
Anticancer Activity of Ω-6 Fatty Acids through Increased 4-HNE in Breast Cancer Cells

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Regulatory Compliance
2.2. Materials
2.3. Cell Culture
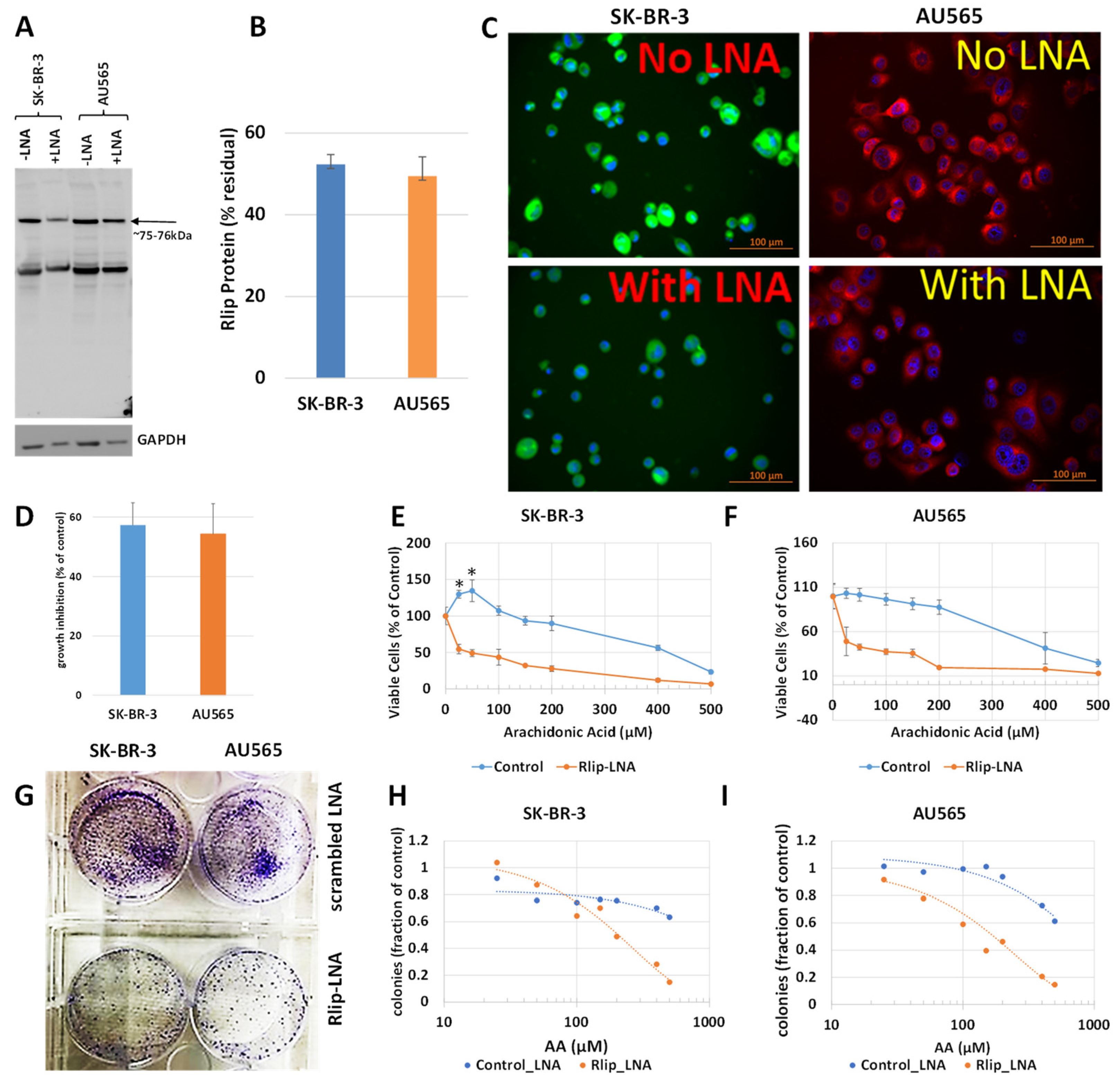
2.4. RLIP Depletion by Rlip Locked Nucleic Acid (Rlip-LNA)
2.5. Cell Treatments
2.6. Cytotoxicity Assay
2.7. Crystal Violet Clonogenic Assay
2.8. Oxidative DNA Damage by TUNEL Assay
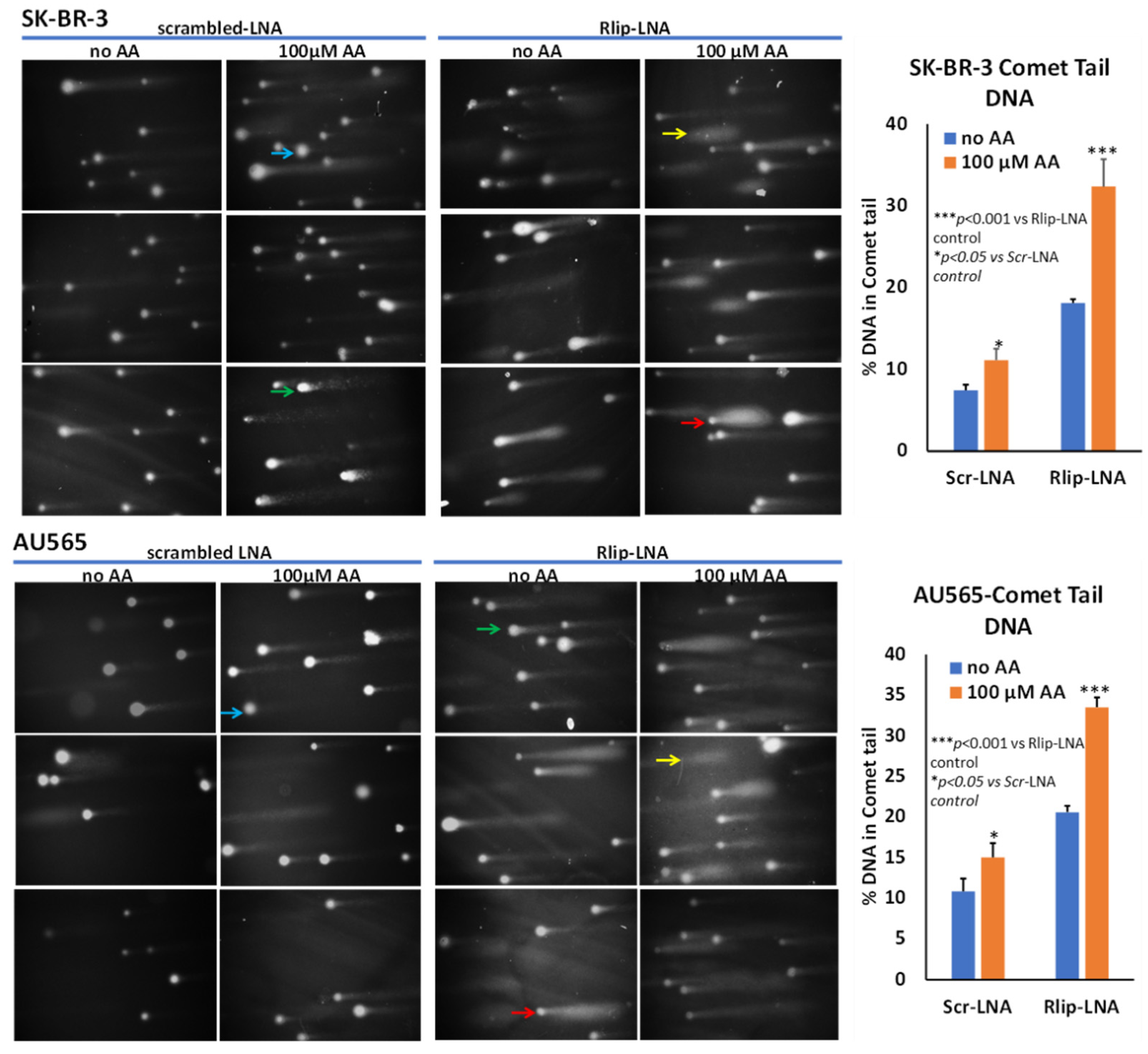
2.9. Comet Assay
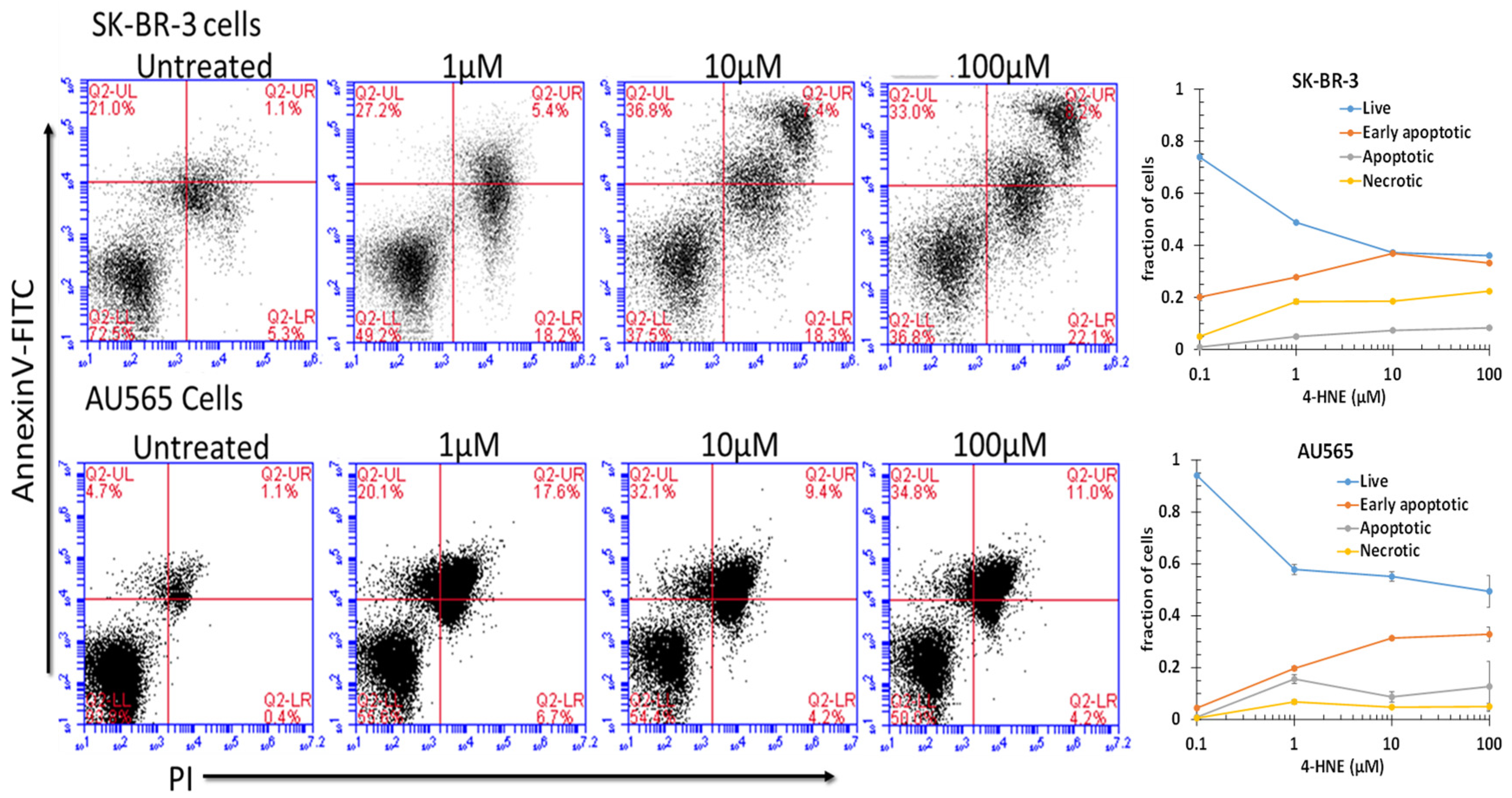
2.10. Apoptosis Assay by Annexin V and PI
2.11. Western Blot Analysis
2.12. Enzyme Assays
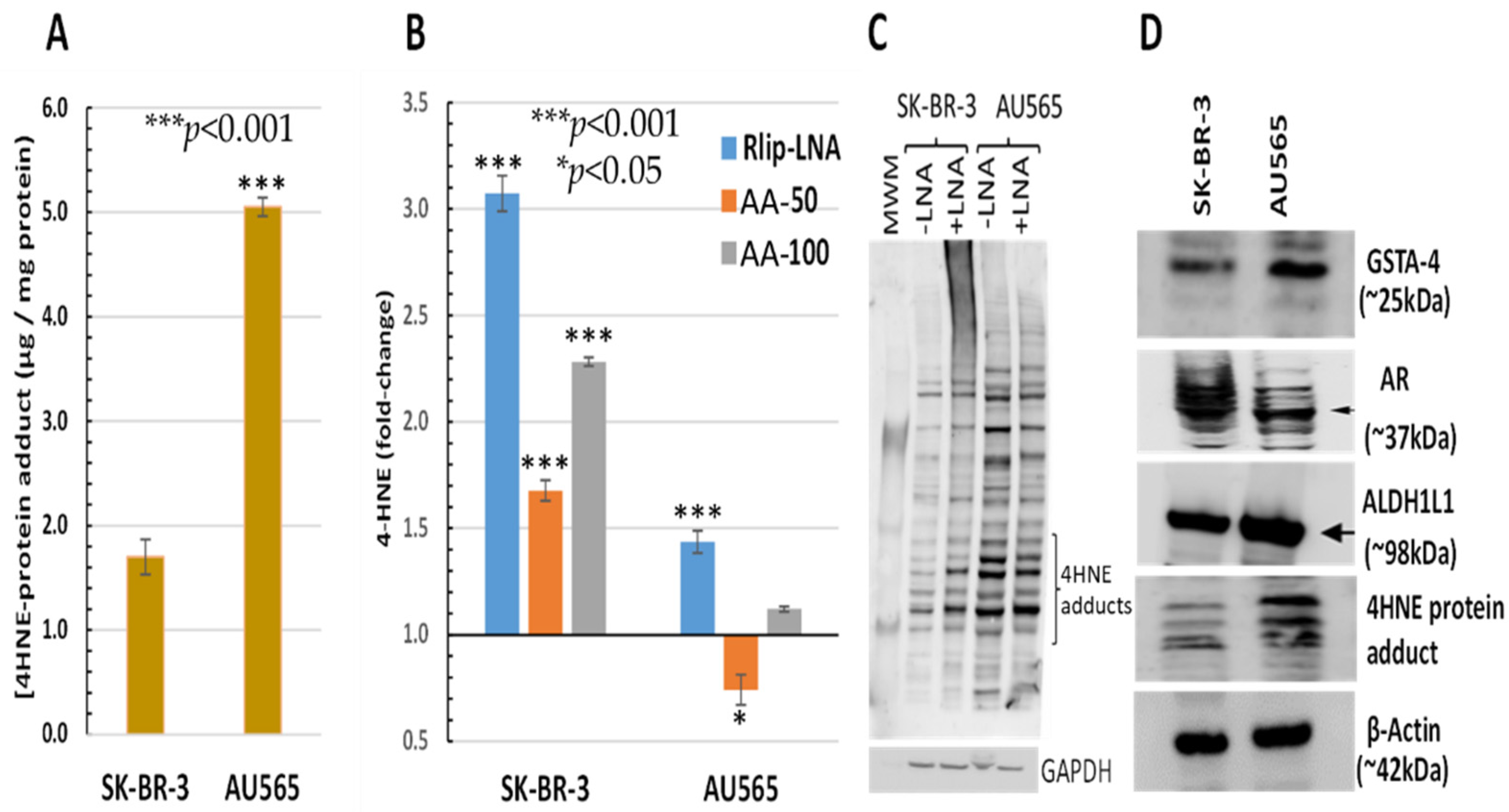
2.13. ELISA Assay for 4-HNE–Protein Adducts
2.14. Intracellular Lipid Peroxidation In Vitro Assay
2.15. Arachidonic Acid Effect on Doxorubicin Efflux
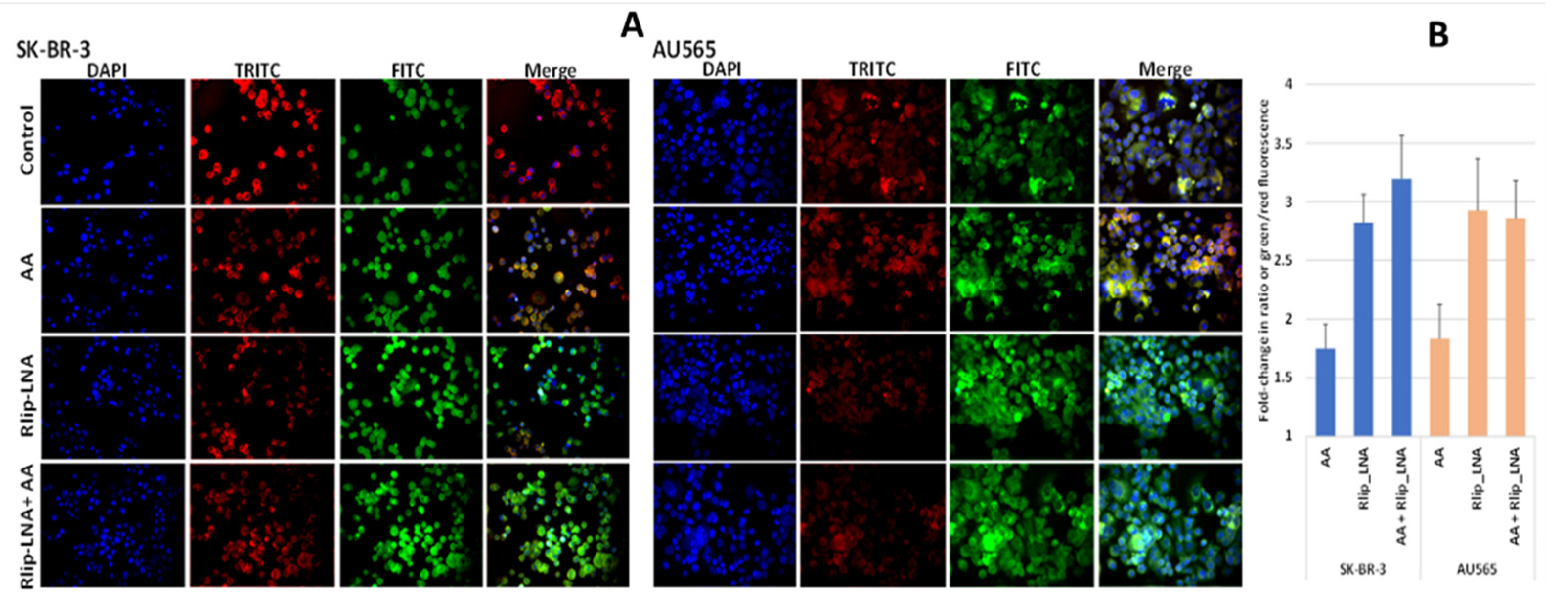
2.16. Effect of Rlip Depletion and AA on EGF Internalization by Immunofluorescence
2.17. Quantitative RT-PCR
2.18. Statistical Analysis
3. Results
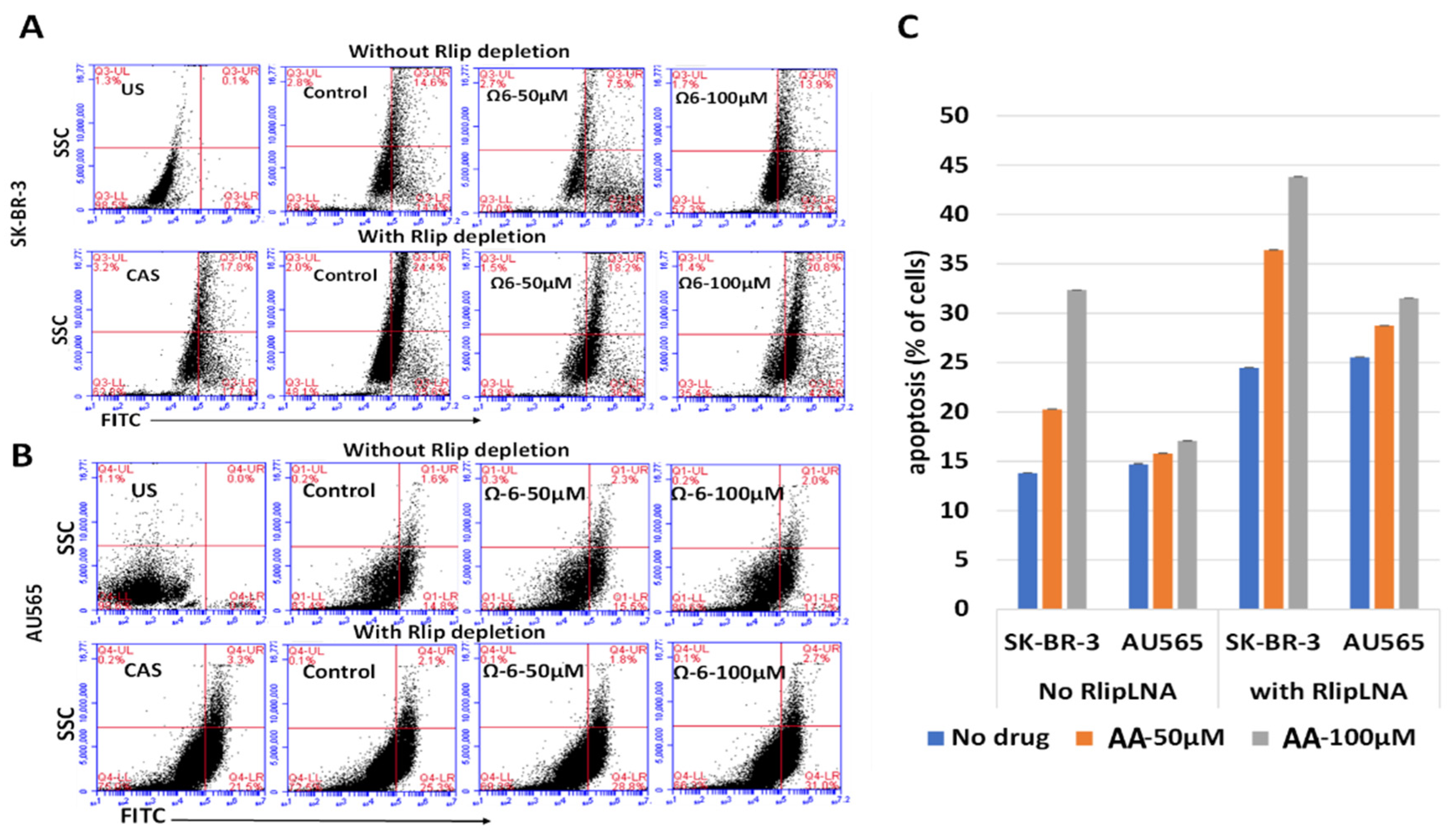
3.1. Effect of Rlip Depletion on AA-Mediated Cytotoxicity
3.2. Effect of AA and Rlip Depletion on Lipid Peroxidation and 4-HNE Levels
3.3. Apoptosis by Rlip Depletion: AA or 4-HNE
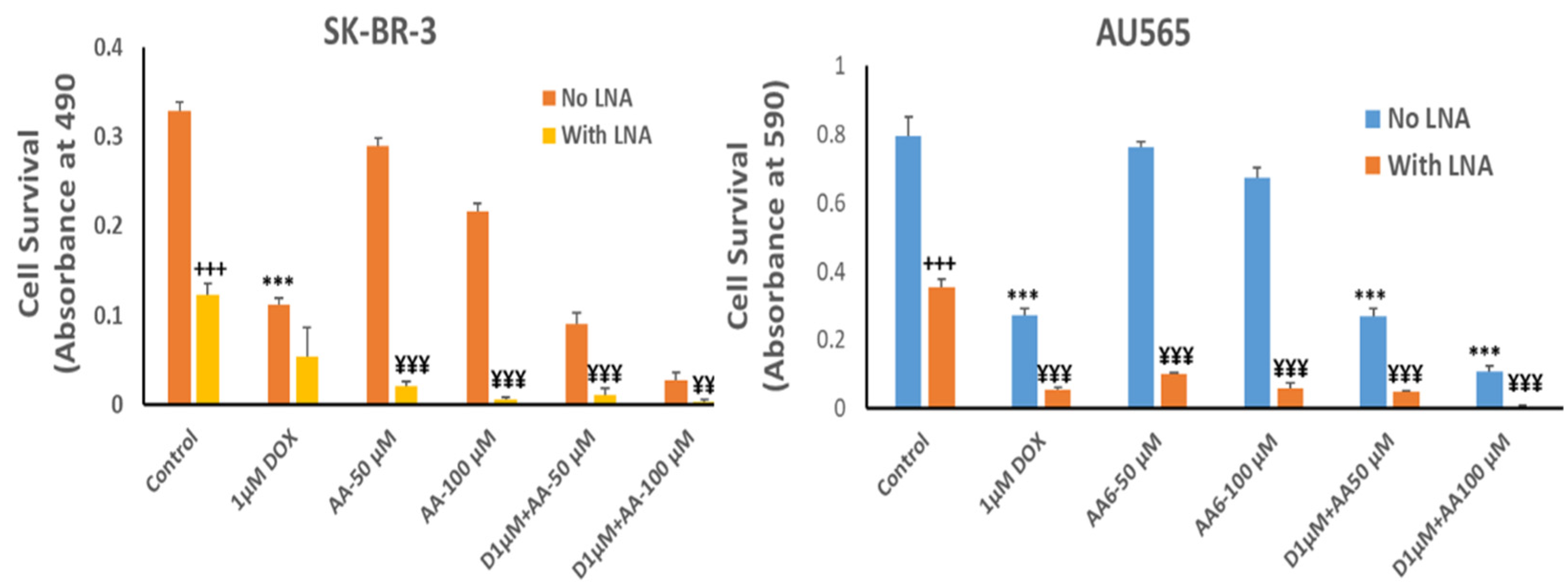
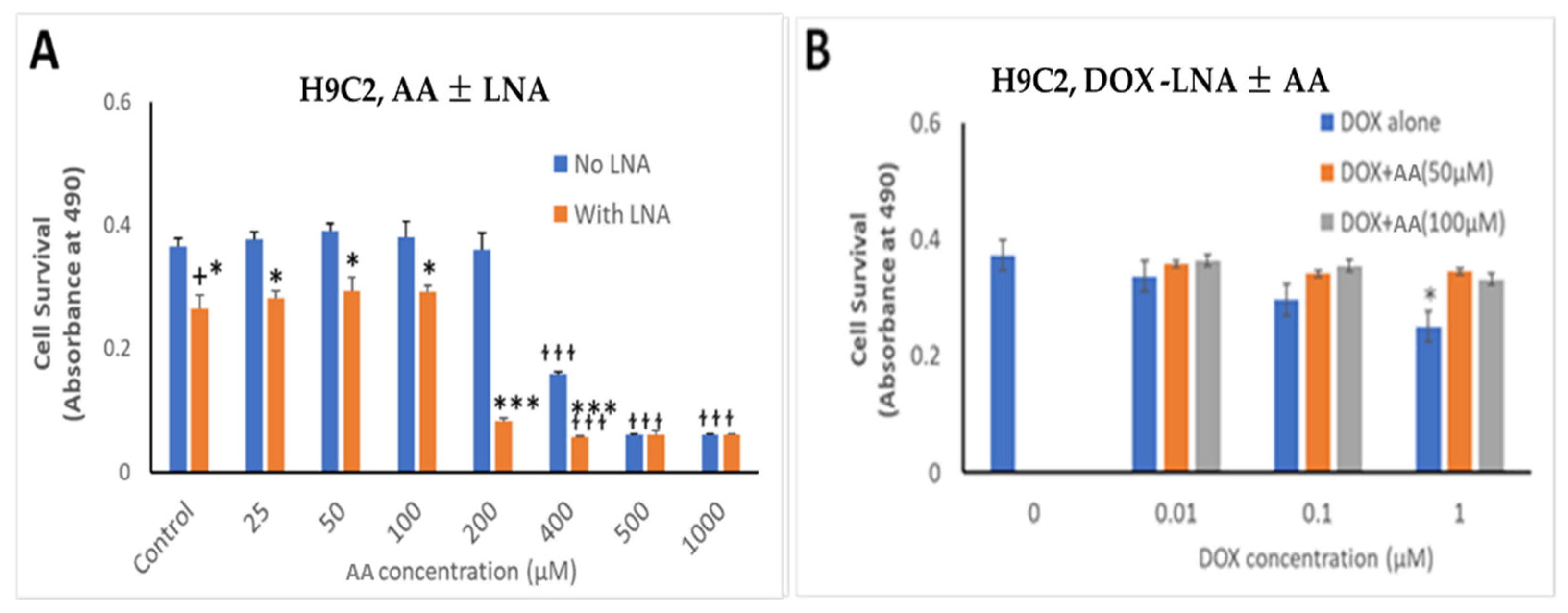
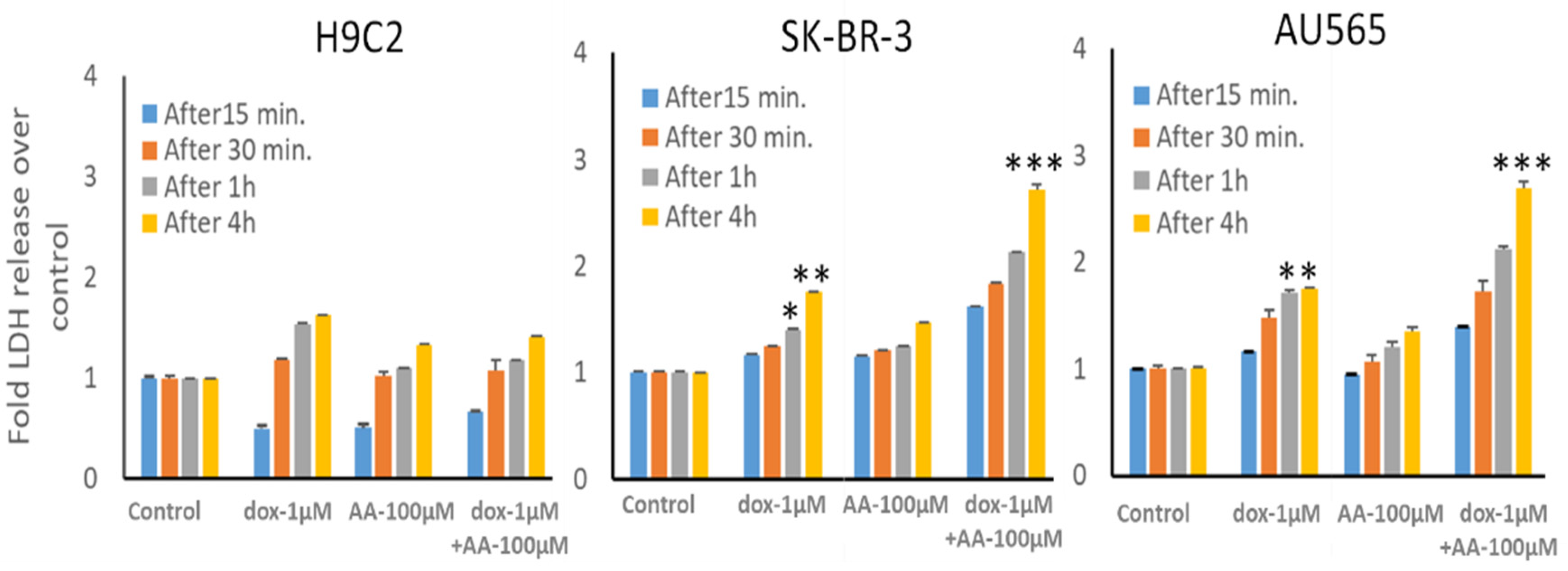
3.4. Effect of AA and Rlip Depletion on Doxorubicin Cytotoxicity in Cancer Cells and Cardiomyocytes
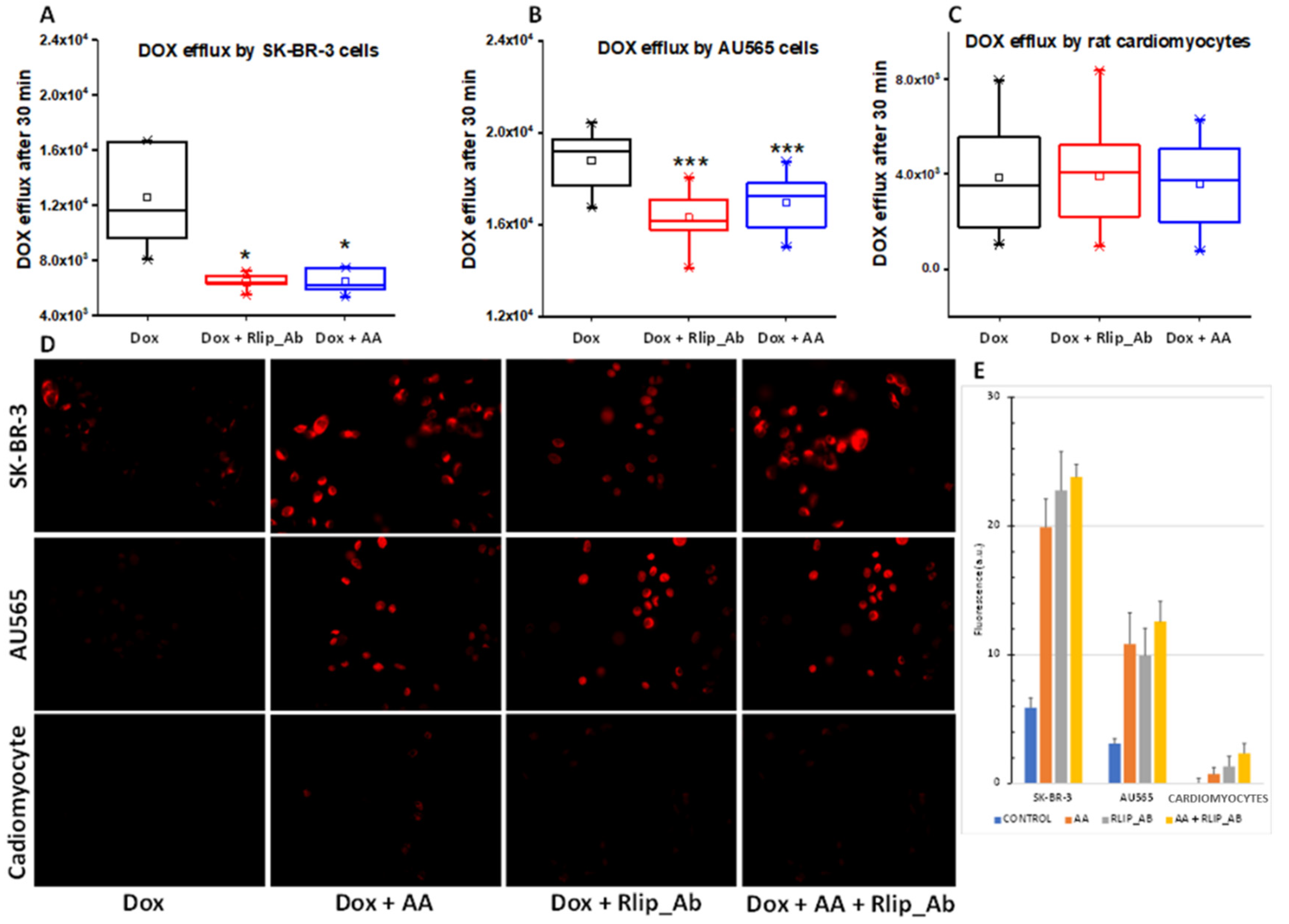
3.5. Effect of AA and Rlip Transport Inhibition on Accumulation and Efflux of Doxorubicin
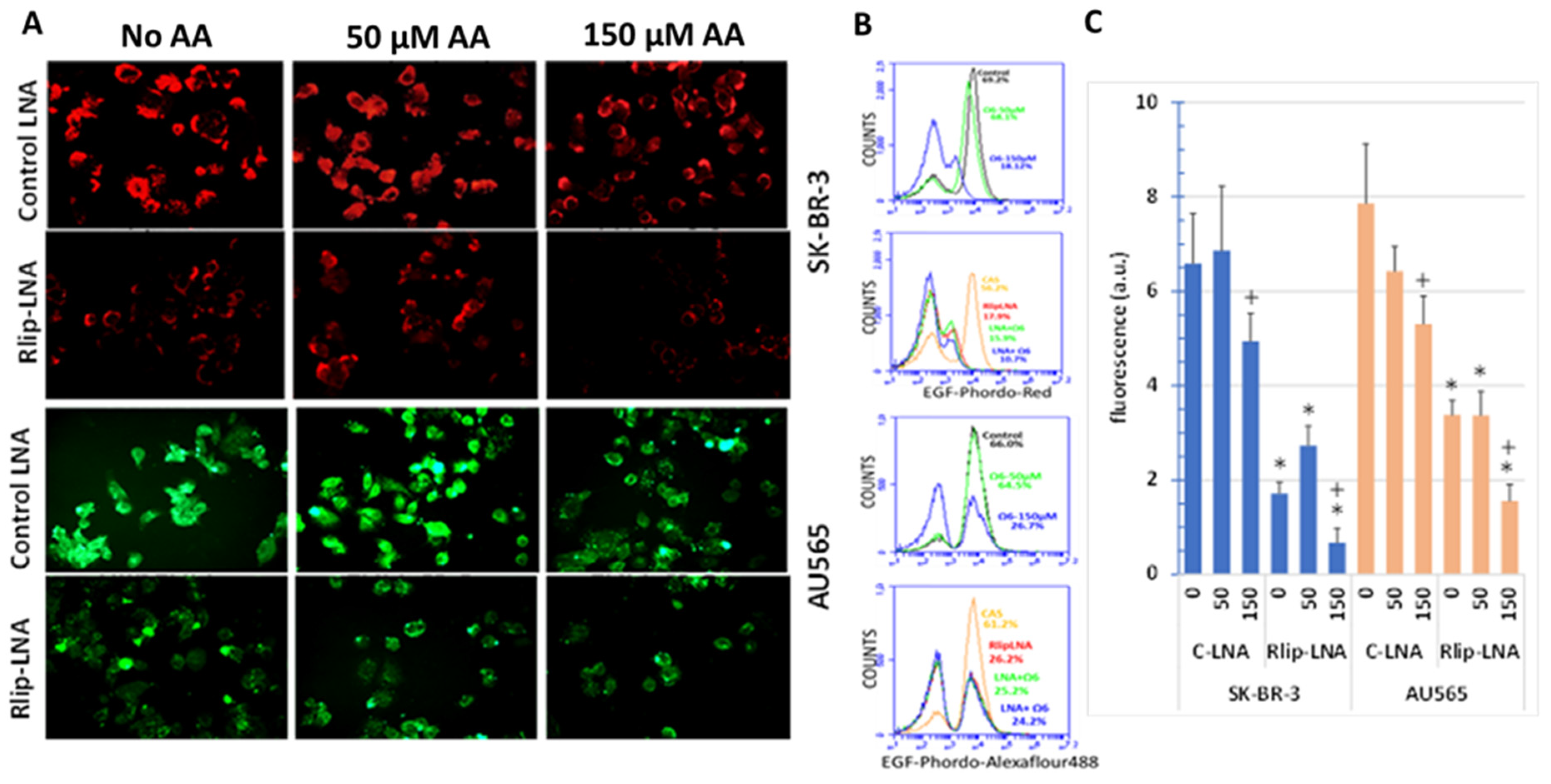
3.6. Effect of AA and Rlip Transport Inhibition on Endocytosis of EGF
3.7. Effect of AA on Expression of Antioxidant Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Awasthi, S.; Singhal, S.S.; Awasthi, Y.C.; Martin, B.; Woo, J.H.; Cunningham, C.C.; Frankel, A.E. RLIP76 and Cancer. Clin. Cancer Res. 2008, 14, 4372–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimniak, P. Relationship of electrophilic stress to aging. Free Radic. Biol. Med. 2011, 51, 1087–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanoaga, O.; Jurj, A.; Raduly, L.; Cojocneanu-Petric, R.; Fuentes-Mattei, E.; Wu, O.; Braicu, C.; Gherman, C.D.; Berindan-Neagoe, I. Implications of dietary ω-3 and ω-6 polyunsaturated fatty acids in breast cancer. Exp. Therap. Med. 2018, 15, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Albert, C.M.; Gordon, D.; Copeland, T.; et al. Marine n-3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N. Engl. J. Med. 2019, 380, 23–32. [Google Scholar] [CrossRef]
- de Lorgeril, M.; Salen, P. New insights into the health effects of dietary saturated and omega-6 and omega-3 polyunsaturated fatty acids. BMC Med. 2012, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Astorg, P. Dietary N-6 and N-3 polyunsaturated fatty acids and prostate cancer risk: A review of epidemiological and experimental evidence. Cancer Causes Control 2004, 15, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Chen, T.; Chen, L.; Mei, N.; McLain, E.; Samokyszyn, V.; Thaden, J.J.; Moore, M.M.; Zimniak, P. Mutagenic effects of 4-hydroxynonenal triacetate, a chemically protected form of the lipid peroxidation product 4-hydroxynonenal, as assayed in L5178Y/Tk+/- mouse lymphoma cells. J. Pharmacol. Exp. Ther. 2005, 313, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Moore, D.R.; Nimmo, S.L.; Lightfoot, S.A.; Huycke, M.M. 4-hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology 2012, 142, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Li, W.; Kong, A.N. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2012, 2, 40. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Niemczyk, M.; Saini, D.; Sadovov, V.; Zimniak, L.; Zimniak, P. Disruption of the mGsta4 gene increases life span of C57BL mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Engle, M.R.; Singh, S.P.; Czernik, P.J.; Gaddy, D.; Montague, D.C.; Ceci, J.D.; Yang, Y.; Awasthi, S.; Awasthi, Y.C.; Zimniak, P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: Generation and analysis of mGsta4 null mouse. Toxicol. Appl. Pharmacol. 2004, 194, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Coronella, J.A.; Benes, H.; Cochrane, B.J.; Zimniak, P. Catalytic function of Drosophila melanogaster glutathione S-transferase DmGSTS1-1 (GST-2) in conjugation of lipid peroxidation end products. Eur. J. Biochem. FEBS 2001, 268, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- McElhanon, K.E.; Bose, C.; Sharma, R.; Wu, L.; Awasthi, Y.C.; Singh, S.P. Gsta4 Null Mouse Embryonic Fibroblasts Exhibit Enhanced Sensitivity to Oxidants: Role of 4-Hydroxynonenal in Oxidant Toxicity. Open J. Apoptosis 2013, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.D.; Prabhu, K.S.; Thompson, J.T.; Reddy, P.S.; Peters, J.M.; Peterson, B.R.; Reddy, C.C.; Vanden Heuvel, J.P. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). Free Radic. Biol. Med. 2007, 42, 1155–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Piper, J.T.; Singhal, S.S.; Salameh, M.S.; Torman, R.T.; Awasthi, Y.C.; Awasthi, S. Mechanisms of anticarcinogenic properties of curcumin: The effect of curcumin on glutathione linked detoxification enzymes in rat liver. Int. J. Biochem. Cell Biol. 1998, 30, 445–456. [Google Scholar] [CrossRef]
- Singhal, S.S.; Yadav, S.; Roth, C.; Singhal, J. RLIP76: A novel glutathione-conjugate and multi-drug transporter. Biochem. Pharmacol. 2009, 77, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Sharma, R.; Sharma, A.; Awasthi, S.; Awasthi, Y.C. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Pol. 2003, 50, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sharma, A.; Sharma, R.; Patrick, B.; Singhal, S.S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: Role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003, 278, 41380–41388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Yang, Y.; Sharma, A.; Dwivedi, S.; Popov, V.L.; Boor, P.J.; Singhal, S.S.; Awasthi, S.; Awasthi, Y.C. Mechanisms and physiological significance of the transport of the glutathione conjugate of 4-hydroxynonenal in human lens epithelial cells. Investig. Ophthalmol. Visual Sci. 2003, 44, 3438–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Sharma, A.; Yang, Y.; Awasthi, S.; Singhal, S.S.; Zimniak, P.; Awasthi, Y.C. Functional reconstitution of Ral-binding GTPase activating protein, RLIP76, in proteoliposomes catalyzing ATP-dependent transport of glutathione conjugate of 4-hydroxynonenal. Acta Biochim. Pol. 2002, 49, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.-Z.; Sharma, R.; Yang, Y.; Singhal, S.S.; Sharma, A.; Saini, M.K.; Singh, S.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Accelerated Metabolism and Exclusion of 4-Hydroxynonenal through Induction of RLIP76 and hGST5.8 Is an Early Adaptive Response of Cells to Heat and Oxidative Stress. J. Biol. Chem. 2001, 276, 41213–41223. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohnert, B.I.; Long, E.K.; Hahn, W.S.; Bernlohr, D.A. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 2014, 63, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammali, R.; Ramana, K.V.; Singhal, S.S.; Awasthi, S.; Srivastava, S.K. Aldose reductase regulates growth factor-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colon cancer cells. Cancer Res. 2006, 66, 9705–9713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonowal, H.; Pal, P.B.; Wen, J.J.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibitor increases doxorubicin-sensitivity of colon cancer cells and decreases cardiotoxicity. Sci. Rep. 2017, 7, 3182. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef] [Green Version]
