
Second Generation Small Molecule Inhibitors of Gankyrin for the Treatment of Pediatric Liver Cancer

 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
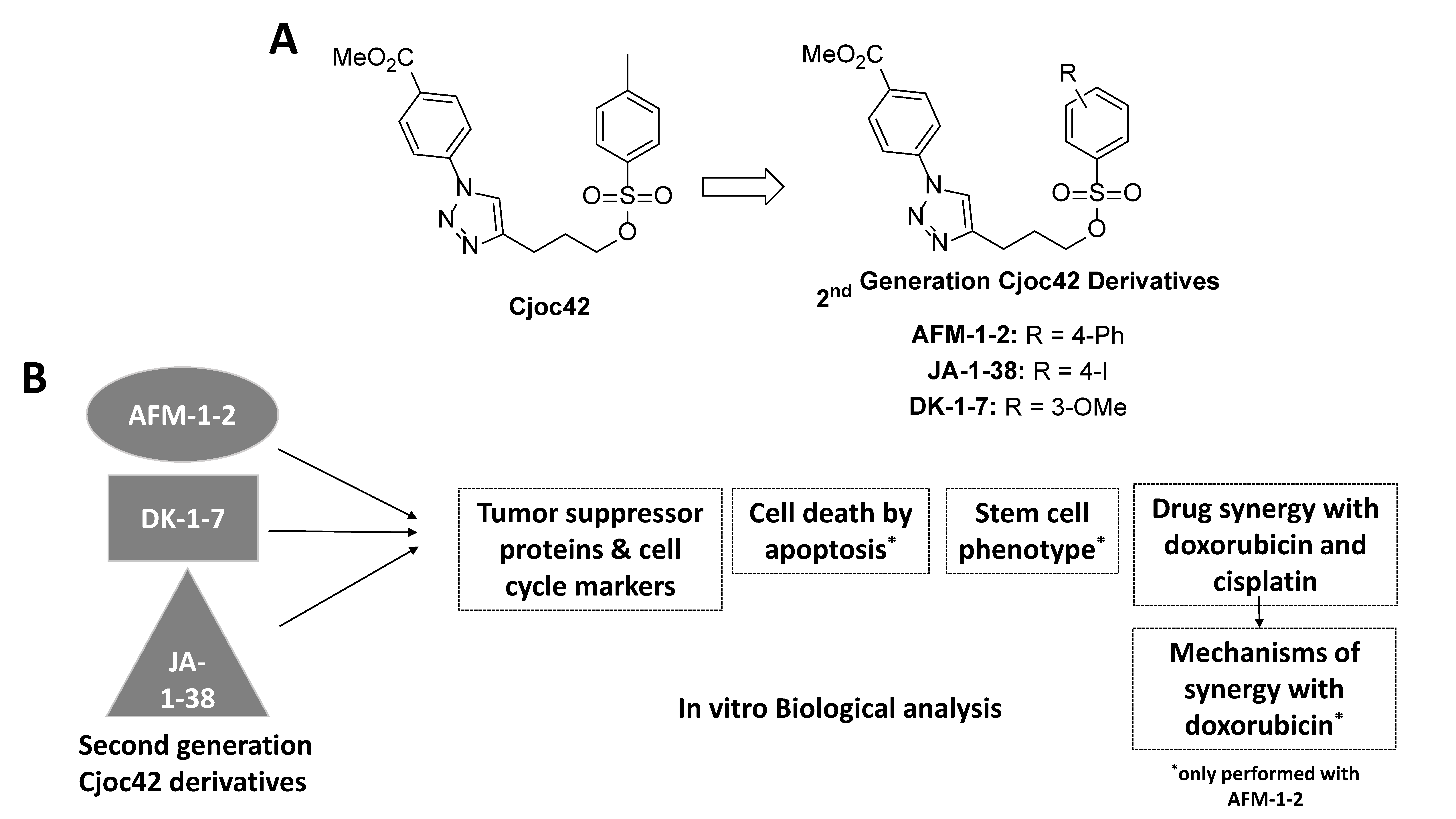
2.1. Cjoc42 Derivatives and Chemotherapy Agents
2.2. Cell Culture Studies
2.3. Proliferation Assays
2.4. Saturation Assay
2.5. Protein Isolation and Western Blotting
