
Progression in Ph-Chromosome-Negative Myeloproliferative Neoplasms: An Overview on Pathologic Issues and Molecular Determinants

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Myelofibrotic Progression
2.1. Pathologic Features
2.2. Predictors of Progression
2.3. Grades of Fibrosis and Prognostic Significance
3. Leukemic Progression
3.1. Pathologic Features
3.2. Predictors of Leukaemic Transformation
3.3. Do Less Than 10% Blasts Bear Prognostic Significance?
4. Atypical/Unusual Types of Progression
4.1. Development of Monocytosis
4.2. Development of Leukocytosis
4.3. Development of Erythrocytosis
5. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Tefferi, A.; Birgegard, G.; Barbui, T.; Gianelli, U.; Barosi, G.; Gisslinger, H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Geneva, Switzerland, 2017; pp. 39–50. [Google Scholar]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Verstovsek, S. Molecular pathways: JAK/STAT pathway: Mutations, inhibitors, and resistance. Clin. Cancer Res. 2013, 19, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Crispino, J.; Stein, B. Myelofibrosis in 2019: Moving beyond JAK2 inhibition. Blood Cancer J. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of CALReticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Rumi, E.; Pietra, D.; Ferretti, V.V.; Klampfl, T.; Harutyunyan, A.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lavu, S.; Mudireddy, M.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Mannarelli, C.; Guglielmelli, P.; et al. JAK2 exon 12 mutated polycythemia vera: Mayo-Careggi MPN Alliance study of 33 consecutive cases and comparison with JAK2 V617F mutated disease. Am. J. Hematol. 2017, 93, E93–E96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 9, 1861–1869. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.M.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Doncombe, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.R.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopulosi, N.; Cantrill, R.; Godfrey, A.J.; Papaemmanuil, E.; Gundemm, G.; MacLean, C.; et al. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Facchetti, F.; Franco, V.; van der Walt, J.; Orazi, A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005, 90, 1128–1132. [Google Scholar] [PubMed]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of CALReticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Rotunno, G.; Fanelli, T.; Pacilli, A.; Brogi, G.; Calabresi, L.; Pancrazzi, A.; Vannucchi, A.M. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015, 5, e360. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information From Karyotype, Platelet Count, and Transfusion Status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Kuykendall, A.T.; Talati, C.; Padron, E.; Sweet, K.; Sallman, D.; List, A.F.; Lancet, J.E.; Komrokji, R.S.; Andrew, K. Genetically inspired prognostic scoring system (GIPSS) outperforms dynamic international prognostic scoring system (DIPSS) in myelofibrosis patients. Am. J. Hematol. 2018, 94, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Guglielmelli, P.; Tefferi, A. Polycythemia vera and essential thrombocythemia: Algorithmic approach. Curr. Opin. Hematol. 2018, 25, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Rumi, E.; Arcaini, L.; Boveri, E.; Elena, C.; Pietra, D.; Boggi, S.; Astori, C.; Bernasconi, P.; Varettoni, M.; et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: A study of 605 patients. Haematologica 2008, 93, 1645–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasserjian, R.P.; Kelley, T.W.; Weinberg, O.K.; A Morgan, E.; Fend, F. Genetic Testing in the Diagnosis and Biology of Myeloid Neoplasms (Excluding Acute Leukemias). Am. J. Clin. Pathol. 2019, 152, 302–321. [Google Scholar] [CrossRef]