- Rousar, T.; Parik, P.; Kucera, O.; Bartos, M.; Cervinkova, Z. Glutathione reductase is inhibited by acetaminophen-glutathione conjugate in vitro. Physiol. Res. 2010, 59, 225–232. [Google Scholar] [CrossRef]
- Awasthi, S.; Srivastava, S.K.; Ahmad, F.; Ahmad, H.; Ansari, G.A. Interactions of glutathione S-transferase-pi with ethacrynic acid and its glutathione conjugate. Biochim. Biophys. Acta 1993, 1164, 173–178. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, A.; Dwivedi, S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal self-limits fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry 2008, 47, 143–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.Z.; Singhal, S.S.; Sharma, A.; Saini, M.; Yang, Y.; Awasthi, S.; Zimniak, P.; Awasthi, Y.C. Transfection of mGSTA4 in HL-60 cells protects against 4-hydroxynonenal-induced apoptosis by inhibiting JNK-mediated signaling. Arch. Biochem. Biophys. 2001, 392, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Z.; Singhal, S.S.; Saini, M.; Singhal, J.; Piper, J.T.; Van Kuijk, F.J.; Zimniak, P.; Awasthi, Y.C.; Awasthi, S. Effects of mGST A4 transfection on 4-hydroxynonenal-mediated apoptosis and differentiation of K562 human erythroleukemia cells. Arch. Biochem. Biophys. 1999, 372, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Chen, G.; Liang, M.; Xie, A.; Li, Q.; Guo, Q.; Sharma, R.; Cheng, J. Reduced Expression of Glutathione S-Transferase α4 Promotes Vascular Neointimal Hyperplasia in CKD. J. Am. Soc. Nephrol. 2018, 29, 505–517. [Google Scholar] [CrossRef]
- Chaudhary, P.; Sharma, R.; Sahu, M.; Vishwanatha, J.K.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces G2/M phase cell cycle arrest by activation of the ataxia telangiectasia mutated and Rad3-related protein (ATR)/checkpoint kinase 1 (Chk1) signaling pathway. J. Biol. Chem. 2013, 288, 20532–20546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008, 480, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Ma, X.; Luo, X.; Zhang, Y.; He, Y.; Dai, Z.; Yang, Y.; Wu, G.; Wu, Z. l-Glutamine Attenuates Apoptosis in Porcine Enterocytes by Regulating Glutathione-Related Redox Homeostasis. J. Nutr. 2018, 148, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.G.; Xu, X.; Xue, Y.M.; Zhao, S.L. Comparison of 4-hydroxynonenal-induced p53-mediated apoptosis in prostate cancer cells LNCaP and DU145. Contemp. Oncol. 2014, 18, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Tammali, R.; Reddy, A.B.; Saxena, A.; Rychahou, P.G.; Evers, B.M.; Qiu, S.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Inhibition of aldose reductase prevents colon cancer metastasis. Carcinogenesis 2011, 32, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.B.; Jin, D.D.; Jiao, Y.J.; Ni, W.K.; Liu, J.X.; Qu, L.S.; Lu, C.H.; Ni, R.Z.; Jiang, F.; Chen, W.C. β2-AR regulates the expression of AKR1B1 in human pancreatic cancer cells and promotes their proliferation via the ERK1/2 pathway. Mol. Biol. Rep. 2018, 45, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Venton, G.; Pérez-Alea, M.; Baier, C.; Fournet, G.; Quash, G.; Labiad, Y.; Martin, G.; Sanderson, F.; Poullin, P.; Suchon, P.; et al. Aldehyde dehydrogenases inhibition eradicates leukemia stem cells while sparing normal progenitors. Blood Cancer J. 2016, 6, e469. [Google Scholar] [CrossRef] [Green Version]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, S.E.; Chen, Y.; Petrovic, N.; Zhang, J.; Bourget, K.; Mackenzie, P.I.; Murray, M. Activation of ALDH1A1 in MDA-MB-468 breast cancer cells that over-express CYP2J2 protects against paclitaxel-dependent cell death mediated by reactive oxygen species. Biochem. Pharmacol. 