
HDAC2- and EZH2-Mediated Histone Modifications Induce PDK1 Expression through miR-148a Downregulation in Breast Cancer Progression and Adriamycin Resistance

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Background
2. Methods
2.1. Cell Lines and Reagents
2.2. Tissue Samples
2.3. Lentiviral Packaging and Stable Cell Line Establishment
2.4. RNA Extraction and qRT-PCR Analysis
2.5. Western Blotting Analysis
2.6. Immunohistochemistry Assay
2.7. Co-Immunoprecipitation Assay
2.8. Luciferase Assay
2.9. Transwell Assay
2.10. Immunofluorescence Assay
2.11. MMP Detection Assays
2.12. Annexin-V/PI Double-Staining Assay
2.13. Tumorigenesis Study
2.14. GSEA
2.15. Correlation Analysis
2.16. Statistical Analysis
3. Results
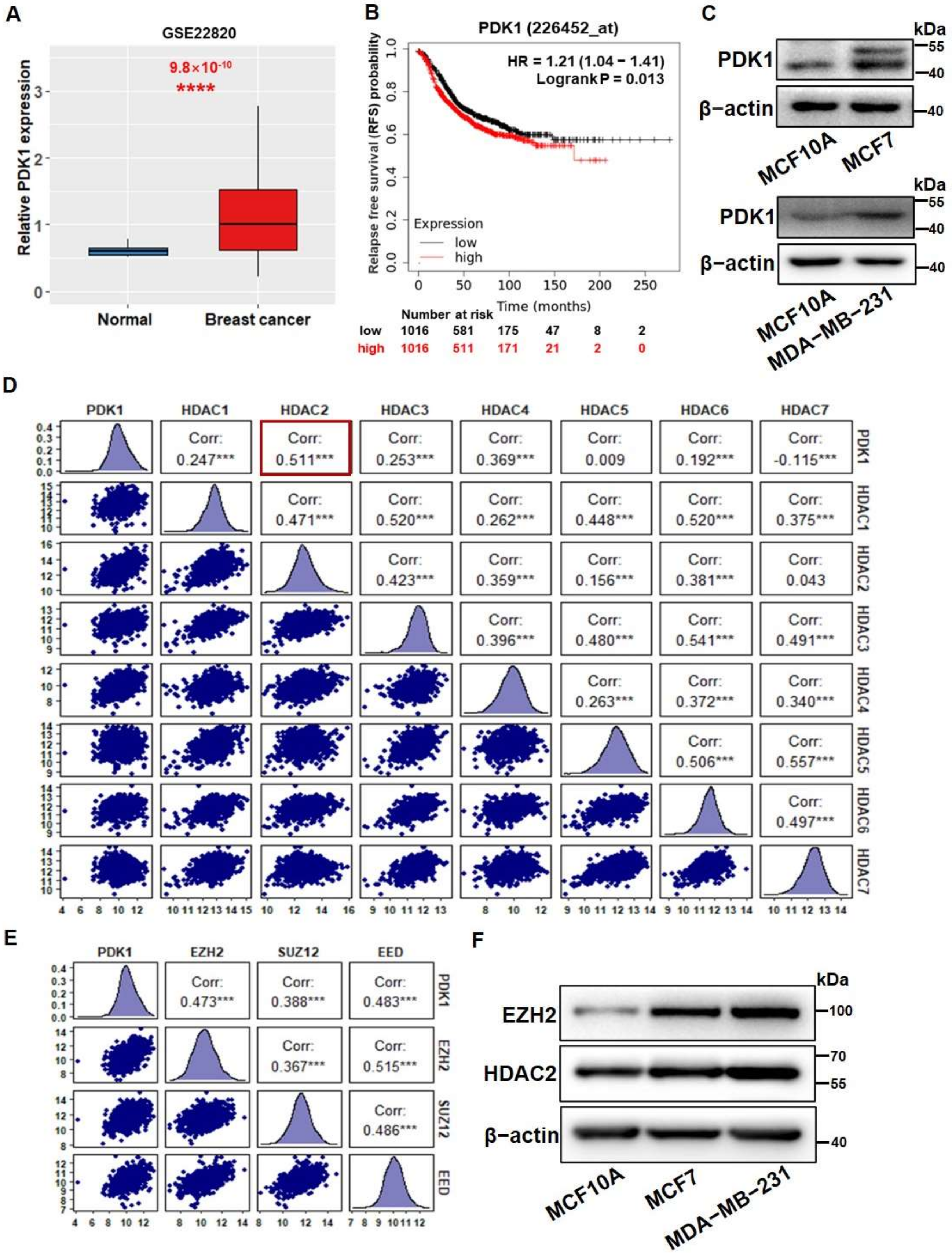
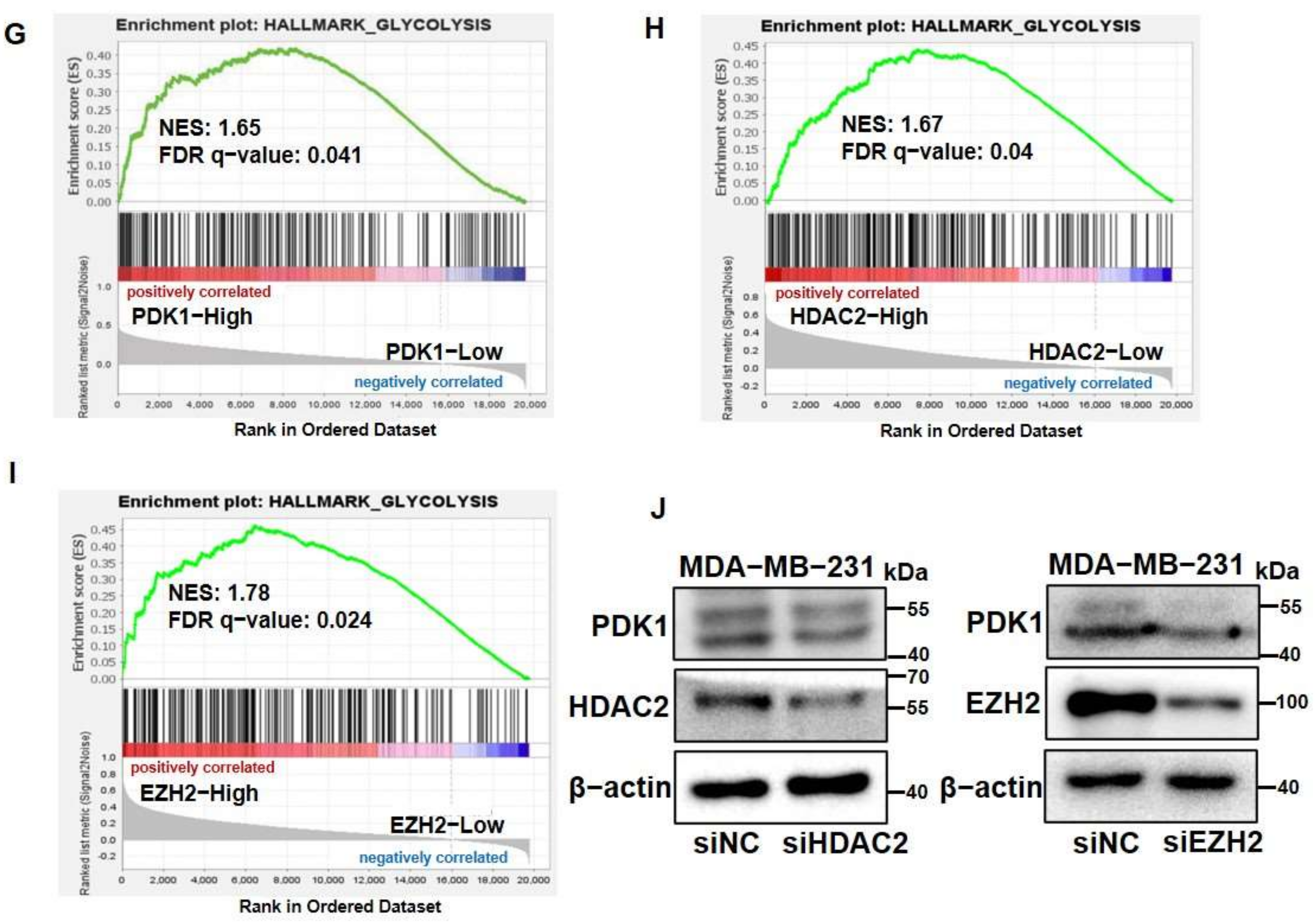
3.1. PDK1 Levels Were Upregulated by HDAC2 and EZH2 Which Are Associated with Glycolysis
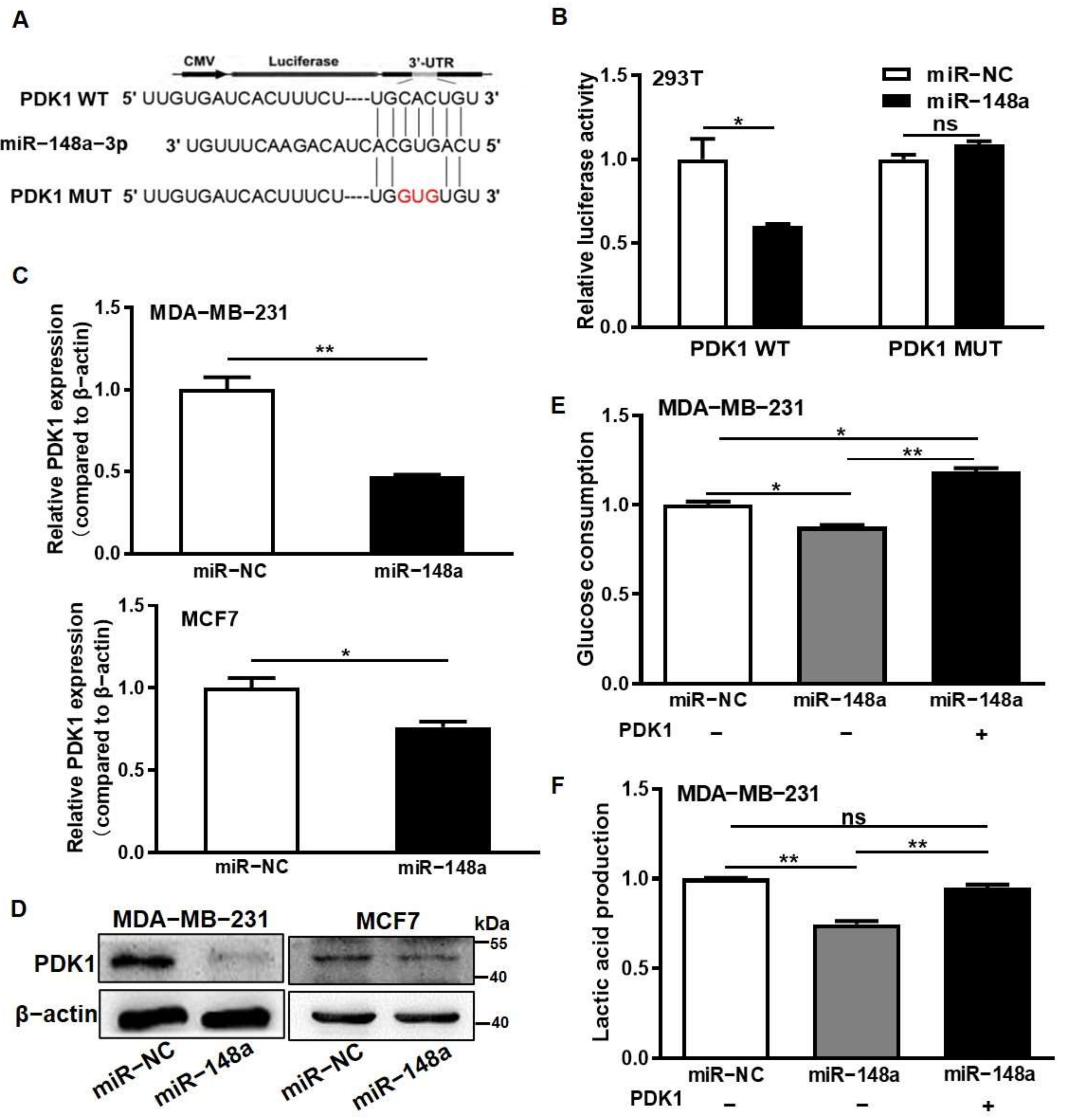
3.2. miR-148a Was Identified for Directly Suppressing PDK1 Expression and Inhibiting Glycometabolism
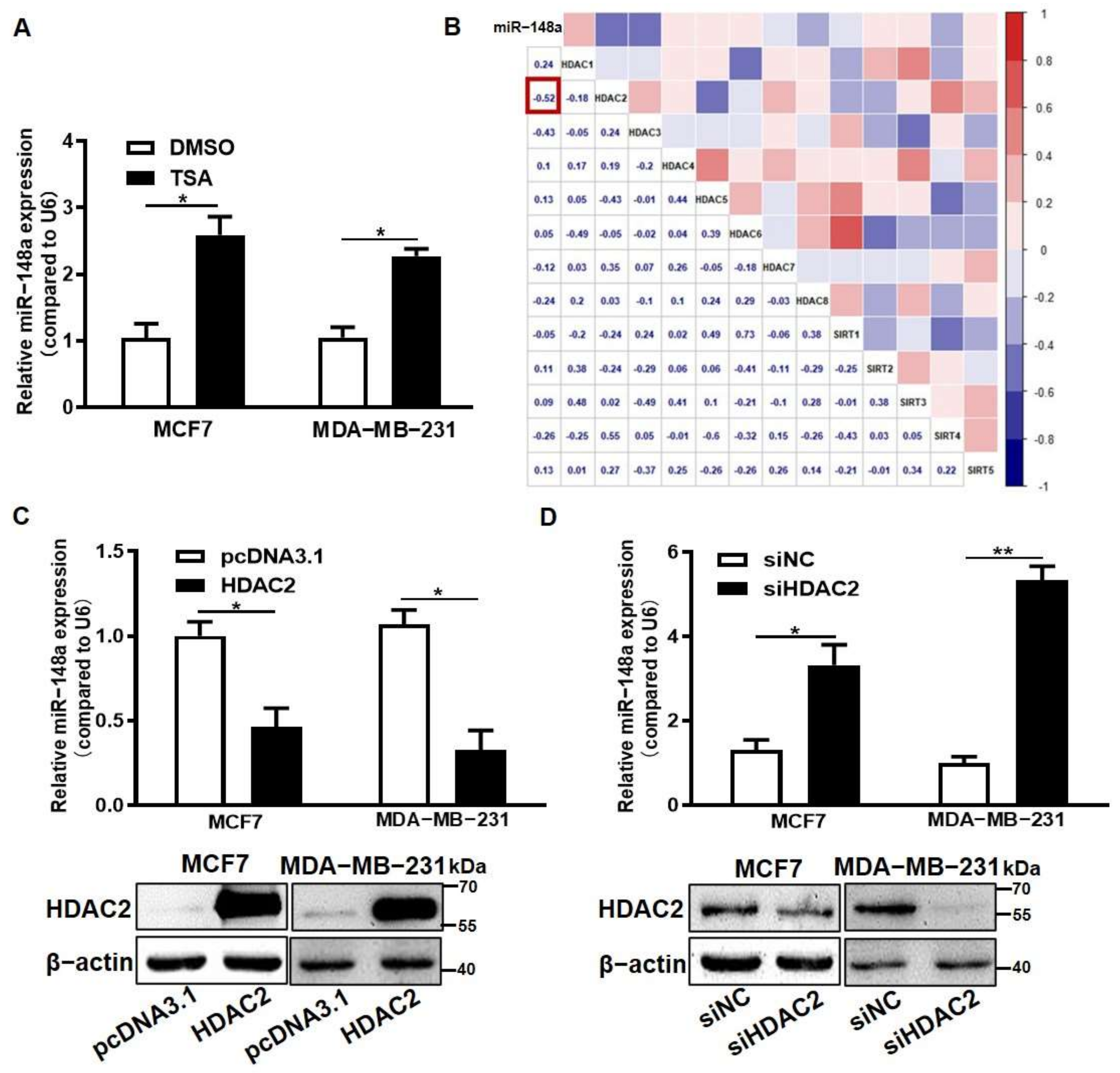
3.3. HDAC2 Overexpression Suppressed miR-148a Expression
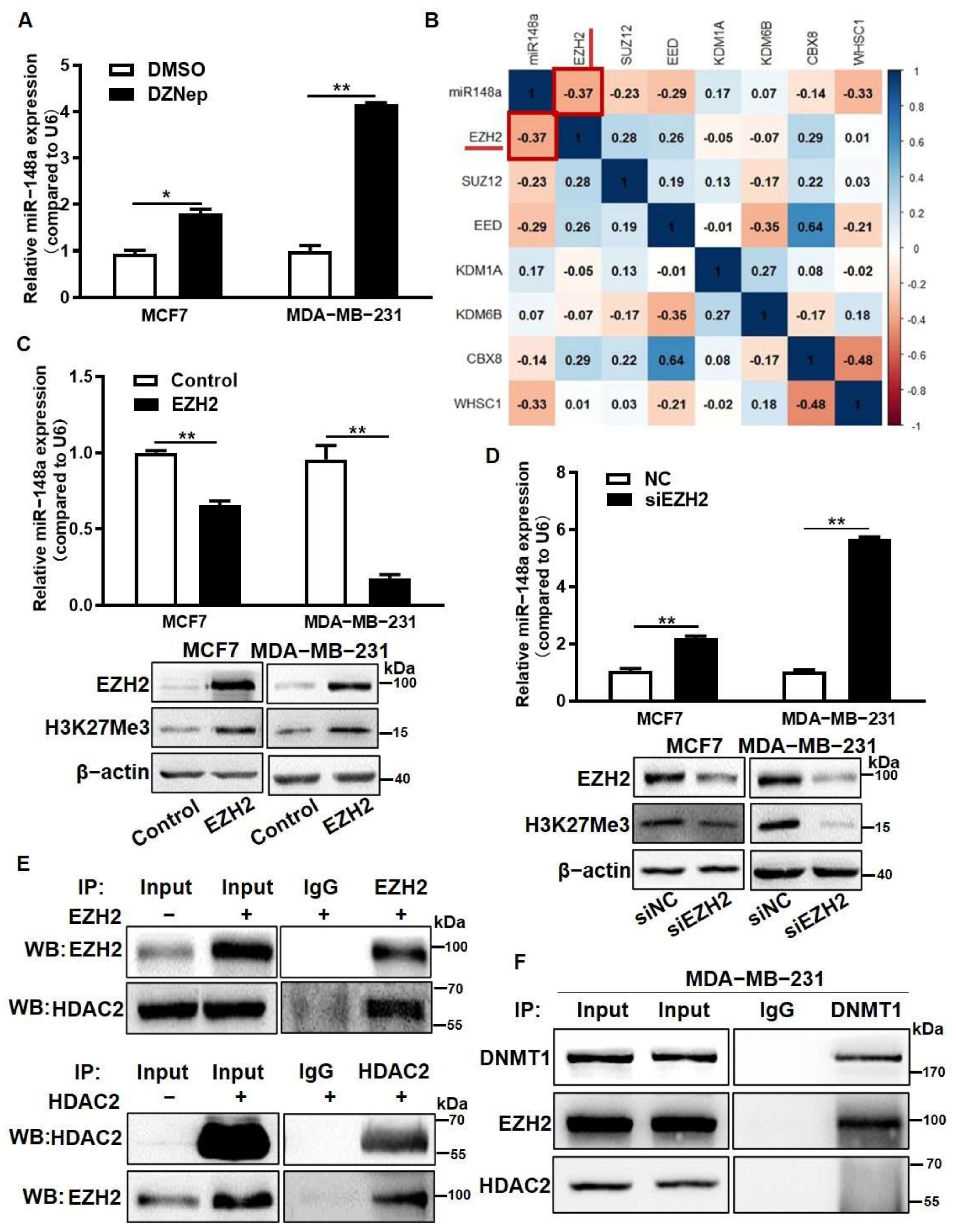
3.4. EZH2 Overexpression Inhibited miR-148a Expression through the Recruitment of HDAC2
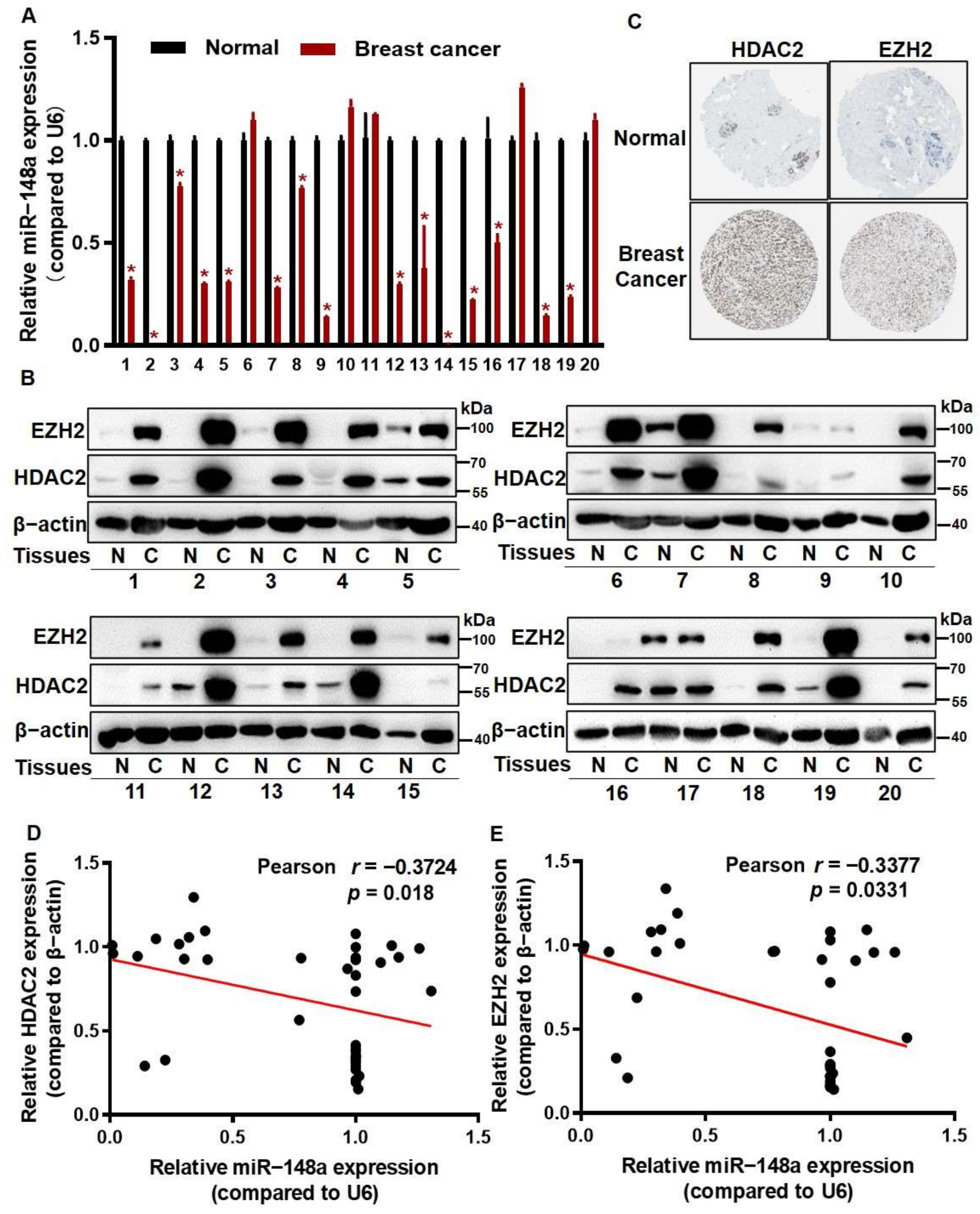
3.5. The Levels of HDAC2 and EZH2 Were Inversely Correlated with miR-148a Levels in Breast Cancer Tissues
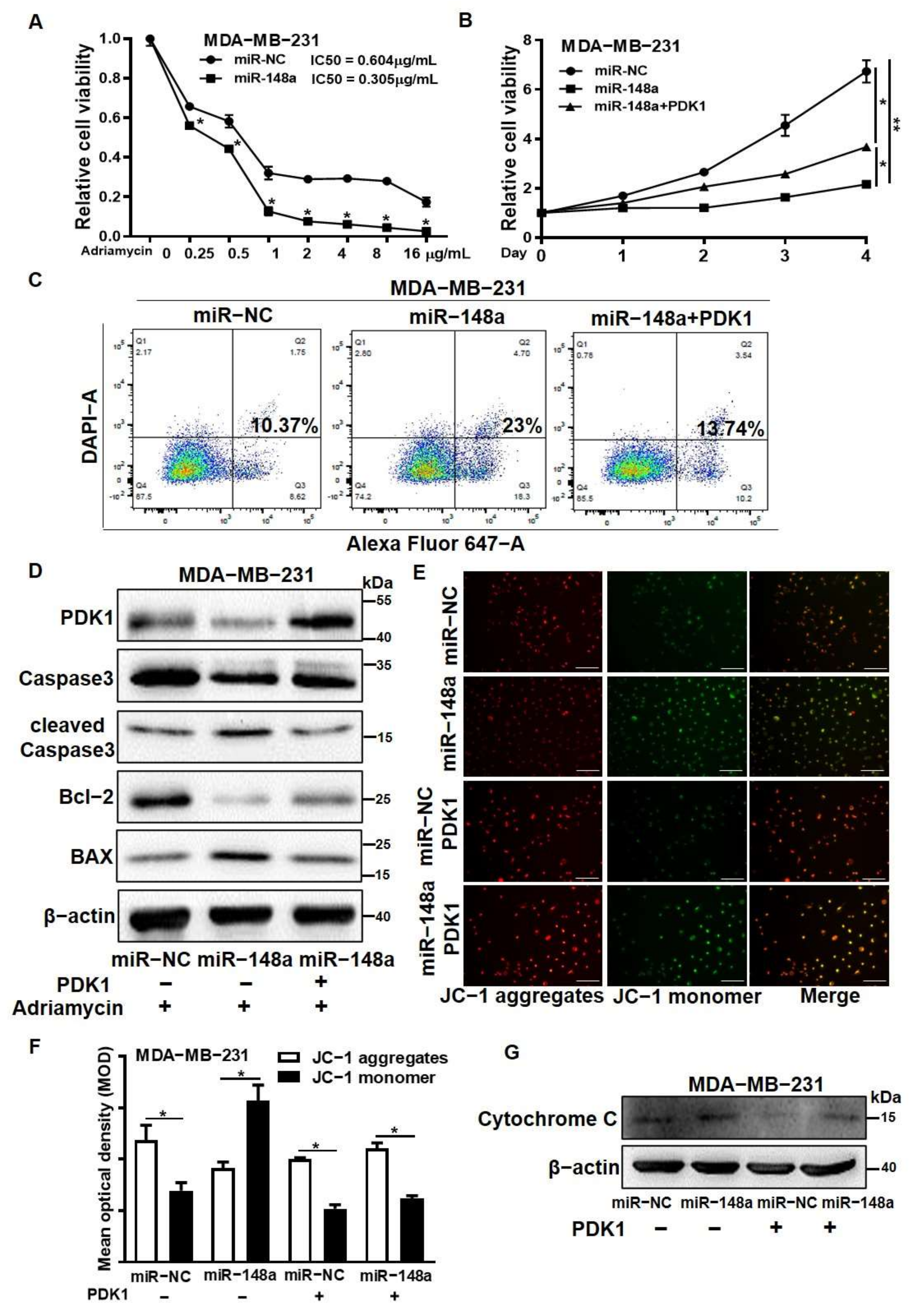
3.6. PDK1 Overexpression Reversed the Effect of miR-148a Decreasing Adriamycin Resistance