- Mina, A.A.; Stein, B. Next-Generation Sequencing in Myeloproliferative Neoplasms: Is This Indicated in All Patients? Curr. Hematol. Malig. Rep. 2019, 14, 137–144. [Google Scholar] [CrossRef]
- Lee, J.-M.; Lee, H.; Eom, K.-S.; Lee, S.-E.; Kim, M.; Kim, Y. Impact of Integrated Genetic Information on Diagnosis and Prognostication for Myeloproliferative Neoplasms in the Next-Generation Sequencing Era. J. Clin. Med. 2021, 10, 1033. [Google Scholar] [CrossRef] [PubMed]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [Green Version]
- Gianelli, U.; Bossi, A.; Cortinovis, I.; Sabattini, E.; Tripodo, C.; Boveri, E.; Moro, A.; Valli, R.; Ponzoni, M.; Florena, M.A.; et al. Reproducibility of the WHO histological criteria for the diagnosis of Philadelphia chromosome-negative myeloproliferative neoplasms. Mod. Pathol. 2014, 27, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Gianelli, U.; Fiori, S.; Cattaneo, D.; Bossi, A.; Cortinovis, I.; Bonometti, A.; Ercoli, G.; Bucelli, C.; Orofino, N.; Bulfamante, G.; et al. Prognostic significance of a comprehensive histological evaluation of reticulin fibrosis, collagen deposition and osteosclerosis in primary myelofibrosis patients. Histopathology 2017, 71, 897–908. [Google Scholar] [CrossRef]
- Kvaniscka, H.M.; Beham-Scmid, C.; Roshanak, B.; Dirhofer, S.; Hussein, K.; Kreipe, H.; Kremer, M.; Shmitt-Graeff, A.; Schwarz, S.; Thiele, J.; et al. Problems and pitfalls in grading of bone marrow fibrosis, collagen deposition and osteosclerosis—A consensus-based study. Histopathology 2016, 68, 905–915. [Google Scholar]
- Boiocchi, L.; Mathew, S.; Gianelli, U.; Iurlo, A.; Radice, T.; Barouk-Fox, S.; Knowles, D.M.; Orazi, A. Morphologic and cytogenetic differences between post-polycythemic myelofibrosis and primary myelofibrosis in fibrotic stage. Mod. Pathol. 2013, 26, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Tiribelli, M.; Barraco, D.; De Marchi, F.; Marin, L.; Medeot, M.; Damiani, D.; Fanin, R. Clinical factors predictive of myelofibrotic evolution in patients with polycythemia vera. Ann. Hematol. 2014, 94, 873–874. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Boveri, E.; Arcaini, L.; Roncoroni, E.; Astori, C.; Merli, M.; Boggi, S.; et al. A prospective study of 338 patients with polycythemia vera: The impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 2010, 24, 1574–1579. [Google Scholar] [CrossRef] [Green Version]
- Benton, C.B.; Tanaka, M.; Wilson, C.; Tanaka, M.; Wilson, C.; Piercce, S.; Zhou, L.; Cortes, J.; Kantarjian, H.; Verstovsek, S. Increased likelihood of post-polycythemia era myelofibrosis in Ph-negative MPN patients with chromosome 12 abnormalities. Leuk. Res. 2015, 39, 419–423. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Randi, M.L.; Bertozzi, I.; Marino, F.; Vannucchi, A.M.; Pieri, L.; et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood 2012, 119, 2239–2241. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Guglielmelli, P.; Bordoni, R.; Casetti, I.; Milanesi, C.; Sant’Antonio, E.; Ferretti, V.; Pancrazzi, A.; Rotunno, G.; et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013, 121, 4388–4395. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Kantarjian, H.M.; Kiladjian, J.