2017, 143, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, Y.C.; Sharma, R.; Cheng, J.Z.; Yang, Y.; Sharma, A.; Singhal, S.S.; Awasthi, S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol. Asp. Med. 2003, 24, 219–230. [Google Scholar] [CrossRef]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Warnke, M.M.; Wanigasekara, E.; Singhal, S.S.; Singhal, J.; Awasthi, S.; Armstrong, D.W. The determination of glutathione-4-hydroxynonenal (GSHNE), E-4-hydroxynonenal (HNE), and E-1-hydroxynon-2-en-4-one (HNO) in mouse liver tissue by LC-ESI-MS. Anal. Bioanal. Chem. 2008, 392, 1325–1333. [Google Scholar] [CrossRef]
- Singhal, S.S.; Yadav, S.; Singhal, J.; Sahu, M.; Sehrawat, A.; Awasthi, S. Diminished drug transport and augmented radiation sensitivity caused by loss of RLIP76. FEBS Lett. 2008, 582, 3408–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Tompkins, J.; Singhal, J.; Riggs, A.D.; Yadav, S.; Wu, X.; Singh, S.; Warden, C.; Liu, Z.; Wang, J.; et al. Rlip depletion prevents spontaneous neoplasia in TP53 null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 3918–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Lee, J.; Bose, C.; Li, H.; Yuan, Y.-C.; Hindle, A.; Singhal, S.S.; Kopel, J.; Palade, P.T.; Jones, C.; et al. Haploinsufficiency Interactions between RALBP1 and p53 in ERBB2 and PyVT Models of Mouse Mammary Carcinogenesis. Cancers 2021, 13, 3329. [Google Scholar] [CrossRef]
- Singhal, J.; Singhal, S.S.; Yadav, S.; Suzuki, S.; Warnke, M.M.; Yacoub, A.; Dent, P.; Bae, S.; Sharma, R.; Awasthi, Y.C.; et al. RLIP76 in defense of radiation poisoning. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Singhal, S.S.; Yadav, S.; Singhal, J.; Drake, K.; Nadkar, A.; Zajac, E.; Wickramarachchi, D.; Rowe, N.; Yacoub, A.; et al. RLIP76 is a major determinant of radiation sensitivity. Cancer Res. 2005, 65, 6022–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Haque, A.K.; Awasthi, S.; Singh, S.V.; Piper, J.T.; Awasthi, Y.C. Differential carcinogenicity of benzo[a]pyrene in male and female CD-1 mouse lung. J. Toxicol. Environ. Health 1997, 52, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Bose, C.; Yadav, S.; Singhal, S.S.; Singhal, J.; Hindle, A.; Lee, J.; Cheedella, N.K.S.; Rehman, S.; Rahman, R.L.; Jones, C.; et al. Rlip Depletion Suppresses Growth of Breast Cancer. Cancers 2020, 12, 1446. [Google Scholar] [CrossRef]
- Singhal, J.; Chikara, S.; Horne, D.; Salgia, R.; Awasthi, S.; Singhal, S.S. RLIP inhibition suppresses breast-to-lung metastasis. Cancer Lett. 2019, 447, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.; Singh, S.P.; Igid, H.; Green, W.C.; Singhal, S.S.; Lee, J.; Palade, P.T.; Rajan, A.; Ball, S.; Tonk, V.; et al. Topical 2'-Hydroxyflavanone for Cutaneous Melanoma. Cancers 2019, 11, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Singhal, J.; Figarola, J.; Horne, D.; Awasthi, S. RLIP76 Targeted Therapy for Kidney Cancer. Pharm. Res. 2015, 32, 3123–3136. [Google Scholar] [CrossRef] [Green Version]
- Singhal, J.; Kulkarni, P.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S.S. Prevention of mammary carcinogenesis in MMTV-neu mice by targeting RLIP. Mol. Carcinog. 2021. [Google Scholar] [CrossRef]
- Sun, A.; Cheng, Y.; Zhang, Y.; Zhang, Q.; Wang, S.; Tian, S.; Zou, Y.; Hu, K.; Ren, J.; Ge, J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J. Mol. Cell Cardiol. 2014, 71, 92–104. [Google Scholar] [CrossRef]