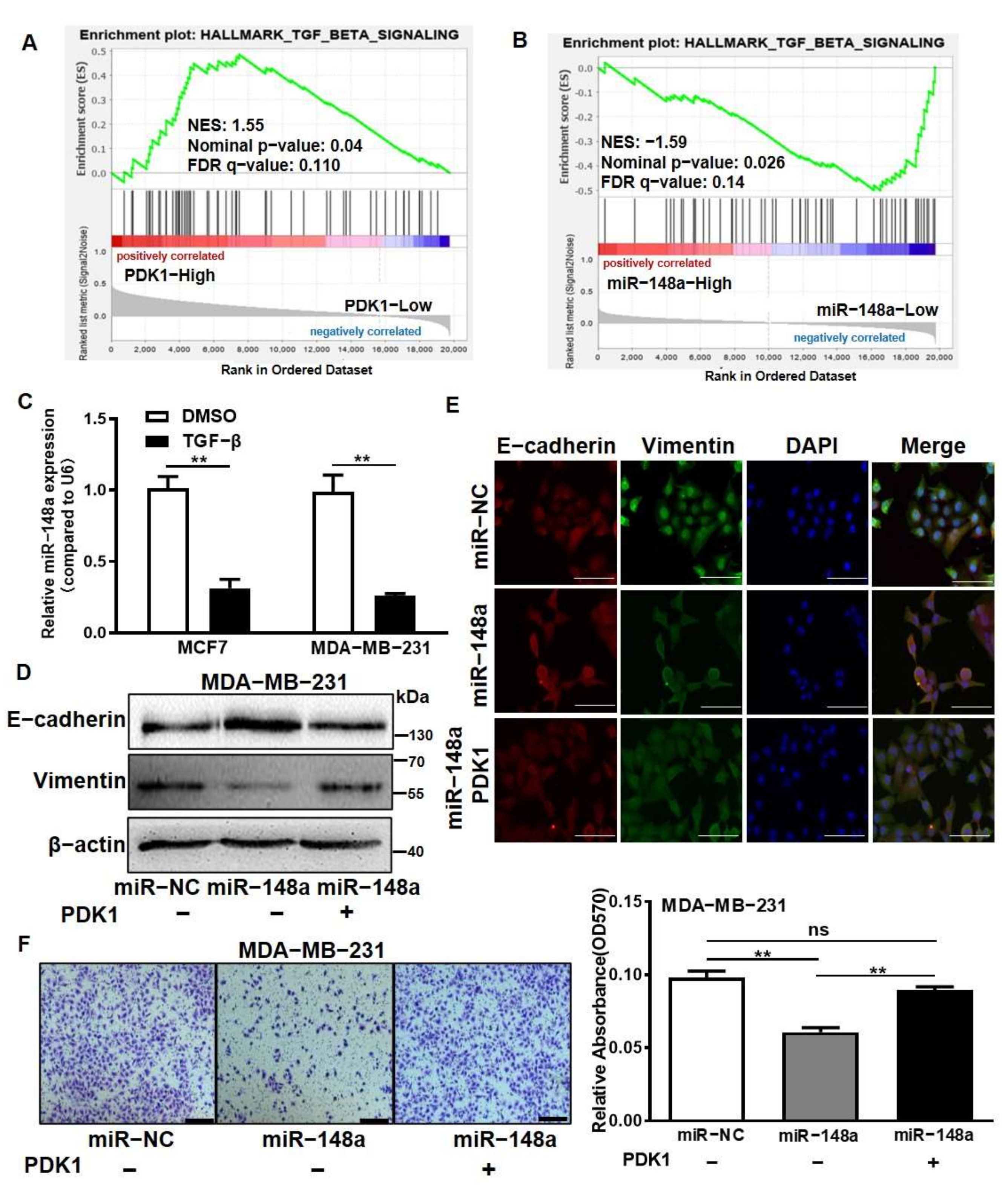
3.7. PDK1 Suppression by miR-148a Inhibited Epithelial–Mesenchymal Transition and Cell Migration
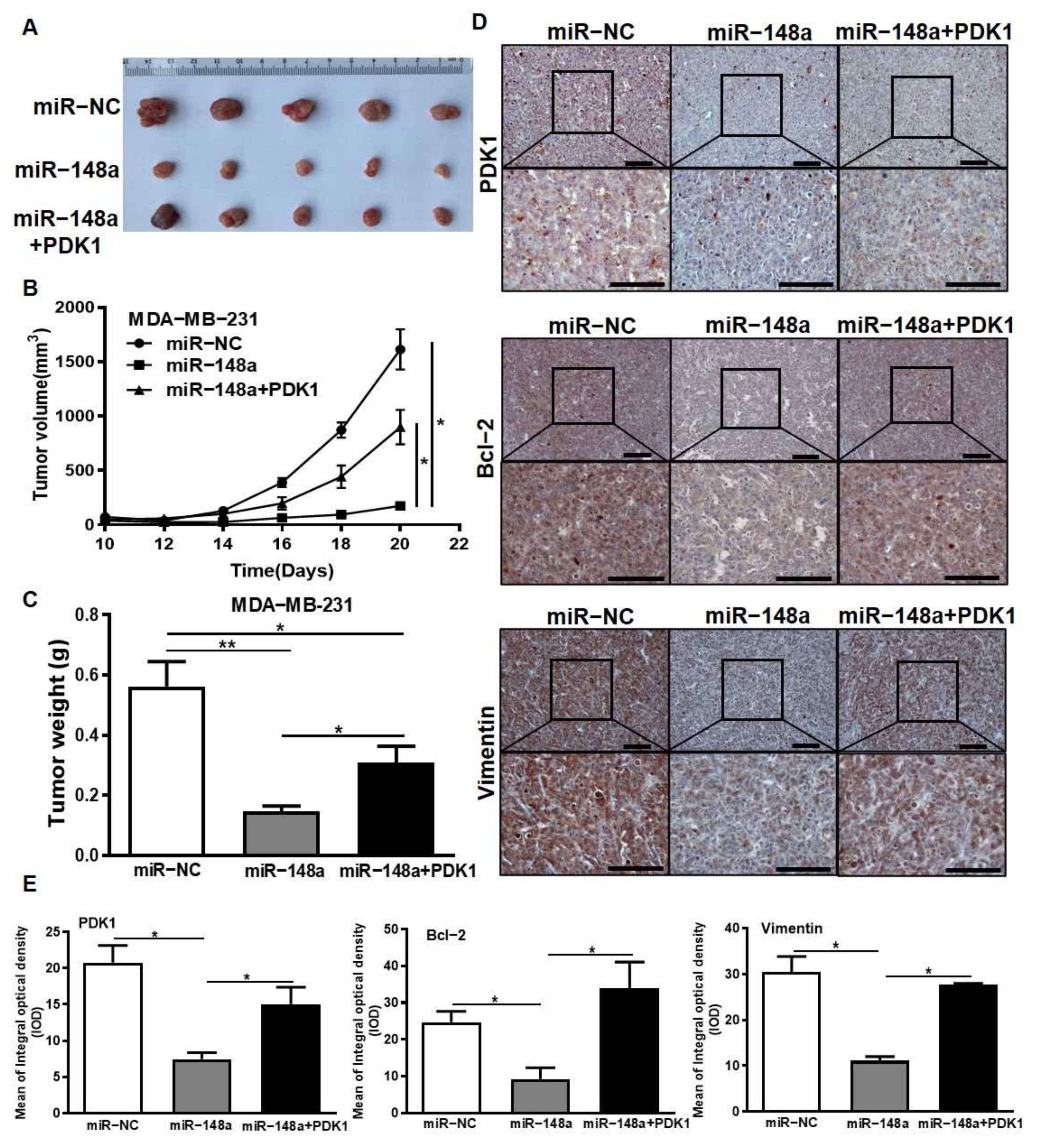
3.8. PDK1 Partially Restores the Inhibitory Effect of miR-148a on Tumor Growth
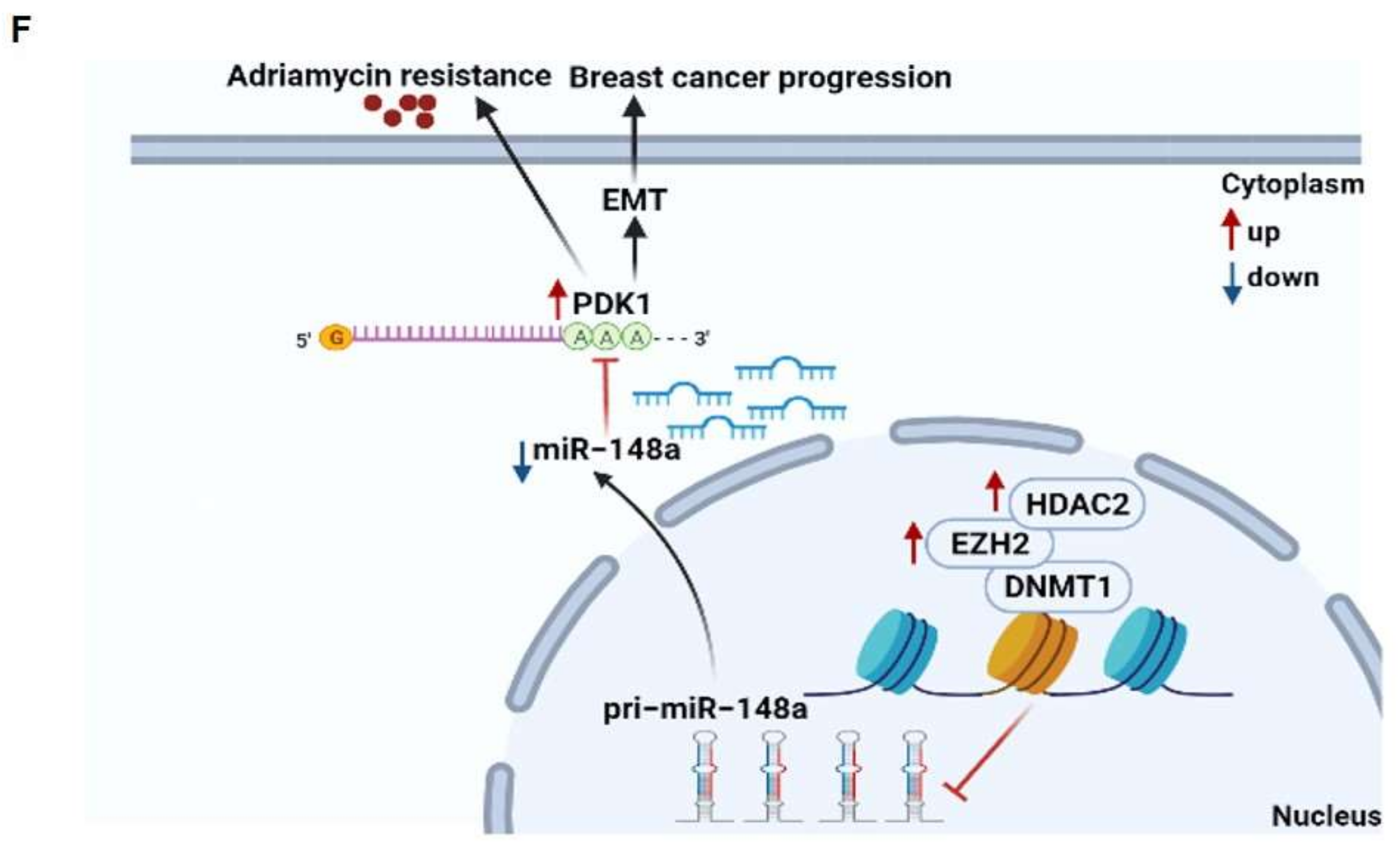
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADR | Adriamycin |
| HDACs | histone deacetylases |
| PRC2 | polycomb repressive complex 2 |
| HDAC2 | histone deacetylase 2 |
| HDAC7 | histone deacetylase 7 |
| HDAC3 | histone deacetylase 3 |
| EZH2 | enhancer of zeste homologue 2 |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| DNMT1 | DNA methyltransferase 1 |
| EMT | epithelial-mesenchymal transition |
| CARM1 | coactivator associated arginine methyltransferase 1 |
| GLUTs | glucose transporters |
| HK | hexokinase |
| PFK | phosphofructokinase |
| PK | pyruvate kinase |
| PKM2 | pyruvate kinase M2 isoform |
| TCA | tricarboxylic acid |
| DMEM | Dulbecco’s modified Eagle’s medium |
| RFP | red fluorescent protein |
| OS | overall survival |
| MUT | mutation |
| WT | wild type |
| Co-IP | Co-immunoprecipitation |
| IF | immunofluorescence |
| MMP | mitochondrial membrane potential |
| MOD | mean optical density |
| IHC | immunohistochemistry |
| PDH | pyruvate dehydrogenase |
References
- Chun, K.-H.; Park, J.H.; Fan, S. Predicting and Overcoming Chemotherapeutic Resistance in Breast Cancer. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2017; Volume 1026, pp. 59–104. [Google Scholar] [CrossRef]