-J.; Gotlib, J.; Cervantes, F.; Mesa, R.A.; Sarlis, N.J.; Peng, W.; Sandor, V.; Gopalakrishna, P.; et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica 2015, 100, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Mora, B.; Giorgino, T.; Guglielmelli, P.; Cazzola, M.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. Driver mutations’ effect in secondary myelofibrosis: An international multicenter study based on 781 patients. Leukemia 2016, 31, 970–973. [Google Scholar] [CrossRef]
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Phenotype variability of patients with post polycythemia vera and post essential thrombocythemia myelofibrosis is associated with the time to progression from polycythemia vera and essential thrombocythemia. Leuk. Res. 2018, 69, 100–102. [Google Scholar] [CrossRef]
- Vener, C.; Fracchiolla, N.S.; Gianelli, U.; Calori, R.; Radaelli, F.; Iurlo, A.; Caberlon, S.; Gerli, G.; Boiocchi, L.; Deliliers, G.L. Prognostic implications of the European consensus for grading of bone marrow fibrosis in chronic idiopathic myelofibrosis. Blood 2008, 111, 1862–1865. [Google Scholar] [CrossRef] [Green Version]
- Gianelli, U.; Vener, C.; Bossi, A.; Cortinovis, I.; Iurlo, A.; Fracchiolla, N.S.; Savi, F.; Moro, A.; Grifoni, F.; De Philippis, C.; et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod. Pathol. 2012, 25, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Iurlo, A.; Elli, E.M.; Palandri, F.; Cattaneo, D.; Bossi, A.; Cortinovis, I.; Bucelli, C.; Orofino, N.; Brioschi, F.; Auteri, G. Integrating clinical, morphological and molecular data to assess prognosis in patients with primary myelofibrosis at diagnosis: A practical apporoach. Hematol Oncol. 2019, 37, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Rotunno, G.; Pacilli, A.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pieri, L.; et al. Prognostic impact of bone marrow fibrosis in primary myelofibrosis. A study of the AGIMM group on 490 patients. Am. J. Hematol. 2016, 91, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Pacilli, A.; Rotunno, G.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017, 129, 3227–3236. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Boluda, J.C.; Pereira, A.; Gómez, M.; Boqué, C.; Ferrer-Marin, F.; Raya, J.-M.; García-Gutiérrez, V.; Kerguelen, A.; Xicoy, B.; Barba, P.; et al. The International Prognostic Scoring System does not accurately discriminate different risk categories in patients with post-essential thrombocythemia and post-polycythemia vera myelofibrosis. Haematologica 2014, 99, e55–e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Saeed, L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Application of current prognostic models for primary myelofibrosis in the setting of post-polycythemia vera or post-essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2851–2852. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef]
- Hernández-Boluda, J.C.; Pereira, A.; Correa, J.G.; Alvarez-Larrain, A.; Ferrrer-Marin, F.; Raya, J.M.; Martinez-Lopez, J.; Encinas, P.M.; Estrada, N.; Velez, P.; et al. Performance of the myelofibrosis secondary to PV and ET-prognostic model (MYSEC-PM) in a series of 262 patients from the Spanish registry of myelofibrosis. Leukemia 2017, 32, 553–555. [Google Scholar] [CrossRef]