2.6. RNA Extraction and Real-Time Quantitative Reverse Transcriptase PCR
2.7. Wound Healing Assay
2.8. Quantification of Apoptosis by Flow Cytometry
2.9. Apoptosis Detection by Apotracker Green
2.10. Synergy Assays
2.11. Statistical Analysis
3. Results
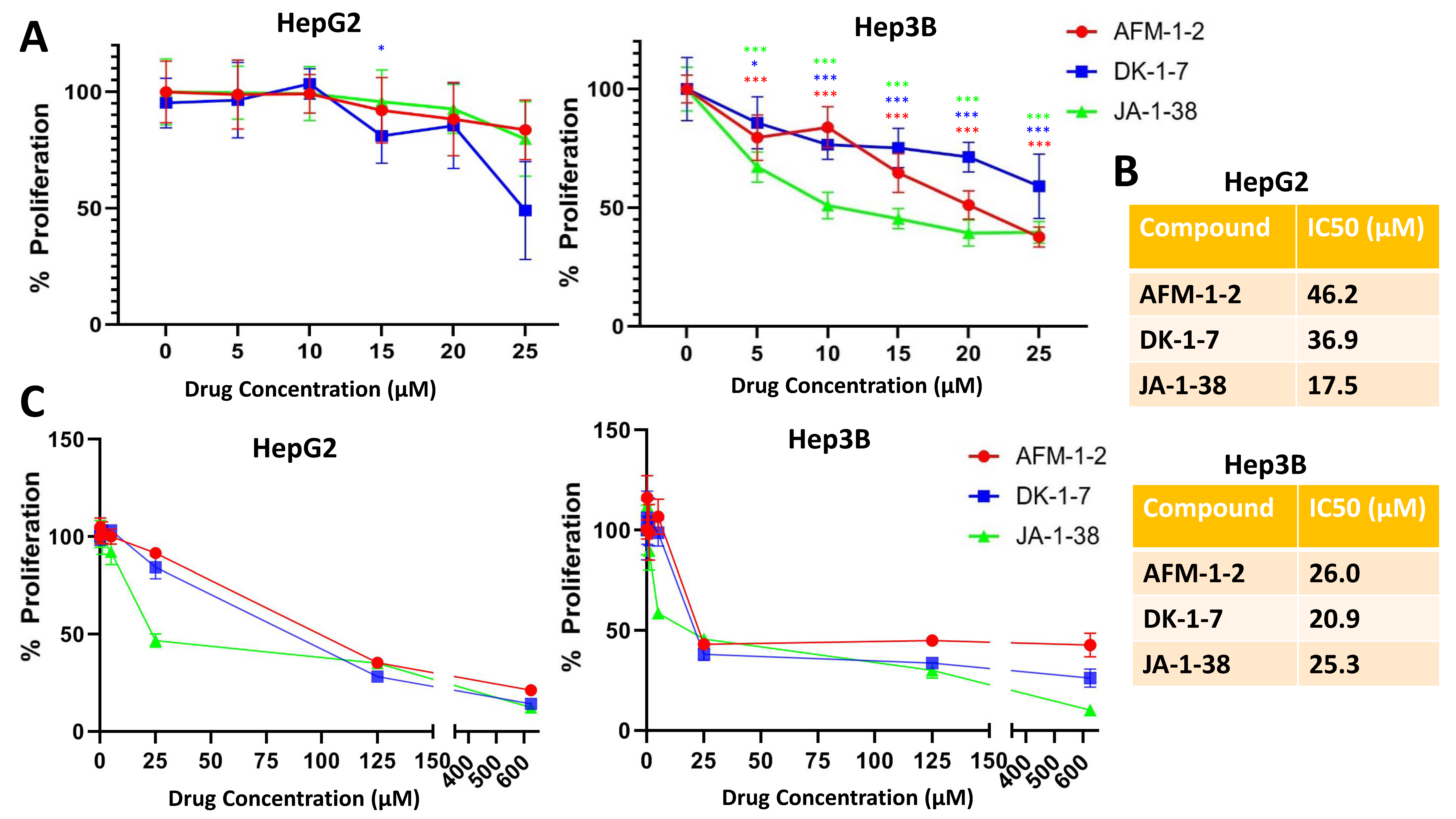