- Gao, P.; Zhou, G.-Y.; Guo, L.-L.; Zhang, Q.-H.; Zhen, J.-H.; Fang, A.-J.; Lin, X.-Y. Reversal of drug resistance in breast carcinoma cells by anti-mdr1 ribozyme regulated by the tumor-specific MUC-1 promoter. Cancer Lett. 2007, 256, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wu, J.; Zhao, Q.; Fu, S.; Jin, J. Emerging roles of aerobic glycolysis in breast cancer. Clin. Transl. Oncol. 2019, 22, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Konno, Y.; Ihira, K.; Kobayashi, N.; Yue, J.; Watari, H. Long non-coding RNA DLEU2 drives EMT and glycolysis in endometrial cancer through HK2 by competitively binding with miR-455 and by modulating the EZH2/miR-181a pathway. J. Exp. Clin. Cancer Res. 2021, 40, 216. [Google Scholar] [CrossRef]
- Liu, F.; Ma, F.; Wang, Y.; Hao, L.; Zeng, H.; Jia, C.; Wang, Y.; Liu, P.; Ong, I.M.; Li, B.; et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 2017, 19, 1358–1370. [Google Scholar] [CrossRef]
- Hitosugi, T.; Fan, J.; Chung, T.-W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine Phosphorylation of Mitochondrial Pyruvate Dehydrogenase Kinase 1 Is Important for Cancer Metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Yin, H. PDK1 promotes tumor cell proliferation and migration by enhancing the Warburg effect in non-small cell lung cancer. Oncol. Rep. 2016, 37, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.-H.; Fan, W.-J.; Meng, Y.-T.; Li, M.-M.; Li, T.-T.; Cui, B.; Wang, H.