. Free. Radic. Biol. Med. 2004, 37, 1290–1299. [Google Scholar] [CrossRef]
- Hassan, H.M.; Varney, M.L.; Chaturvedi, N.K.; Joshi, S.S.; Weisenburger, D.D.; Singh, R.K.; Dave, B.J. Modulation of p73 isoforms expression induces anti-proliferative and pro-apoptotic activity in mantle cell lymphoma independent of p53 status. Leuk. Lymphoma 2016, 57, 2874–2889. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. BioMed Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, C.; Slingerland, J. Deregulation of the cell cycle in cancer. Cancer Detect. Prev. 2000, 24, 107–118. [Google Scholar]
- Kohrman, A.Q.; Matus, D.Q. Divide or Conquer: Cell Cycle Regulation of Invasive Behavior. Trends Cell Biol. 2017, 27, 12–25. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef]
- Senderowicz, A.M. The Cell Cycle as a Target for Cancer Therapy: Basic and Clinical Findings with the Small Molecule Inhibitors Flavopiridol and UCN-01. Oncologist 2002, 7, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Pegram, M.D.; Konecny, G.E.; O’Callaghan, C.; Beryt, M.; Pietras, R.; Slamon, D.J. Rational Combinations of Trastuzumab With Chemotherapeutic Drugs Used in the Treatment of Breast Cancer. JNCI J. Natl. Cancer Inst. 2004, 96, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Pomatico, G.; De Placido, S.; Bianco, A.R.; Tortora, G. Antitumor Effect and Potentiation of Cytotoxic Drugs Activity in Human Cancer Cells by ZD-1839 (Iressa), an Epidermal Growth Factor Receptor-selective Tyrosine Kinase Inhibitor. Clin. Cancer Res. 2000, 6, 2053. [Google Scholar] [PubMed]
- Sparreboom, A.; de Jonge, M.J.A.; Verweij, J. The use of oral cytotoxic and cytostatic drugs in cancer treatment. Eur. J. Cancer 2002, 38, 18–22. [Google Scholar] [CrossRef]
- Amanullah, A.; Upadhyay, A.; Joshi, V.; Mishra, R.; Jana, N.R.; Mishra, A. Progressing neurobiological strategies against proteostasis failure: Challenges in neurodegeneration. Prog. Neurobiol. 2017, 159, 1–38. [Google Scholar] [CrossRef]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2007, 9, 767–776. [Google Scholar] [CrossRef]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In Vitro and In Vivo Anticancer Activities of Synthetic Macrocyclic Ketone Analogues of Halichondrin B. Cancer Res. 2001, 61, 1013. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Szabo, C. Inventing new therapies without reinventing the wheel: The power of drug repurposing. Br. J. Pharmacol. 2018, 175, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Fox, A.; Gentry, C.; Patel, S.; Kesingland, A.; Bevan, S. Comparative activity of the anti-convulsants oxcarbazepine, carbamazepine, lamotrigine and gabapentin in a model of neuropathic pain in the rat and guinea-pig. Pain 2003, 105, 355–362. [Google Scholar] [CrossRef]
- Friedman, B.; Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt. Bone Spine 2018, 86, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Cecere, F.; Iuliano, A.; Albano, F.; Zappelli, C.; Castellano, I.; Grimaldi, P.; Masullo, M.; De Vendittis, E.; Ruocco, M.R. Diclofenac-Induced Apoptosis in the Neuroblastoma Cell Line SH-SY5Y: Possible Involvement of the Mitochondrial Superoxide Dismutase. J. Biomed. Biotechnol. 2010, 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Krischak, G.D.; Augat, P.; Blakytny, R.; Claes, L.; Kinzl, L.; Beck, A. The non-steroidal anti-inflammatory drug diclofenac reduces appearance of osteoblasts in bone defect healing in rats. Arch. Orthop. Trauma Surg. 2007, 127, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Seliger, C.; Jachnik, B.; Welz, T.; Leukel, P.; Vollmann-Zwerenz, A.; Bogdahn, U.; Kreutz, M.; Grauer, O.M.; Hau, P. Ibuprofen and Diclofenac Restrict Migration and Proliferation of Human Glioma Cells by Distinct Molecular Mechanisms. PLoS ONE 2015, 10, e0140613. [Google Scholar] [CrossRef]
- Hamoya, T.; Fujii, G.; Miyamoto, S.; Takahashi, M.; Totsuka, Y.; Wakabayashi, K.; Toshima, J.; Mutoh, M. Effects of NSAIDs on the risk factors of colorectal cancer: A mini review. Genes Environ. Off. J. Jpn. Environ. Mutagen Soc. 2016, 38, 6. [Google Scholar] [CrossRef]