- Masarova, L.; Bose, P.; Pemmaraju, N.; Daver, N.; Cortes, J.E.; Estrov, Z.; Kantarjian, H.M.; Verstovsek, S. Validation of the myelofibrosis secondary to PV and ET-prognostic model in patients with post-polycythemia vera and post-essential thrombocythemia myelofibrosis. MD Anderson Cancer Center. Blood 2017, 130. [Google Scholar] [CrossRef]
- Palandri, F.; Palumbo, G.A.; Iurlo, A.; Polverelli, N.; Benevol, O.G.; Breccia, M.; Abruzzese, E.; Tiribelli, M.; Bonifacio, M.; Tieghi, A.; et al. Differences in presenting features, outcome and prognostic models in patients with primary myelofibrosis and postpolycythemia vera and/or post-essential thrombocythemia myelofibrosis treated with ruxolitinib. New perspective of the MYSEC-PM in a large multicenter study. Semin. Hematol. 2018, 55, 248–255. [Google Scholar] [PubMed]
- Pozdnyakova, O.; Hasserjian, R.P.; Verstovsek, S.; Orazi, A. Impact of bone marrow pathology on the clinical management of Philadelphiachromosome-negative myeloproliferative neoplasms. Clin. Lymphoma. Myeloma. Leuk. 2015, 15, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Masarova, L.; Bose, P.; Daver, N.; Pemmaraju, N.; Newberry, K.J.; Manshouri, T.; Cortes, J.; Kantarjian, H.M.; Verstovsek, S. Patients with post-essential thrombocythemia and post-polycythemia vera differ from patients with primary myelofibrosis. Leuk. Res. 2017, 59, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Verstovsek, S. The evolving understanding of prognosis in post-essential thrombocythemia myelofibrosis and post-polycythemia vera myelofibrosis vs. primary myelofibrosis. Clin. Adv. Hematol Oncol. 2019, 17, 299–307. [Google Scholar]
- Masarova, L.; Bose, P.; Pemmaraju, N.; Newberry, K.J.; Bueso-Ramos, C.E.; Cortes, J.E.; Kantarjian, H.M.; Verstovsek, S. Clinical associations of cytogenetic abnormalities in patients with primary and post-essential thrombocythemia and post-polycythemia vera myelofibrosis. Blood 2016, 128, 4265. [Google Scholar] [CrossRef]
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.-J.; et al. Value of cytogenetic abnormalities in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study of the MYSEC project. Haematologica 2018, 103, e392–e394. [Google Scholar] [CrossRef]
- Rumi, E.; Harutyunyan, A.; Elena, C.; Pietra, D.; Klampfl, T.; Bagienski, K.; Berg, T.; Casetti, I.; Pascutto, C.; Passamonti, F.; et al. Identification of genomic aberrations associated with disease transformation by means of high-resolution SNP array analysis in patients with myeloproliferative neoplasm. Am. J. Hematol. 2011, 86, 974–979. [Google Scholar] [CrossRef]
- Rotunno, G.; Pacilli, A.; Artusi, V.; Rumi, E.; Maffioli, M.; Delaini, F.; Brogi, G.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: A study on 359 patients of the AGIMM group. Am. J. Hematol. 2016, 91, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Lancman, G.; Brunner, A.; Hoffman, R.; Mascarenhas, J.; Hobbs, G. Outcomes and predictors of survival in blast phase myeloproliferative neoplasms. Leuk. Res. 2018, 70, 49–55. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Czader, M.; Orazi, A. Acute Myeloid Leukemia and Other Types of Disease Progression in Myeloproliferative Neoplasms. Am. J. Clin. Pathol. 2015, 144, 188–206. [Google Scholar] [CrossRef] [Green Version]
- Coltro, G.; Mannelli, F.; Vergoni, F.; Santi, R.; Massi, D.; Siliani, L.M.; Marzullo, A.; Bonifacio, S.; Pelo, E.; Pacilli, A.; et al. Extramedullary blastic transformation of primary myelofibrosis in the form of disseminated myeloid sarcoma: A case report and review of the literature. Clin. Exp. Med. 2020, 20, 313–320. [Google Scholar] [CrossRef]