-F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2017, 37, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- NBSP; Ropero, S.; Cancer Epigenetics Laboratory, M.P.P.; Spanish National Cancer Centre; Esteller, M.; Spanish National Cancer Centre (CNIO). The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Liu, T.; Wan, Y.; Xiao, Y.; Xia, C.; Duan, G. Dual-Target Inhibitors Based on HDACs: Novel Antitumor Agents for Cancer Therapy. J. Med. Chem. 2020, 63, 8977–9002. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Zhao, Y.; He, J.; Yang, L.; Luo, Q.; Liu, Z. Histone Deacetylase-3 Modification of MicroRNA-31 Promotes Cell Proliferation and Aerobic Glycolysis in Breast Cancer and Is Predictive of Poor Prognosis. J. Breast Cancer 2018, 21, 112–123. [Google Scholar] [CrossRef]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.-Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Leithner, K. Epigenetic Marks Repressing Gluconeogenesis in Liver and Kidney Cancer. Cancer Res. 2020, 80, 657–658. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Cao, M.; Luo, X.; Li, L.; Wang, K.; Wang, S.; Wang, H.; Tang, Y.; Liang, X. EZH2 promotes invasion and tumour glycolysis by regulating STAT3 and FoxO1 signalling in human OSCC cells. J. Cell. Mol. Med. 2019, 23, 6942–6954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P.; et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2016, 24, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Lin, X.; Ren, S.; Wu, S.; Wang, X.; Ma, L.; Xiang, H. Development of novel tetrahydroisoquinoline-hydroxamate conjugates as potent dual SERDs/HDAC inhibitors for the treatment of breast cancer. Eur. J. Med. Chem. 2021, 226, 113870. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.F.; Xie, Y.X.; Qian, Y.C.; Wang, M.; Liu, L.Z.; Shu, Y.Q.; Bai, X.-M.; Jiang, B.-H. TBX15/miR-152/KIF2C pathway regulates breast cancer doxorubicin resistance via promoting PKM2 ubiquitination. Cancer Cell Int. 2021, 21, 542. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-M.; Wang, X.-F.; Qian, X.; Tao, T.; Wang, L.; Chen, Q.-D.; Wang, X.-R.; Cao, L.; Wang, Y.-Y.; Zhang, J.-X.; et al. MiRNA-181b suppresses IGF-1R and functions as a tumor suppressor gene in gliomas. RNA 2013, 19, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M. Irizarry affy–analysis of Affymetrix GeneChip data at the probe lev Bioinformatics. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Xu, Q.; Jiang, Y.; Yin, Y.; Li, Q.; He, J.; Jing, Y.; Qi, Y.-T.; Xu, Q.; Li, W.; Lu, B.; et al. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J. Mol. Cell Biol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of MYC-Mediated miR-29 by HDAC3 and EZH2 as a Therapeutic Target of Histone Modification in Aggressive B-Cell Lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Xu, H.; He, Y.; Ma, J.; Zhao, Y.; Liu, Y.; Sun, L.; Su, J. Inhibition of pyruvate dehydrogenase kinase-1 by dicoumarol enhances the sensitivity of hepatocellular carcinoma cells to oxaliplatin via metabolic reprogramming. Int. J. Oncol. 