- Hofer, M.; Hoferova, Z.; Fedorocko, P.; Mackova, N.O. Hematopoiesis-stimulating and anti-tumor effects of repeated administration of diclofenac in mice with transplanted fibrosarcoma cells. Physiol. Res. 2002, 51, 629–632. [Google Scholar]
- Mayorek, N.; Naftali-Shani, N.; Grunewald, M. Diclofenac inhibits tumor growth in a murine model of pancreatic cancer by modulation of VEGF levels and arginase activity. PLoS ONE 2010, 5, e12715. [Google Scholar] [CrossRef]
- Wakimoto, N.; Wolf, I.; Yin, D.; Kelly, J.; Akagi, T.; Abramovitz, L.; Black, K.L.; Tai, H.-H.; Koeffler, H.P. Nonsteroidal Anti-inflammatory Drugs Suppress Glioma via 15-Hydroxyprostaglandin Dehydrogenase. Cancer Res. 2008, 68, 6978. [Google Scholar] [CrossRef]
- Brooks, G.; Yu, X.M.; Wang, Y.; Crabbe, M.J.; Shattock, M.J.; Harper, J.V. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit vascular smooth muscle cell proliferation via differential effects on the cell cycle. J. Pharm. Pharmacol. 2003, 55, 519–526. [Google Scholar] [CrossRef]
- Rana, C.; Piplani, H.; Vaish, V.; Nehru, B.; Sanyal, S.N. Downregulation of telomerase activity by diclofenac and curcumin is associated with cell cycle arrest and induction of apoptosis in colon cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 5999–6010. [Google Scholar] [CrossRef]
- Engelmann, D.; Pützer, B.M. The Dark Side of E2F1: In Transit beyond Apoptosis. Cancer Res. 2012, 72, 571. [Google Scholar] [CrossRef] [PubMed]
- Valle, B.L.; D’Souza, T.; Becker, K.G.; Wood, W.H.; Zhang, Y.; Wersto, R.P.; Morin, P.J. Non-Steroidal Anti-inflammatory Drugs Decrease E2F1 Expression and Inhibit Cell Growth in Ovarian Cancer Cells. PLoS ONE 2013, 8, e61836. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010, 215158. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Lindskog, M.; Ponthan, F.; Pettersen, I.; Elfman, L.; Orrego, A.; Sveinbjornsson, B.; Kogner, P. Cyclooxygenase-2 is expressed in neuroblastoma, and nonsteroidal anti-inflammatory drugs induce apoptosis and inhibit tumor growth in vivo. Cancer Res. 2004, 64, 7210–7215. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, A.; Yonekura, S.; Zhao, Q.L.; Wei, Z.L.; Takasaki, I.; Tabuchi, Y.; Wang, L.L.; Hasuike, S.; Nomura, T.; Tachibana, A.; et al. Mitochondria-targeted superoxide dismutase (SOD2) regulates radiation resistance and radiation stress response in HeLa cells. J. Radiat. Res. 2012, 53, 58–71. [Google Scholar] [CrossRef]
- Petrache, I.; Medler, T.R.; Richter, A.T.; Kamocki, K.; Chukwueke, U.; Zhen, L.; Gu, Y.; Adamowicz, J.; Schweitzer, K.S.; Hubbard, W.C.; et al. Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L44–L53. [Google Scholar] [CrossRef]
- Nzeako, U.C.; Guicciardi, M.E.; Yoon, J.-H.; Bronk, S.F.; Gores, G.J. COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology 2003, 35, 552–559. [Google Scholar] [CrossRef]
- Singh, R.; Cadeddu, R.P.; Frobel, J.; Wilk, C.M.; Bruns, I.; Zerbini, L.F.; Prenzel, T.; Hartwig, S.; Brunnert, D.; Schroeder, T.; et al. The non-steroidal anti-inflammatory drugs Sulindac sulfide and Diclofenac induce apoptosis and differentiation in human acute myeloid leukemia cells through an AP-1 dependent pathway. Apoptosis Int. J. Program. Cell Death 2011, 16, 889–901. [Google Scholar] [CrossRef]
- Fredriksson, L.; Herpers, B.; Benedetti, G.; Matadin, Q.; Puigvert, J.C.; de Bont, H.; Dragovic, S.; Vermeulen, N.P.; Commandeur, J.N.; Danen, E.; et al. Diclofenac inhibits tumor necrosis factor-alpha-induced nuclear factor-kappaB activation causing synergistic hepatocyte apoptosis. Hepatology 2011, 53, 2027–2041. [Google Scholar] [CrossRef]
- Li, H.; Fan, T.-J.; Zou, P.; Xu, B. Diclofenac Sodium Triggers p53-Dependent Apoptosis in Human Corneal Epithelial Cells via ROS-Mediated Crosstalk. Chem. Res. Toxicol. 2021, 34, 70–79. [Google Scholar] [CrossRef]