- Casalini, P.; Botta, L.; Menard, S. Role of p53 in HER2-induced proliferation or apoptosis. J. Biol. Chem. 2001, 276, 12449–12453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, S.S.; Dane, M.; Chin, K.; Tatarova, Z.; Liu, M.; Liby, T.; Thompson, W.; Smith, R.; Nederlof, M.; Bucher, E.; et al. Microenvironment-Mediated Mechanisms of Resistance to HER2 Inhibitors Differ between HER2+ Breast Cancer Subtypes. Cell Syst. 2018, 6, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Burns, D.J.; Rich, B.E.; MacNeil, I.A.; Dandapat, A.; Soltani, S.M.; Myhre, S.; Sullivan, B.F.; Lange, C.A.; Furcht, L.T.; et al. Development of a test that measures real-time HER2 signaling function in live breast cancer cell lines and primary cells. BMC Cancer 2017, 17, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Horne, D.; Singhal, J.; Vonderfecht, S.; Salgia, R.; Awasthi, S. Synergistic efficacy of RLIP inhibition and 2'-hydroxyflavanone against DMBA-induced mammary carcinogenesis in SENCAR mice. Mol. Carcinog. 2019. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Sharma, R.; Sharma, A.; Chaudhary, P.; Pearce, V.; Vatsyayan, R.; Singh, S.V.; Awasthi, S.; Awasthi, Y.C. Role of lipid peroxidation in cellular responses to D,L-sulforaphane, a promising cancer chemopreventive agent. Biochemistry 2010, 49, 3191–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, G.B.; Satyanarayana, A.; Balakrishna, N.; Ayyagari, R.; Padma, M.; Viswanath, K.; Petrash, J.M. Erythrocyte aldose reductase activity and sorbitol levels in diabetic retinopathy. Mol. Vis. 2008, 14, 593–601. [Google Scholar]
- Singhal, S.S.; Zimniak, P.; Awasthi, S.; Piper, J.T.; He, N.G.; Teng, J.I.; Petersen, D.R.; Awasthi, Y.C. Several closely related glutathione S-transferase isozymes catalyzing conjugation of 4-hydroxynonenal are differentially expressed in human tissues. Arch. Biochem. Biophys. 1994, 311, 242–250. [Google Scholar] [CrossRef]
- Xiao, B.; Singh, S.P.; Nanduri, B.; Awasthi, Y.C.; Zimniak, P.; Ji, X. Crystal structure of a murine glutathione S-transferase in complex with a glutathione conjugate of 4-hydroxynon-2-enal in one subunit and glutathione in the other: Evidence of signaling across the dimer interface. Biochemistry 1999, 38, 11887–11894. [Google Scholar] [CrossRef]
- Singh, S.P.; Niemczyk, M.; Zimniak, L.; Zimniak, P. Fat accumulation in Caenorhabditis elegans triggered by the electrophilic lipid peroxidation product 4-hydroxynonenal (4-HNE). Aging 2008, 1, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Nagaprashantha, L.; Li, H.; Yuan, Y.C.; Liu, Z.; Berz, D.; Igid, H.; Green, W.C.; et al. Anticancer activity of 2′-hydroxyflavanone towards lung cancer. Oncotarget 2018, 9, 36202–36219. [Google Scholar] [CrossRef]
- Chen, Z.H.; Niki, E. 4-hydroxynonenal (4-HNE) has been widely accepted as an inducer of oxidative stress. Is this the whole truth about it or can 4-HNE also exert protective effects? IUBMB Life 2006, 58, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.M.; Gorringe, K.L.; Chin, S.F.; Orsetti, B.; Besret, C.; Courtay-Cahen, C.; Roberts, I.; Theillet, C.; Caldas, C.; Edwards, P.A. Molecular cytogenetic analysis of breast cancer cell lines. Br. J. Cancer 2000, 83, 1309–1317. [Google Scholar] [CrossRef] [Green Version]
- Koppen, G.; Azqueta, A.; Pourrut, B.; Brunborg, G.; Collins, A.R.; Langie, S.A.S. The next three decades of the comet assay: A report of the 11th International Comet Assay Workshop. Mutagenesis 2017, 32, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Creasey, W.A.; McIntosh, L.S.; Brescia, T.; Odujinrin, O.; Aspnes, G.T.; Murray, E.; Marsh, J.C. Clinical effects and pharmacokinetics of different dosage schedules of adriamycin. Cancer Res. 1976, 36, 216–221. [Google Scholar] [PubMed]
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986, 261, 3068–3074. [Google Scholar] [CrossRef]
- Sharma, R.; Singhal, S.S.; Cheng, J.; Yang, Y.; Sharma, A.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. RLIP76 is the major ATP-dependent transporter of glutathione-conjugates and doxorubicin in human erythrocytes. Arch. Biochem. Biophys. 2001, 391, 171–179. [Google Scholar] [CrossRef]