3.1. Cjoc42 Derivatives Showed Antiproliferative Effects in Hep3B and HepG2 Cells
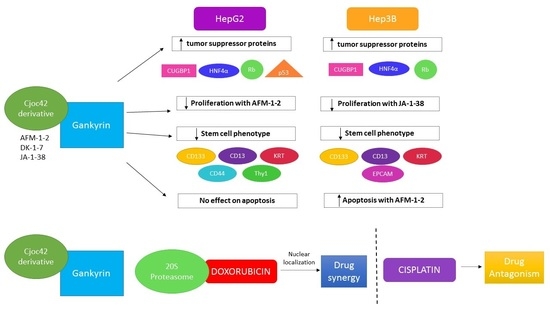
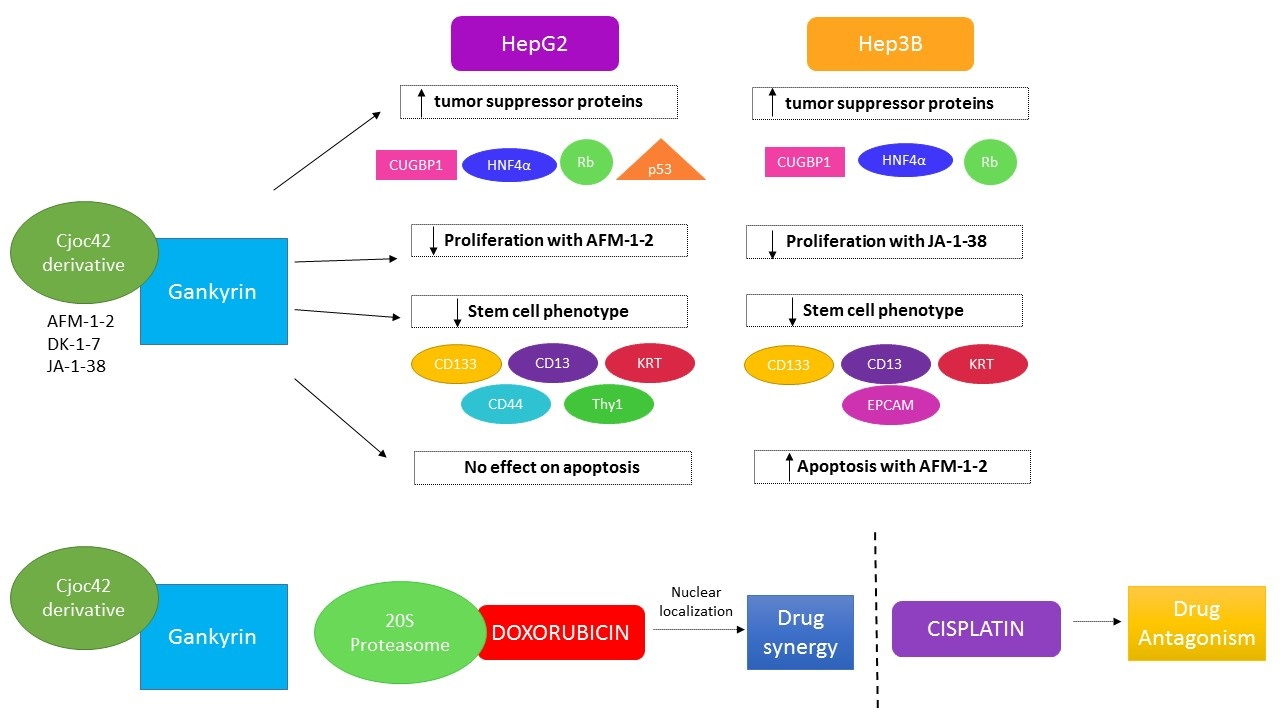
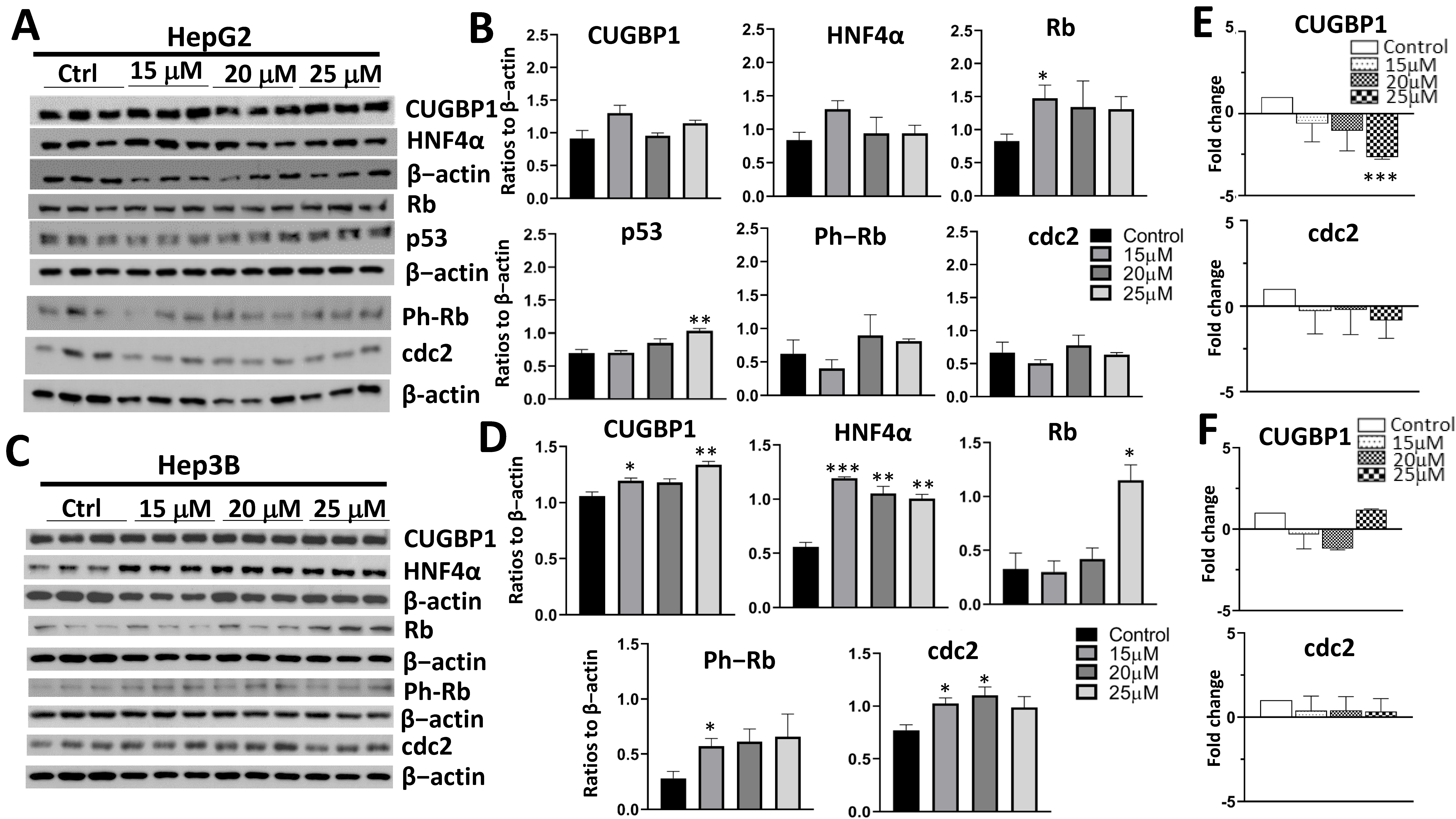
3.2. Treatment with AFM-1 and -2 Resulted in the Rescue of Tumor Suppressor Proteins CUGBP1, HNF4α, and Rb and a Decrease in Cell Cycle Markers in HepG2