- Aboubakr, M.; Abdelkader, A.; Habotta, O.A.; Adel, N.; Emam, M.A.; Abdelhiee, E.Y.; Shanab, O.; Shoghy, K.; Elnoury, H.; Soliman, M.M.; et al. Cefepime and diclofenac sodium combined treatment-potentiated multiple organ injury: Role of oxidative damage and disrupted lipid metabolism. J. Biochem. Mol. Toxicol. 2021, 35, e22929. [Google Scholar] [CrossRef]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Dettmer, K.; Unger, P.; Schönhammer, G.; Renner, K.; Peter, K.; Siska, P.J.; Berneburg, M.; Herr, W.; Oefner, P.J.; et al. Topical Diclofenac Reprograms Metabolism and Immune Cell Infiltration in Actinic Keratosis. Front. Oncol. 2019, 9, 605. [Google Scholar] [CrossRef] [PubMed]
- Breuer, S.; Maimon, O.; Appelbaum, L.; Peretz, T.; Hubert, A. TL-118-anti-angiogenic treatment in pancreatic cancer: A case report. Med. Oncol. 2013, 30, 585. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Patel, C.; Sarkar, D.; Acharya, S. Lactobacillus Rhamnosus UBLR-58 and Diclofenac Potentiate the Anti- Alzheimer Activity of Curcumin in Mice. Curr. Enzym. Inhib. 2021, 17, 49–56. [Google Scholar] [CrossRef]
- Hoer, A.; Gothe, H.; Schiffhorst, G.; Sterzel, A.; Grass, U.; Haussler, B. Comparison of the effects of diclofenac or other non-steroidal anti-inflammatory drugs (NSAIDs) and diclofenac or other NSAIDs in combination with proton pump inhibitors (PPI) on hospitalisation due to peptic ulcer disease. Pharmacoepidemiol. Drug Saf. 2007, 16, 854–858. [Google Scholar] [CrossRef]
- Aycan, İ.Ö.; Elpek, Ö.; Akkaya, B.; Kıraç, E.; Tuzcu, H.; Kaya, S.; Coşkunfırat, N.; Aslan, M. Diclofenac induced gastrointestinal and renal toxicity is alleviated by thymoquinone treatment. Food Chem. Toxicol. 2018, 118, 795–804. [Google Scholar] [CrossRef]
- Tamaddonfard, E.; Samadi, F.; Egdami, K. The effects of vitamin B(12) and diclofenac and their combination on cold and mechanical allodynia in a neuropathic pain model in rats. Vet. Res. Forum Int. Q. J. 2013, 4, 19–24. [Google Scholar]
- Shep, D.; Khanwelkar, C.; Gade, P.; Karad, S. Efficacy and safety of combination of curcuminoid complex and diclofenac versus diclofenac in knee osteoarthritis: A randomized trial. Medicine 2020, 99, e19723. [Google Scholar] [CrossRef]
- Tateishi, Y.; Ohe, T.; Ogawa, M.; Takahashi, K.; Nakamura, S.; Mashino, T. Development of Novel Diclofenac Analogs Designed to Avoid Metabolic Activation and Hepatocyte Toxicity. ACS Omega 2020, 5, 32608–32616. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.M.; Vavia, P.R. Diclofenac-loaded biopolymeric nanosuspensions for ophthalmic application. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Pireddu, R.; Caddeo, C.; Valenti, D.; Marongiu, F.; Scano, A.; Ennas, G.; Lai, F.; Fadda, A.M.; Sinico, C. Diclofenac acid nanocrystals as an effective strategy to reduce in vivo skin inflammation by improving dermal drug bioavailability. Colloids Surf. B Biointerfaces 2016, 143, 64–70. [Google Scholar] [CrossRef]
- Will, O.M.; Purcz, N.; Chalaris, A.; Heneweer, C.; Boretius, S.; Purcz, L.; Nikkola, L.; Ashammakhi, N.; Kalthoff, H.; Gluer, C.C.; et al. Increased survival rate by local release of diclofenac in a murine model of recurrent oral carcinoma. Int. J. Nanomed. 2016, 11, 5311–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amanullah, A.; Upadhyay, A.; Dhiman, R.; Singh, S.; Kumar, A.; Ahirwar, D.K.; Gutti, R.K.; Mishra, A. Development and Challenges of Diclofenac-Based Novel Therapeutics: Targeting Cancer and Complex Diseases. Cancers 2022, 14, 4385. https://doi.org/10.3390/cancers14184385
Amanullah A, Upadhyay A, Dhiman R, Singh S, Kumar A, Ahirwar DK, Gutti RK, Mishra A. Development and Challenges of Diclofenac-Based Novel Therapeutics: Targeting Cancer and Complex Diseases. Cancers. 2022; 14(18):4385. https://doi.org/10.3390/cancers14184385
Chicago/Turabian StyleAmanullah, Ayeman, Arun Upadhyay, Rohan Dhiman, Sarika Singh, Amit Kumar, Dinesh Kumar Ahirwar, Ravi Kumar Gutti, and Amit Mishra. 2022. "Development and Challenges of Diclofenac-Based Novel Therapeutics: Targeting Cancer and Complex Diseases" Cancers 14, no. 18: 4385. https://doi.org/10.3390/cancers14184385