- Hirose, Y.; Masaki, Y.; Shimoyama, K.; Sugai, S.; Nojima, T. Granulocytic sarcoma of megacariyoblastic differentiation in the lymph nodes terminating as acute megacaryoblastic leukemia in a case of chronic idiopathic myelofibrosis persisting for 16 years. Eur. J. Haematol. 2001, 67, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Li, S.; Meloni, G.; Schnittger, S.; Gattenlohner, S.; Liso, A.; Di Ianni, M.; Martelli, M.P.; Pescarmona, E.; Foa, R.; et al. NPM1 mutated acute myeloid leukaemia occurring in JAK2-V617F primary myelofibrosis: De-novo origin? Leukemia 2008, 22, 1459–1463. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Chi, H.-S.; Shim, H.; Jang, S.; Park, C.-J. Two novelNPM1mutations in an acute myeloid leukemia patient transformed from primary myelofibrosis. Int. J. Lab. Hematol. 2012, 35, e1–e3. [Google Scholar] [CrossRef]
- Hidalgo López, J.E.; Carballo-Zarate, A.; Verstovsek, S.; Wang, S.A.; Hu, S.; Li, S.; Xu, J.; Zuo, W.; Tang, Z.; Yin, C.C.; et al. Bone marrow findings in blast phase of polcytemia vera. Ann. Hematol. 2018, 97, 425–434. [Google Scholar] [CrossRef]
- Dunbar, A.J.; Rampal, R.K.; Levine, R.L. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70. [Google Scholar] [CrossRef]
- Barosi, G.; Bergamaschi, G.; Marchetti, M.; Vannucchi, A.M.; Guglielmelli, P.; Elisabetta Antonioli, E.; Massa, M.; Rosti, V.; Campanelli, R.; Villani, L.; et al. JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood 2007, 110, 4030–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Coco, F.L.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009, 114, 1477–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Baxter, E.J.; Beer, P.A.; Scott, L.M.; Bench, A.J.; Huntly, B.J.P.; Erber, W.N.; Kusec, R.; Larsen, T.S.; Giraudier, S.; et al. Mutation of JAK2 in the myeloproliferative disorders: Timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood 2006, 108, 3548–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharides, A.; Boissinot, M.; Girodon, F.; Garand, R.; Teo, S.S.; Lippert, P.; Talmabnt, P.; Tuichelli, A.; Hwermoyet, S.; Skida, C. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 2007, 110, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Aynardi, J.; Manur, R.; Hess, P.R.; Chekol, S.; Morrissette, J.J.D.; Babushok, D.; Hexner, E.; Rogers, H.J.; Hsi, E.D.; Margolskee, E.; et al. JAK2 V617F-positive acute myeloid leukaemia (AML): A comparison between de novo AML and secondary AML transformed from an underlying myeloproliferative neoplasm. A study from the Bone Marrow Pathology Group. Br. J. Haematol. 2018, 182, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Benton, C.B.; Boddu, P.C.; DiNardo, C.D.; Bose, P.; Wang, F.; Assi, R.; Pemmaraju, N.; Kc, D.; Pierce, S.; Patel, K.; et al. Janus kinase 2 variants associated with the transformation of muyeloproliferative neoplasm into acute myeloid leukemia. Cancer 2019, 125, 1855–1866. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.-P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.; et al. Differential association of CALReticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2014, 29, 249–252. [Google Scholar] [CrossRef]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbuonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Cottin, L.; Riou, J.; Orvain, C.; Ianotto, J.C.; Boyer, F.; Renard, M.; Truchan-Graczyk, M.; Murati, A.; Jouanneau-Courville, R.; Allangba, O.; et al. Sequential mutational evaluation of CALR -mutated myeloproliferative neoplasms with thrombocytosis reveals an association between CALR allele burden evolution and disease progression. Br. J. Haematol. 2019, 188, 935–944. [Google Scholar] [CrossRef]