- Awasthi, S.; Cheng, J.Z.; Singhal, S.S.; Pandya, U.; Sharma, R.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Functional reassembly of ATP-dependent xenobiotic transport by the N- and C-terminal domains of RLIP76 and identification of ATP binding sequences. Biochemistry 2001, 40, 4159–4168. [Google Scholar] [CrossRef]
- Awasthi, S.; Cheng, J.; Singhal, S.S.; Saini, M.K.; Pandya, U.; Pikula, S.; Bandorowicz-Pikula, J.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry 2000, 39, 9327–9334. [Google Scholar] [CrossRef]
- Magdy, T.; Burmeister, B.T.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Vatsyayan, R.; Lelsani, P.C.; Awasthi, S.; Singhal, S.S. RLIP76: A versatile transporter and an emerging target for cancer therapy. Biochem. Pharmacol. 2010, 79, 1699–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, K.J.; Singhal, J.; Yadav, S.; Nadkar, A.; Pungaliya, C.; Singhal, S.S.; Awasthi, S. RALBP1/RLIP76 mediates multidrug resistance. Int. J. Oncol. 2007, 30, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Singhal, S.S.; Wickramarachchi, D.; Awasthi, Y.C.; Awasthi, S. RLIP76 (RALBP1)-mediated transport of leukotriene C4 (LTC4) in cancer cells: Implications in drug resistance. Int. J. Cancer 2004, 112, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Salgia, R.; Verma, N.; Horne, D.; Awasthi, S. RLIP controls receptor-ligand signaling by regulating clathrin-dependent endocytosis. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188337. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Song, Q.; Cai, Y.; Wang, P.; Wang, M.; Zhang, D. RLIP76-dependent suppression of PI3K/AKT/Bcl-2 pathway by miR-101 induces apoptosis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 463, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cai, Z.; Liang, Q.; Wang, Q.; Lu, Y.; Hu, L.; Hu, G. RLIP76 Depletion Enhances Autophagic Flux in U251 Cells. Cell. Mol. Neurobiol. 2017, 37, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K. Class II phosphatidylinositol 3-kinase isoforms in vesicular trafficking. Biochem. Soc. Trans. 2021, 49, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yoshioka, K.; Aki, S.; Takuwa, Y. Class II phosphatidylinositol 3-kinase-C2α is essential for Notch signaling by regulating the endocytosis of γ-secretase in endothelial cells. Sci. Rep. 2021, 11, 5199. [Google Scholar] [CrossRef]
- Singhal, S.S.; Wickramarachchi, D.; Yadav, S.; Singhal, J.; Leake, K.; Vatsyayan, R.; Chaudhary, P.; Lelsani, P.; Suzuki, S.; Yang, S.; et al. Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol. Cancer Therap. 2011, 10, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Hyuga, S.; Takiguchi, S.; Hyuga, M.; Itoh, K.; Hanawa, T. Ephedrae herba stimulates hepatocyte growth factor-induced MET endocytosis and downregulation via early/late endocytic pathways in gefitinib-resistant human lung cancer cells. Int. J. Oncol. 2016, 48, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Zaoui, K.; Duhamel, S.; Parachoniak, C.A.; Park, M. CLIP-170 spatially modulates receptor tyrosine kinase recycling to coordinate cell migration. Traffic 2019, 20, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jullien-Flores, V.; Mahe, Y.; Mirey, G.; Leprince, C.; Meunier-Bisceuil, B.; Sorkin, A.; Camonis, J.H. RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: Involvement of the Ral pathway in receptor endocytosis. J. Cell Sci. 2000, 113, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Pandya, U.; Singhal, S.S.; Lin, J.T.; Thiviyanathan, V.; Seifert, W.E., Jr.; Awasthi, Y.C.; Ansari, G.A. Curcumin-glutathione interactions and the role of human glutathione S-transferase P1-1. Chem. Biol. Interact. 2000, 128, 19–38. [Google Scholar] [CrossRef]
- Schramm, V.L.; McCluskey, R.; Emig, F.A.; Litwack, G. Kinetic studies and active site-binding properties of glutathione S-transferase using spin-labeled glutathione, a product analogue. J. Biol. Chem. 1984, 259, 714–722. [Google Scholar] [CrossRef]