2020, 57, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cong, Q.; Zhang, X.; Zhang, M.; Lu, Y.; Xu, C. Pyruvate dehydrogenase kinase 1 contributes to cisplatin resistance of ovarian cancer through EGFR activation. J. Cell. Physiol. 2018, 234, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lin, C.; Yang, X.; Xie, Y.; Hu, P.; Li, H.; Zhu, W.; Hu, H. Mitochondrial targeting nanodrugs self-assembled from 9-O-octadecyl substituted berberine derivative for cancer treatment by inducing mitochondrial apoptosis pathways. J. Control. Release 2018, 294, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; Ten Dijke, P. TGF-Beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef]
- Sradhanjali, S.; Reddy, M.M. Inhibition of Pyruvate Dehydrogenase Kinase as a Therapeutic Strategy against Cancer. Curr. Top. Med. Chem. 2018, 18, 444–453. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, C.; Yang, J.; Zhao, Y.; Jia, H.; Xue, M.; Xu, D.; Yang, F.; Fu, D.; Wang, C.; et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct. Target Ther. 2020, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.D.; Noothi, S.K.; Pullum, D.A.; Lawrie, C.H.; Pallapati, R.; Potluri, V.; Kuntzen, C.; Khan, S.; Plas, D.R.; Orlowski, R.Z.; et al. Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat. Commun. 2015, 6, 8428. [Google Scholar] [CrossRef] [Green Version]