- Parenti, S.; Rontauroli, S.; Carretta, C.; Mallia, S.; Genovese, E.; Chiereghin, C.; Peano, C.; Tavernari, L.; Bianchi, E.; Fantini, S.; et al. Mutated clones driving leukemic transformation are alredy detectable at the single -cell leve CD34-positive cells in the chronic phase of primary myelofibrosis. NPJ Precis. Oncol. 2021, 5, 1–11. [Google Scholar]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [Green Version]
- Shahin, O.A.; Chifotides, H.T.; Bose, P.; Masarova, L.; Verstovsek, S. Accelerated Phase of Myeloproliferative Neoplasms. Acta Haematol. 2021, 21, 1–16. [Google Scholar]
- Boiocchi, L.; Espinal-Witter, R.; Geyer, J.T.; Steinhilber, J.; Bonzheim, I.; Knowles, D.M.; Fend, F.; Orazi, A. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod. Pathol. 2013, 26, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Shah, S.; Mudireddy, M.; Lasho, T.L.; Barraco, D.; Hanson, C.A.; Ketterling, R.P.; Elliott, M.A.; Patnaik, M.S.; Pardanani, A.; et al. Monocytosis is a powerful and independent predictor of inferior survival in primary myelofibrosis. Br. J. Haematol. 2018, 183, 835–838. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, J.; Pasca, S.; Teodorescu, P.; Selicean, C.; Rus, I.; Zdrenghea, M.; Bojan, A.; Trifa, A.; Fetica, B.; Petrushev, B.; et al. Persistent Basophilia May Suggest an “Accelerated Phase” in the Evolution of CALR-Positive Primary Myelofibrosis Toward Acute Myeloid Leukemia. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- Vallapureddy, R.R.; Mudireddy, M.; Penna, D.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Begna, K.H.; Gangat, N.; Pardanani, A.; et al. Leukemic transformation among 1306 patients with primary myelofibrosis: Risk factors and development of a predictive model. Blood Cancer J. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef]
- Bartels, S.; Vogtmann, J.; Schipper, E.; Büsche, G.; Schlue, J.; Lehmann, U.; Kreipe, H. Combination of myeloproliferative neoplasm driver gene activation with mutations of splice factor or epigenetic modifier genes increases risk of rapid blastic progression. Eur. J. Haematol. 2021, 106, 520–528. [Google Scholar] [CrossRef]
- McKenney, A.S.; Lau, A.N.; Somasundara, A.V.H.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant–driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef]
- Geyer, J.T.; Margolskee, E.; Krichevsky, S.A.; Cattaneo, D.; Boiocchi, L.; Ronchi, P.; Lunghi, F.; Scandura, J.M.; Ponzoni, M.; Hasserjian, R.P.; et al. Disease progression in myeloproliferative neoplasms: Comparing patients in accelerated phase with those in chronic phase with increased blasts (<10%) or with other types of disease progression. Haematologica 2020, 105, e221–e224. [Google Scholar] [PubMed]
- Masarova, L.; Bose, P.; Pemmaraju, N.; Daver, N.G.; Zhou, L.; Pierce, S.; Sasaki, K.; Kantarjian, H.M.; Estrov, Z.; Verstovsek, S. Prognostic value of blasts in peripheral blood in myelofibrosis in the ruxolitinib era. Cancer 2020, 126, 4322–4331. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Bose, P.; Pemmaraju, N.; Daver, N.; Zhou, L.; Pierce, S.; Kantarjian, H.; Estrov, Z.; Verstovsek, S. Clinical Significance of Bone Marrow Blast Percentage in Patients With Myelofibrosis and the Effect of Ruxolitinib Therapy. Clin. Lymphoma Myeloma Leuk. 2021, 21, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, C.-Y.; Mesa, R.A.; Wu, W.; Hanson, C.A.; Pardanani, A.; Tefferi, A. Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer 2008, 112, 2726–2732. [Google Scholar] [CrossRef]