- Jochheim, C.M.; Baillie, T.A. Selective and irreversible inhibition of glutathione reductase in vitro by carbamate thioester conjugates of methyl isocyanate. Biochem. Pharmacol. 1994, 47, 1197–1206. [Google Scholar] [CrossRef]
- Chen, J.; Xia, Q.; Jiang, B.; Chang, W.; Yuan, W.; Ma, Z.; Liu, Z.; Shu, X. Prognostic Value of Cancer Stem Cell Marker ALDH1 Expression in Colorectal Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0145164. [Google Scholar] [CrossRef]
- Tammali, R.; Srivastava, S.K.; Ramana, K.V. Targeting aldose reductase for the treatment of cancer. Curr. Cancer Drug Targets 2011, 11, 560–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonowal, H.; Ramana, K.V. Development of Aldose Reductase Inhibitors for the Treatment of Inflammatory Disorders and Cancer: Current Drug Design Strategies and Future Directions. Curr. Med. Chem. 2021, 28, 3683–3712. [Google Scholar] [CrossRef]
- Banerjee, S. Aldo Keto Reductases AKR1B1 and AKR1B10 in Cancer: Molecular Mechanisms and Signaling Networks. Adv. Exp. Med. Biol. 2021. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, R.; McElhanon, K.; Allen, C.D.; Megyesi, J.K.; Benes, H.; Singh, S.P. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic. Biol. Med. 2015, 86, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsari, A.; Casalini, P.; Bufalino, R.; Berrino, F.; Ménard, S. Role of hormonal risk factors in HER2-positive breast carcinomas. Br. J. Cancer 2003, 88, 1032–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Bidu, C.; Bellenger, S.; Narce, M.; Bellenger, J. n-3 polyunsaturated fatty acids and HER2-positive breast cancer: Interest of the fat-1 transgenic mouse model over conventional dietary supplementation. Biochimie 2014, 96, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Devi, B.C.R.; Tang, T.S.; Rosenberg, P.S.; Anderson, W.F.; Yang, X.R. Divergent breast cancer incidence trends by hormone receptor status in the state of Sarawak, Malaysia. Int. J. Cancer 2020, 147, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Au Yeung, S.L.; Schooling, C.M. Associations of Arachidonic Acid Synthesis with Cardiovascular Risk Factors and Relation to Ischemic Heart Disease and Stroke: A Univariable and Multivariable Mendelian Randomization Study. Nutrients 2021, 13, 1489. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Pikula, S.; Piper, J.T.; Srivastava, S.K.; Torman, R.T.; Bandorowicz-Pikula, J.; Lin, J.T.; Singh, S.V.; Zimniak, P.; et al. ATP-Dependent human erythrocyte glutathione-conjugate transporter. II. Functional reconstitution of transport activity. Biochemistry 1998, 37, 5239–5248. [Google Scholar] [CrossRef]
- Sissung, T.M.; Gardner, E.R.; Piekarz, R.L.; Howden, R.; Chen, X.; Woo, S.; Franke, R.; Clark, J.A.; Miller-DeGraff, L.; Steinberg, S.M.; et al. Impact of ABCB1 allelic variants on QTc interval prolongation. Clin. Cancer Res. 2011, 17, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniowska, B.; Tylutki, Z.; Wyszogrodzka, G.; Polak, S. Drug-drug interactions and QT prolongation as a commonly assessed cardiac effect—Comprehensive overview of clinical trials. BMC Pharmacol. Toxicol. 2016, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Bauer, J.; Pagani, O.; Gerard, B.; Cerny, T.; Sessa, C.; Bastian, G.; Sarkany, M.; Schläpfer, J.; Giroux, B.; et al. Ventricular arrhythmia and torsade de pointe: Dose limiting toxicities of the MDR-modulator S9788 in a phase I trial. Ann. Oncol. 1998, 9, 1233–1242. [Google Scholar] [CrossRef]