3.3. Treatment of Cancer Cells with cjoc42 Derivatives DK-1–7 Demonstrated an Increase in Several Tumor Suppressor Proteins but Variable Changes in Cell Cycle Markers
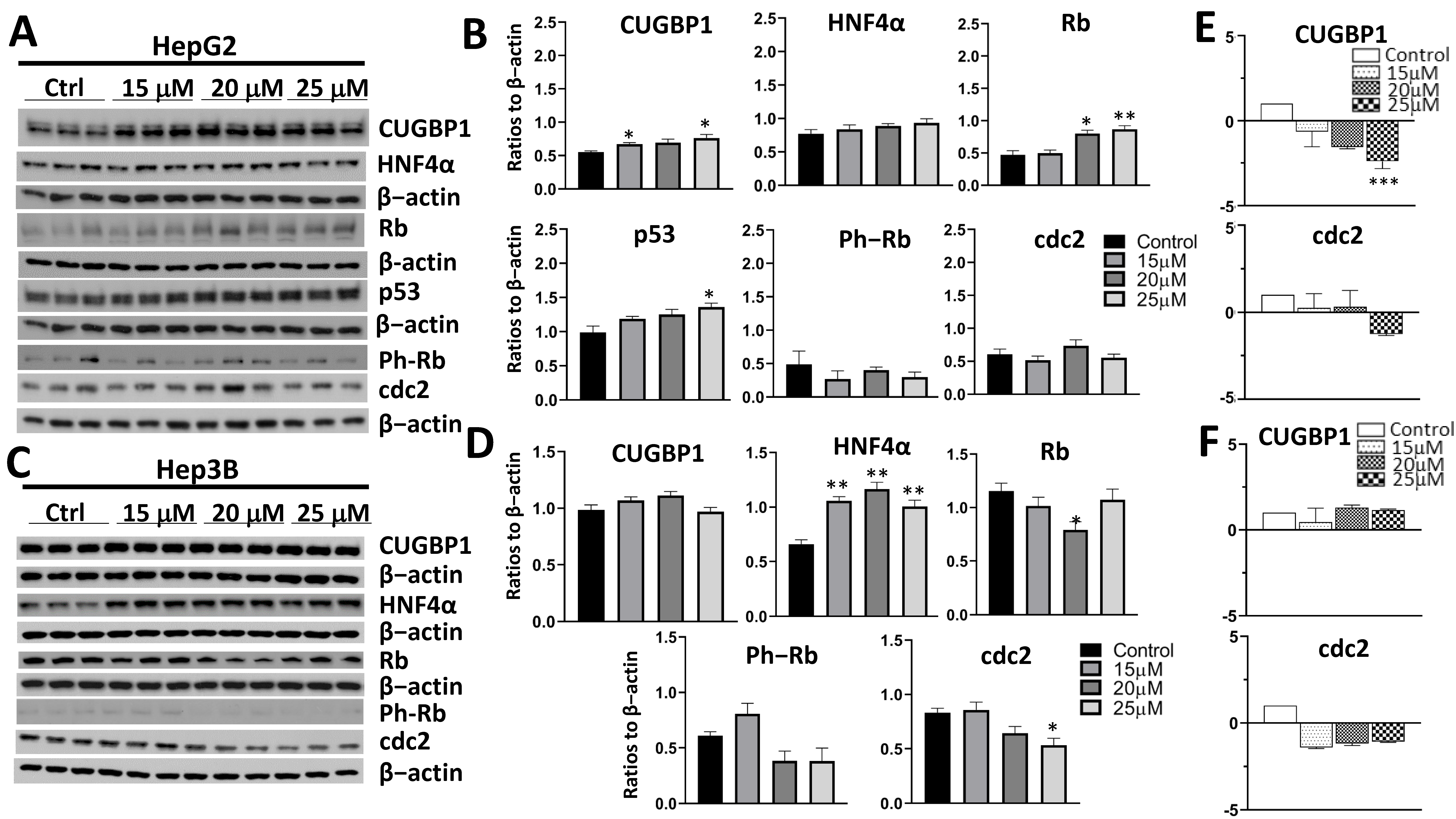
3.4. Treatment of Cancer Cells with JA-1–38 Demonstrated an Increase in Tumor Suppressor Proteins and a Decrease in Cell Cycle Markers
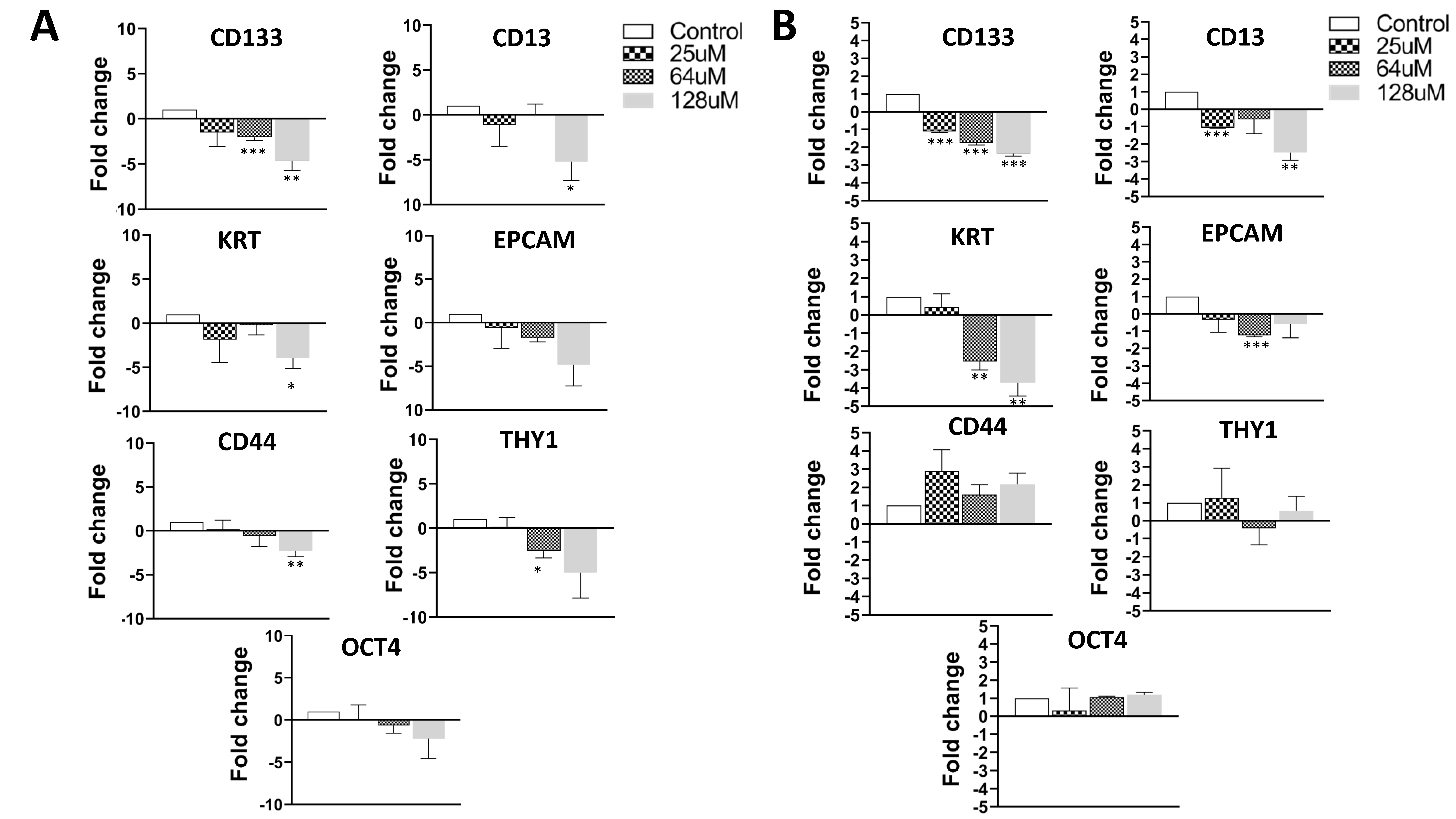