- Björkholm, M.; Derolf, R.; Hultcrantz, M.; Kristinsson, S.Y.; Ekstrand, C.; Goldin, L.R.; Andreasson, B.; Birgegård, G.; Linder, O.; Malm, C.; et al. Treatment-Related Risk Factors for Transformation to Acute Myeloid Leukemia and Myelodysplastic Syndromes in Myeloproliferative Neoplasms. J. Clin. Oncol. 2011, 29, 2410–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Ramos, C.E.B.; Medeiros, L.J.; Zhao, C.; Yin, C.C.; Li, S.; Hu, S.; Wang, W.; Thakral, B.; Xu, J.; et al. Utility of JAK2 V617F allelic borden in distinguishing myelomonocytic leukemia from primary myelofibrosis with monocytosis. Hum. Pathol. 2019, 85, 290–298. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Timm, M.M.; Vallapureddy, R.; Lasho, T.L.; Ketterling, R.P.; Gangat, N.; Shi, M.; Tefferi, A.; Solary, E.; Reichard, K.K.; et al. Flow cytometry based monocyte subset analysis accurately distinguishes chronic myelomonocytic leukemia from myeloproliferative neoplasms with associated monocytosis. Blood Cancer J. 2017, 7, e584. [Google Scholar] [CrossRef]
- Beran, M.; Shen, Y.; Onida, F.; Wen, S.; Kantarjian, H.; Estey, E. Prognostic significance of monocytosis in patients with myeloproliferative disorders. Leuk. Lymphoma 2006, 47, 417–423. [Google Scholar] [CrossRef]
- Chapman, J.; Geyer, J.T.; Khanlari, M.; Moul, A.; Casas, C.; Connor, S.T.; Fan, Y.-S.; Watts, J.M.; Swords, R.T.; Vega, F.; et al. Myeloid neoplasms with features intermediate between primary myelofibrosis and chronic myelomonocytic leukemia. Mod. Pathol. 2017, 31, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Barraco, D.; Cerquozzi, S.; Gangat, N.; Patnaik, M.M.; Lasho, T.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Monocytosis in polycythemia vera: Clinical and molecular correlates. Am. J. Hematol. 2017, 92, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, M.; Verstovsek, S.; Dingli, D.; Schwager, S.; Mesa, R.; Li, C.; Tefferi, A. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk. Res. 2007, 31, 1503–1509. [Google Scholar] [CrossRef]
- Gangat, N.; Wolanskyj, A.P.; Schwager, S.M.; Hanson, C.A.; Tefferi, A. Leukocytosis at diagnosis and the risk of subsequent thrombosis in patients with low-risk essential thrombocythemia and polycythemia vera. Cancer 2009, 115, 5740–5745. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Strand, J.; Li, C.Y.; Wu, W.; Pardanani, A.; Tefferi, A. Leucocytosis in polycythaemia vera predicts both inferior survbival and leukaemic transformation. Br. J. Haemathol. 2007, 138, 354–358. [Google Scholar] [CrossRef]
- Boiocchi, L.; Gianelli, U.; Iurlo, A.; Fend, F.; Bonzheim, I.; Cattaneo, D.; Knowles, D.M.; Orazi, A. Neutrophilic leukocytosis in advanced stage polycythemia vera: Hematopathologic features and prognostic implications. Mod. Pathol. 2015, 28, 1448–1457. [Google Scholar] [CrossRef]
- Rotunno, G.; Mannarelli, C.; Guglielmelli, P.; Pacilli, A.; Pancrazzi, A.; Pieri, L.; Fanelli, T.; Bosi, A.; Vannucchi, A.M. Impact of CALReticulin mutations on clinical and hematological phenotype and outcome in esseential thrombocythemia. Blood 2014, 6, 123–1552. [Google Scholar]