- Gaynor, E.R.; Unger, J.M.; Miller, T.P.; Grogan, T.M.; White, L.A., Jr.; Mills, G.M.; Balcerzak, S.P.; Varterasian, M.; LeBlanc, M.; Fisher, R.I. Infusional CHOP chemotherapy (CVAD) with or without chemosensitizers offers no advantage over standard CHOP therapy in the treatment of lymphoma: A Southwest Oncology Group Study. J. Clin. Oncol. 2001, 19, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Warner, E.; Hedley, D.; Andrulis, I.; Myers, R.; Trudeau, M.; Warr, D.; Pritchard, K.I.; Blackstein, M.; Goss, P.E.; Franssen, E.; et al. Phase II study of dexverapamil plus anthracycline in patients with metastatic breast cancer who have progressed on the same anthracycline regimen. Clin. Cancer Res. 1998, 4, 1451–1457. [Google Scholar]
- Martinez-Saez, O.; Prat, A. Current and Future Management of HER2-Positive Metastatic Breast Cancer. JCO Oncol. Pract. 2021, 17, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Lavaud, P.; Andre, F. Strategies to overcome trastuzumab resistance in HER2-overexpressing breast cancers: Focus on new data from clinical trials. BMC Med. 2014, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, C.; Romieu, G.; Salvat, J.; Chaigneau, L.; Merrouche, Y.; N’Guyen, T.; Vuillemin, A.T.; Demarchi, M.; Dobi, E.; Pivot, X. Phase II study assessing lapatinib added to letrozole in patients with progressive disease under aromatase inhibitor in metastatic breast cancer-Study BES 06. Target. Oncol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Oliveira, M.; Isakoff, S.J.; Im, S.A.; Espié, M.; Blau, S.; Tan, A.R.; Saura, C.; Wongchenko, M.J.; Xu, N.; et al. Final results of the double-blind placebo-controlled randomized phase 2 LOTUS trial of first-line ipatasertib plus paclitaxel for inoperable locally advanced/metastatic triple-negative breast cancer. Breast Cancer Ress. Treat. 2021, 189, 377–386. [Google Scholar] [CrossRef]
- Turner, S.; Chia, S.; Kanakamedala, H.; Hsu, W.C.; Park, J.; Chandiwana, D.; Ridolfi, A.; Yu, C.L.; Zarate, J.P.; Rugo, H.S. Effectiveness of Alpelisib + Fulvestrant Compared with Real-World Standard Treatment Among Patients with HR+, HER2-, PIK3CA-Mutated Breast Cancer. Oncologist 2021, 26, e1133–e1142. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity/Level | SK-BR-3 | AU565 | p-Value |
|---|---|---|---|
| Aldose Reductase 1 | 43.2 ± 10.2 | 27.7 ± 6.3 | <0.001 |
| GSTA4 1 | 6.0 ± 0.8 | 22.9 ± 2.0 | <0.0001 |
| GSH 2 | 23.0 ± 0.6 | 20.7 ± 0.5 | <0.05 |
| SK-BR-3 | AU565 | |
|---|---|---|
| Genes for Antioxidant Enzymes | ||
| CAT | 0.96 ± 0.01 n.s. | 0.99 ± 0.03 n.s. |
| SOD1 | 1.42 ± 0.03 *** | 1.18 ± 0.13 n.s. |
| SOD2 | 0.81 ± 0.02 *** | 0.93 ± 0.04 n.s. |
| GPX1 | 2.77 ± 0.09 *** | 1.28 ± 0.33 n.s |
| Genes for Anti-Electrophile Enzymes | ||
| GSTA4 | 0.98 ± 0.07 n.s. | 1.12 ± 0.12 n.s. |
| AKR1C3 | 3.84 ± 0.18 *** | 2.29 ± 0.04 *** |
| AKR7A2 | 0.66 ± 0.01 * | 1.07 ± 0.04 n.s. |
| Genes for Glutathione Synthesis and Loss | ||
| GCLC | 1.92 ± 0.02 *** | 1.32 ± 0.06 * |
| GCLM | 1.83 ± 0.03 *** | 1.60 ± 0.01 *** |
| Transcriptional Regulator of Antioxidant Genes | ||
| NFE2L2 | 1.09 ± 0.10 n.s. | 0.97 ± 0.03 n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bose, C.; Hindle, A.; Lee, J.; Kopel, J.; Tonk, S.; Palade, P.T.; Singhal, S.S.; Awasthi, S.; Singh, S.P. Anticancer Activity of Ω-6 Fatty Acids through Increased 4-HNE in Breast Cancer Cells. Cancers 2021, 13, 6377. https://doi.org/10.3390/cancers13246377
Bose C, Hindle A, Lee J, Kopel J, Tonk S, Palade PT, Singhal SS, Awasthi S, Singh SP. Anticancer Activity of Ω-6 Fatty Acids through Increased 4-HNE in Breast Cancer Cells. Cancers. 2021; 13(24):6377. https://doi.org/10.3390/cancers13246377
Chicago/Turabian StyleBose, Chhanda, Ashly Hindle, Jihyun Lee, Jonathan Kopel, Sahil Tonk, Philip T. Palade, Sharad S. Singhal, Sanjay Awasthi, and Sharda P. Singh. 2021. "Anticancer Activity of Ω-6 Fatty Acids through Increased 4-HNE in Breast Cancer Cells" Cancers 13, no. 24: 6377. https://doi.org/10.3390/cancers13246377