3.5. Treatment of Cancer Cells with AFM-1 and -2 Leads to a Reduction in Stem Cell Phenotype
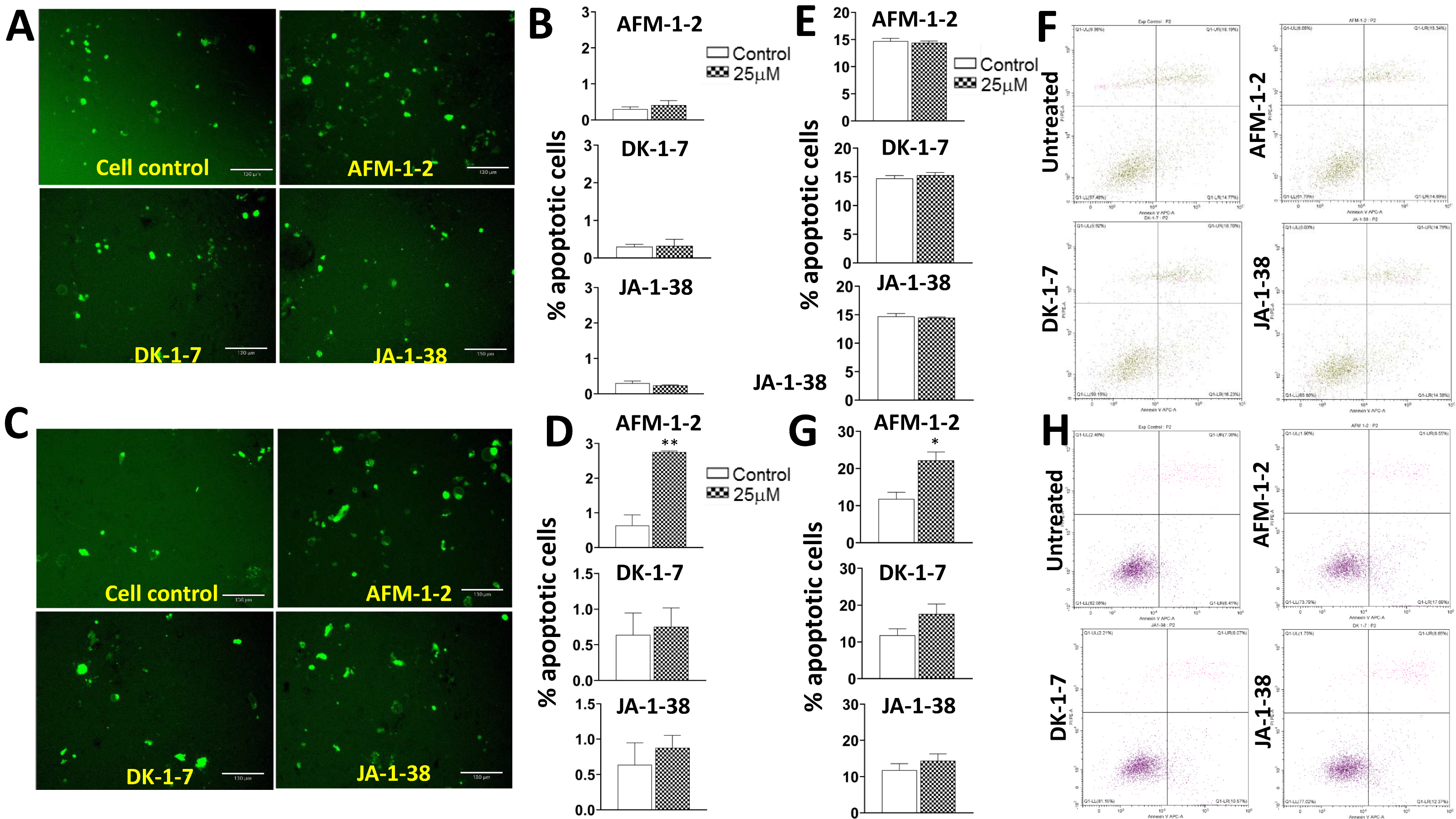
3.6. An Increase in Apoptosis Was Detected in Hep3B Cells Treated with AFM-1 and -2
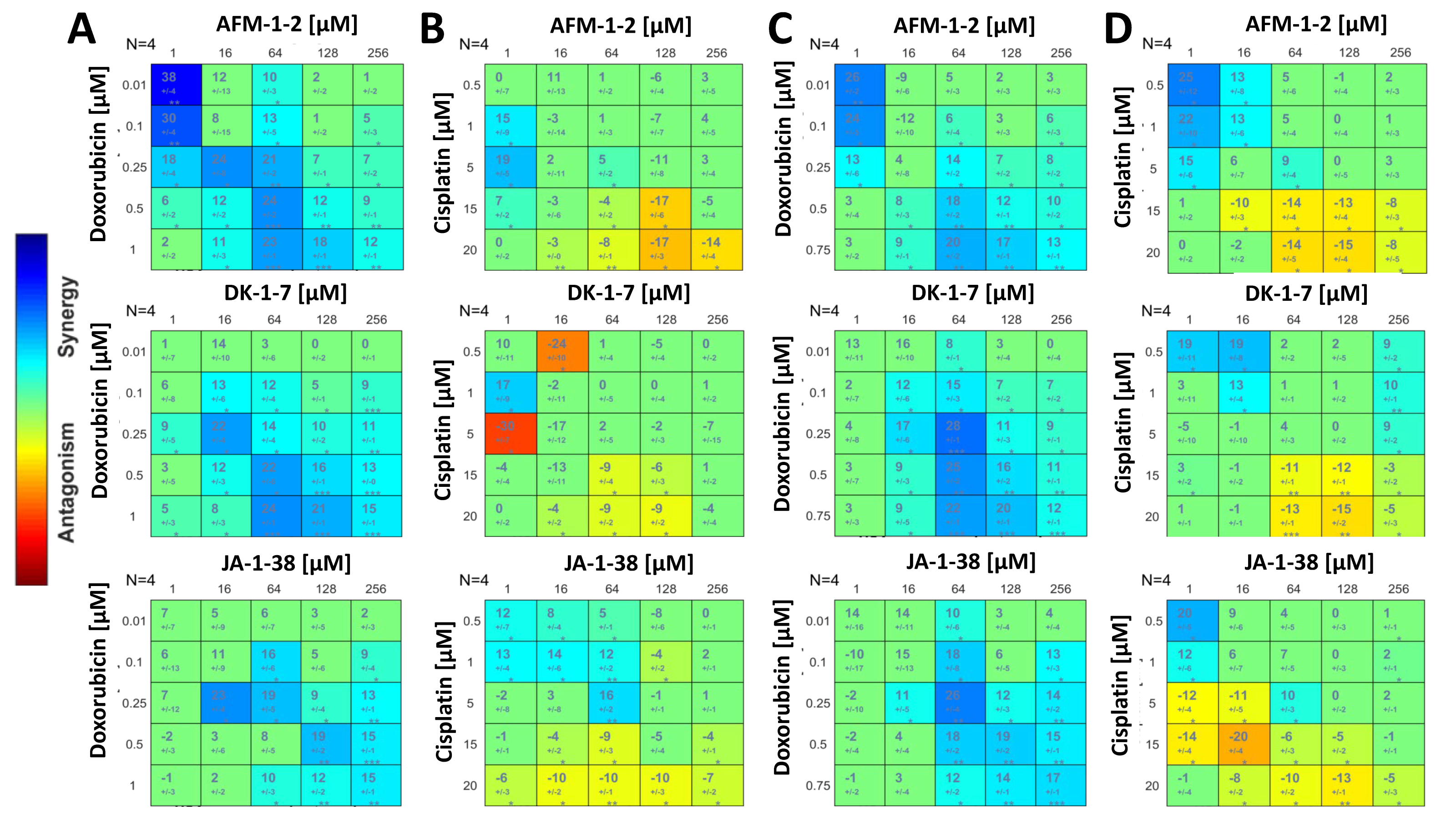
3.7. A Synergistic Effect Was Found between cjoc42 Derivatives and Doxorubicin, While an Antagonistic Effect Was Seen with Cisplatin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.; et al. SEER Cancer Statistics Review, 1975–2018, National Cancer Institute. Bethesda, MD, Based on November 2020 SEER Data Submission, Posted to the SEER Web Site, April 2021. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 1 June 2022).