- Barbui, T.; Thiele, J.; Carobbio, A.; Guglielmelli, P.; Rambaldi, A.; Vannucchi, A.M.; Tefferi, A. Discriminating between essential thrombocythemia and masked polycythemia vera inJAK2mutated patients. Am. J. Hematol. 2014, 89, 588–590. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Carobbio, A.; Vannucchi, A.M.; Tefferi, A. The rate of transformation from JAK2-mutated ET to PV is influenced by an accurate WHO-defined clinico-morphological diagnosis. Leukemia 2015, 29, 992–993. [Google Scholar] [CrossRef] [PubMed]
- Sobieralski, P.; Leszczyńska, A.; Bieniaszewska, M. Late polycytemic transformation in JAK2 mutated essential Thrombocythemia patients-characteristic along with a validation of 2016 WHO criteria. Eur. J. Haematol. 2019, 103, 558–563. [Google Scholar] [CrossRef]


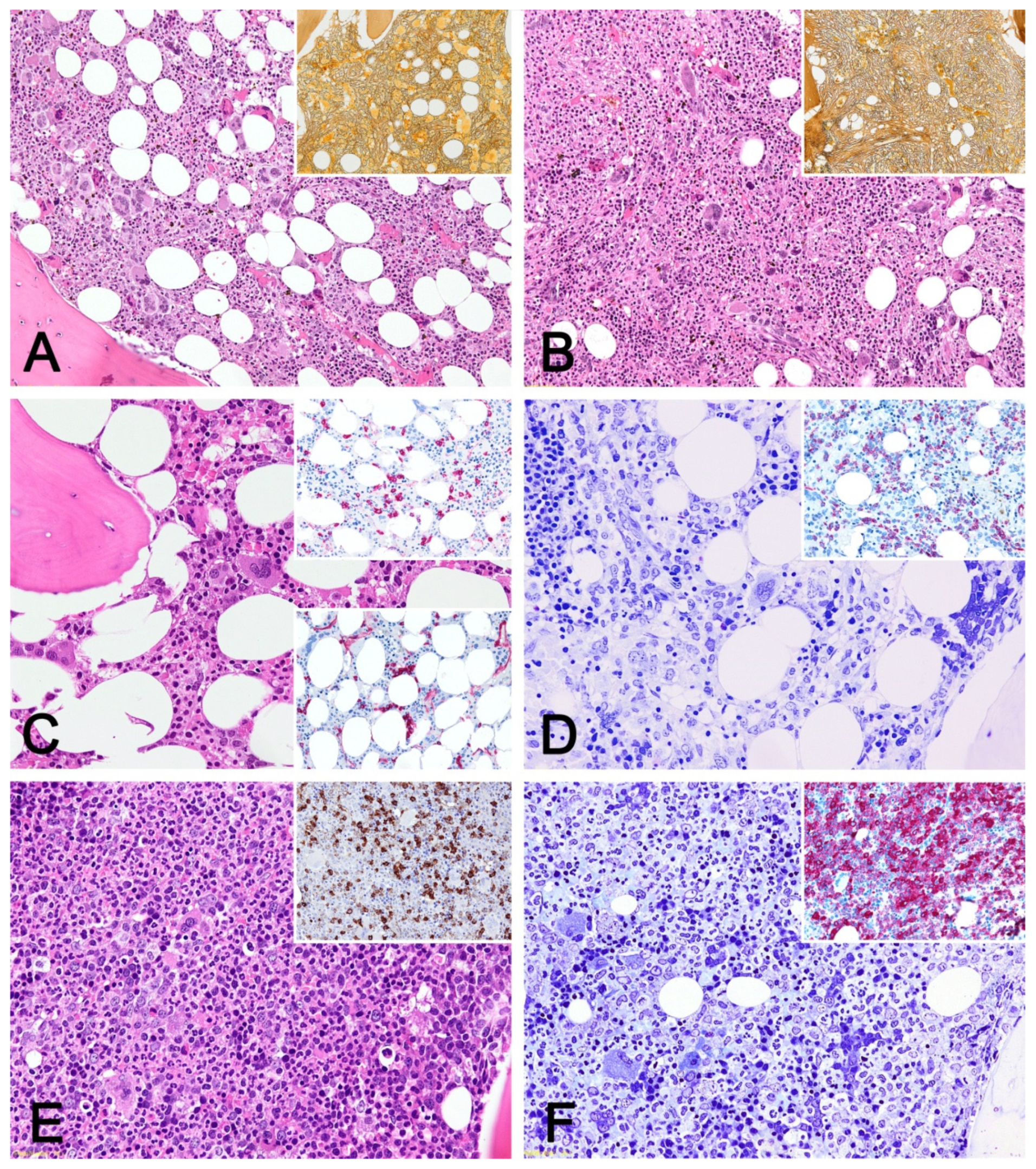{kind=link}
| (A) Diagnostic Criteria for Post-Essential Thrombocythemia Myelofibrosis | |
|---|---|
| Required criteria | Documentation of previous diagnosis of WHO-defined ET |
| Bone marrow fibrosis of grade 2–3 on 0–3 scale or 3–4 on a 0–4 scale | |
| Additional criteria (2 are required) | Anemia (i.e., below the reference range given age, sex, and altitude considerations) and a >2 g/dL decrease from baseline hemoglobin concentration |
| Leukoerythroblastosis | |
| Increasing splenomegaly, defined as either an increase in palpable splenomegaly of >5 cm from baseline (distance from the left costal margin) or the development of a newly palpable splenomegaly | |
| Elevated lactate dehydrogenase level (above the reference range) | |
| Development of any two (or all three) of the following constitutional symptoms: >10% weight loss in 6 months, night sweats, unexplained fever (>37.5 °C) | |
| (B) Diagnostic Criteria for Post-Polycythemia Vera Myelofibrosis | |
| Required criteria | Documentation of previous diagnosis of WHO-defined ET |
| Bone marrow fibrosis of grade 2–3 on 0–3 scale or 3–4 on a 0–4 scale | |
| Additional criteria (2 are required) | Anemia (i.e., below the reference range given age, sex, and altitude considerations) or sustained loss of requirement of either phlebotomy (in the absence of cytoreductive therapy) or cytoreductive treatment for erythrocytosis |
| Leukoerythroblastosis | |
| Increasing splenomegaly, defined as either an increase in palpable splenomegaly of >5 cm from baseline (distance from the left costal margin) or the development of a newly palpable splenomegaly | |
| Development of any two (or all three) of the following constitutional symptoms: >10% weight loss in 6 months, night sweats, unexplained fever (>37.5 °C) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabattini, E.; Pizzi, M.; Agostinelli, C.; Bertuzzi, C.; Sagramoso Sacchetti, C.A.; Palandri, F.; Gianelli, U. Progression in Ph-Chromosome-Negative Myeloproliferative Neoplasms: An Overview on Pathologic Issues and Molecular Determinants. Cancers 2021, 13, 5531. https://doi.org/10.3390/cancers13215531
Sabattini E, Pizzi M, Agostinelli C, Bertuzzi C, Sagramoso Sacchetti CA, Palandri F, Gianelli U. Progression in Ph-Chromosome-Negative Myeloproliferative Neoplasms: An Overview on Pathologic Issues and Molecular Determinants. Cancers. 2021; 13(21):5531. https://doi.org/10.3390/cancers13215531
Chicago/Turabian StyleSabattini, Elena, Marco Pizzi, Claudio Agostinelli, Clara Bertuzzi, Carlo Alberto Sagramoso Sacchetti, Francesca Palandri, and Umberto Gianelli. 2021. "Progression in Ph-Chromosome-Negative Myeloproliferative Neoplasms: An Overview on Pathologic Issues and Molecular Determinants" Cancers 13, no. 21: 5531. https://doi.org/10.3390/cancers13215531