- Kabekkodu, S.P.; Shukla, V.; Varghese, V.K.; Adiga, D.; Jishnu, P.V.; Chakrabarty, S.; Satyamoorthy, K. Cluster miRNAs and cancer: Diagnostic, prognostic and therapeutic opportunities. Wiley Interdiscip. Rev. RNA 2019, 11, e1563. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in Cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef]
- Jiang, Q.; He, M.; Ma, M.-T.; Wu, H.-Z.; Yu, Z.-J.; Guan, S.; Jiang, L.-Y.; Wang, Y.; Zheng, D.-D.; Jin, F.; et al. MicroRNA-148a inhibits breast cancer migration and invasion by directly targeting WNT-1. Oncol. Rep. 2015, 35, 1425–1432. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Huang, Q.; Ren, X.; Hu, H.; Sheng, H.; Lai, M. MiR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell Death Differ. 2011, 18, 1702–1710. [Google Scholar] [CrossRef]
- Song, C.; Yang, J.; Jiang, R.; Yang, Z.; Li, H.; Huang, Y.; Lan, X.; Lei, C.; Ma, Y.; Qi, X.; et al. miR-148a-3p regulates proliferation and apoptosis of bovine muscle cells by targeting KLF6. J. Cell. Physiol. 2019, 234, 15742–15750. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, L.Z.; Yin, Y.; He, J.; Li, Q.; Qian, X.; You, Y.; Lu, Z.; Perper, S.C.; Shu, Y.; et al. Regulatory circuit of PKM2/NF-kappaB/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene 2015, 34, 5482–5493. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype. Oxid. Med. Cell Longev. 2018, 2018, 1027453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, D.H.; Kim, M.-K.; No, J.H.; Chung, H.H.; Song, Y.S. Metabolic approaches to overcoming chemoresistance in ovarian cancer. Ann. N. Y. Acad. Sci. 2011, 1229, 53–60. [Google Scholar] [CrossRef]