- Katzenstein, H.M. COG Clinical Trials for Children with Liver Cancer. In Proceedings of the International Pediatric Liver Tumor Symposium, Houston, TX, USA, 25–26 February 2016. [Google Scholar]
- Aronson, D.C.; Meyers, R.L. Malignant tumors of the liver in children. Semin. Pediatr. Surg. 2016, 25, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, H.M.; Krailo, M.D.; Malogolowkin, M.H.; Ortega, J.A.; Liu-Mares, W.; Douglass, E.C.; Feusner, J.H.; Reynolds, M.; Quinn, J.J.; Newman, K.D.; et al. Hepatocellular Carcinoma in Children and Adolescents: Results From the Pediatric Oncology Group and the Children’s Cancer Group Intergroup Study. J. Clin. Oncol. 2002, 20, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Czauderna, P.; Mackinlay, G.; Perilongo, G.; Brown, J.; Shafford, E.; Aronson, D.; Pritchard, J.; Chapchap, P.; Keeling, J.; Plaschkes, J.; et al. Hepatocellular Carcinoma in Children: Results of the First Prospective Study of the International Society of Pediatric Oncology Group. J. Clin. Oncol. 2002, 20, 2798–2804. [Google Scholar] [CrossRef] [PubMed]
- Czauderna, P.; Garnier, H. Hepatoblastoma: Current understanding, recent advances, and controversies. F1000Research 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, A.; Tanwar, S.; Kumar, V.; Banarjee, B.D. Present and Future Prospect of Small Molecule & Related Targeted Therapy Against Human Cancer. Vivechan Int. J. Res. 2018, 9, 36–49. [Google Scholar] [PubMed]
- D’Souza, A.M.; Jiang, Y.; Cast, A.; Valanejad, L.; Wright, M.; Lewis, K.; Kumbaji, M.; Shah, S.; Smithrud, D.; Karns, R.; et al. Gankyrin Promotes Tumor-Suppressor Protein Degradation to Drive Hepatocyte Proliferation. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Valanejad, L.; Cast, A.; Wright, M.; Bissig, K.-D.; Karns, R.; Weirauch, M.T.; Timchenko, N. PARP1 activation increases expression of modified tumor suppressors and pathways underlying development of aggressive hepatoblastoma. Commun. Biol. 2018, 1, 67. [Google Scholar] [CrossRef] [PubMed]
- Cast, A.; Valanejad, L.; Wright, M.; Nguyen, P.; Gupta, A.; Zhu, L.; Shin, S.; Timchenko, N. C/EBPα-dependent preneoplastic tumor foci are the origin of hepatocellular carcinoma and aggressive pediatric liver cancer. Hepatology 2017, 67, 1857–1871. [Google Scholar] [CrossRef] [Green Version]
- Higashitsuji, H.; Itoh, K.; Nagao, T.; Dawson, S.; Nonoguchi, K.; Kido, T.; Mayer, R.J.; Arii, S.; Fujita, J. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat. Med. 2000, 6, 96–99. [Google Scholar] [CrossRef]
- Valanejad, L.; Lewis, K.; Wright, M.; Jiang, Y.; D’Souza, A.; Karns, R.; Sheridan, R.; Gupta, A.; Bove, K.; Witte, D.; et al. FXR-Gankyrin axis is involved in development of pediatric liver cancer. Carcinogenesis 2017, 38, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.-Y.; Wang, H.-Y.; Tan, L.; Liu, S.-Q.; Cao, H.-F.; Wu, M.-C. Overexpression of P28/Gankyrin in Human Hepatocellular Carcinoma and Its Clinical Significance. World J. Gastroenterol. 2002, 8, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Komeda, Y.; Nagai, T.; Kamata, K.; Minaga, K.; Yamao, K.; Takenaka, M.; Hagiwara, S.; Watanabe, T.; Nishida, N.; et al. Gankyrin Contributes to Tumorigenesis and Chemoresistance in Sporadic Colorectal Cancer. Digestion 2018, 100, 192–200. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Chen, H.; Yang, P.; Wu, Q.; Zhang, T.; Wang, C.; Wei, J.; Chen, Z.; Hu, H.; Li, W.; et al. Gankyrin sustains PI3K/GSK-3β/β-catenin signal activation and promotes colorectal cancer aggressiveness and progression. Oncotarget 2016, 7, 81156–81171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-P.; Yan, X.-L.; Li, W.-M.; Ni, Y.-F.; Zhao, J.-B.; Lu, Q.; Wang, X.-J.; Sun, Y.; Chen, P.; Yan, B.-Y.; et al. Clinicopathologic features and prognostic implications of Gankyrin protein expression in non-small cell lung cancer. Pathol. Res. Pract. 2015, 211, 939–947. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; O’Connor, C.J.; Zhang, F.; Galvagnion, C.; Galloway, W.R.J.D.; Tan, Y.S.; Stokes, J.E.; Rahman, T.; Verma, C.; Spring, D.R.; et al. Discovery of a small-molecule binder of the oncoprotein gankyrin that modulates gankyrin activity in the cell. Sci. Rep. 2016, 6, 23732. [Google Scholar] [CrossRef] [Green Version]