- Tiwari, N.; Tiwari, V.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schübeler, D.; Van Nimwegen, E.; Christofori, G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling ezh2 expression and epigenetic reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Li, Q.-G.; Xiao, T.; Zhu, W.; Yu, Z.-Z.; Huang, X.-P.; Yi, H.; Lu, S.-S.; Tang, Y.-Y.; Huang, W.; Xiao, Z.-Q. HDAC7 promotes the oncogenicity of nasopharyngeal carcinoma cells by miR-4465-EphA2 signaling axis. Cell Death Dis. 2020, 11, 322. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Shi, Z.; Qian, Y.; Jiang, C.; Liu, W.; Liu, B.; Jiang, B. HDAC2- and EZH2-Mediated Histone Modifications Induce PDK1 Expression through miR-148a Downregulation in Breast Cancer Progression and Adriamycin Resistance. Cancers 2022, 14, 3600. https://doi.org/10.3390/cancers14153600
Xie Y, Shi Z, Qian Y, Jiang C, Liu W, Liu B, Jiang B. HDAC2- and EZH2-Mediated Histone Modifications Induce PDK1 Expression through miR-148a Downregulation in Breast Cancer Progression and Adriamycin Resistance. Cancers. 2022; 14(15):3600. https://doi.org/10.3390/cancers14153600
Chicago/Turabian StyleXie, Yunxia, Zhumei Shi, Yingchen Qian, Chengfei Jiang, Wenjing Liu, Bingjie Liu, and Binghua Jiang. 2022. "HDAC2- and EZH2-Mediated Histone Modifications Induce PDK1 Expression through miR-148a Downregulation in Breast Cancer Progression and Adriamycin Resistance" Cancers 14, no. 15: 3600. https://doi.org/10.3390/cancers14153600