- Kanabar, D.; Farrales, P.; Gnanamony, M.; Almasri, J.; Abo-Ali, E.M.; Otmankel, Y.; Shah, H.; Nguyen, D.; El Menyewi, M.; Dukhande, V.V.; et al. Structural modification of the aryl sulfonate ester of cjoc42 for enhanced gankyrin binding and anti-cancer activity. Bioorg. Med. Chem. Lett. 2019, 30, 126889. [Google Scholar] [CrossRef]
- Kanabar, D.; Farrales, P.; Kabir, A.; Juang, D.; Gnanmony, M.; Almasri, J.; Torrents, N.; Shukla, S.; Gupta, V.; Dukhande, V.V.; et al. Optimizing the aryl-triazole of cjoc42 for enhanced gankyrin binding and anti-cancer activity. Bioorg. Med. Chem. Lett. 2020, 30, 127372. [Google Scholar] [CrossRef]
- Wang, G.-L.; Shi, X.; Haefliger, S.; Jin, J.; Major, A.; Iakova, P.; Finegold, M.; Timchenko, N.A. Elimination of C/EBPα through the ubiquitin-proteasome system promotes the development of liver cancer in mice. J. Clin. Investig. 2010, 120, 2549–2562. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Iakova, P.; Jin, J.; Sullivan, E.; Sharin, V.; Hong, I.-H.; Anakk, S.; Mayor, A.; Darlington, G.; Finegold, M.; et al. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology 2012, 57, 1098–1106. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Suarez-Arnedo, A.; Figueroa, F.T.; Clavijo, C.; Arbeláez, P.; Cruz, J.C.; Muñoz-Camargo, C. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef]
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Flow Cytometry-Based Apoptosis Detection. Apoptosis 2009, 559, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.S.; Schnellmann, R.G.; Wills, L.P. Measurement of Cell Death in Mammalian Cells. Curr. Protoc. Pharmacol. 2004, 25, 12.8.1–12.8.22. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani, P.; Matbou Riahi, M.; Momtazi-Borojeni, A.A.; Jamialahmadi, K. Gankyrin: A novel promising therapeutic target for hepatocellular carcinoma. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1301–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speth, P.A.J.; Van Hoesel, Q.G.C.M.; Haanen, C. Clinical Pharmacokinetics of Doxorubicin. Clin. Pharmacokinet. 1988, 15, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Kiyomiya, K.; Matsuo, S.; Kurebe, M. Mechanism of specific nuclear transport of adriamycin: The mode of nuclear translocation of adriamycin-proteasome complex. Cancer Res. 2001, 61, 2467–2471. [Google Scholar] [PubMed]
- Lund, R.J.; Huhtinen, K.; Salmi, J.; Rantala, J.; Nguyen, E.V.; Moulder, R.; Goodlett, D.R.; Lahesmaa, R.; Carpén, O. DNA methylation and Transcriptome Changes Associated with Cisplatin Resistance in Ovarian Cancer. Sci. Rep. 2017, 7, 1469. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Souza, A.M.; Gnanamony, M.; Thomas, M.; Hanley, P.; Kanabar, D.; de Alarcon, P.; Muth, A.; Timchenko, N. Second Generation Small Molecule Inhibitors of Gankyrin for the Treatment of Pediatric Liver Cancer. Cancers 2022, 14, 3068. https://doi.org/10.3390/cancers14133068
D’Souza AM, Gnanamony M, Thomas M, Hanley P, Kanabar D, de Alarcon P, Muth A, Timchenko N. Second Generation Small Molecule Inhibitors of Gankyrin for the Treatment of Pediatric Liver Cancer. Cancers. 2022; 14(13):3068. https://doi.org/10.3390/cancers14133068
Chicago/Turabian StyleD’Souza, Amber M., Manu Gnanamony, Maria Thomas, Peter Hanley, Dipti Kanabar, Pedro de Alarcon, Aaron Muth, and Nikolai Timchenko. 2022. "Second Generation Small Molecule Inhibitors of Gankyrin for the Treatment of Pediatric Liver Cancer" Cancers 14, no. 13: 3068. https://doi.org/10.3390/cancers14133068