PMEPA1/TMEPAI Is a Unique Tumorigenic Activator of AKT Promoting Proteasomal Degradation of PHLPP1 in Triple-Negative Breast Cancer Cells

 , ,
, ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Analysis of RNA Sequencing Data in TCGA-Invasive Breast Cancer (BRCA) Cases
2.2. Cell Lines and Cell Culture
2.3. Plasmid Construction
2.4. Knockdown of Target Genes
2.5. Colony Formation Assay
2.6. Cell Proliferation Assay
2.7. Tumor Sphere Formation Assay
2.8. Transwell Cell Migration Assay
2.9. Wound Healing Assay
2.10. TMEPAI Re-Expression
2.11. Western Blot Analysis
2.12. Reverse Transcription-qPCR
2.13. Co-Immunoprecipitation (Co-IP)
2.14. Xenograft Mouse Model for Tumorigenicity Studies
2.15. Immunohistochemical Staining
2.16. Statistical Analysis
3. Results
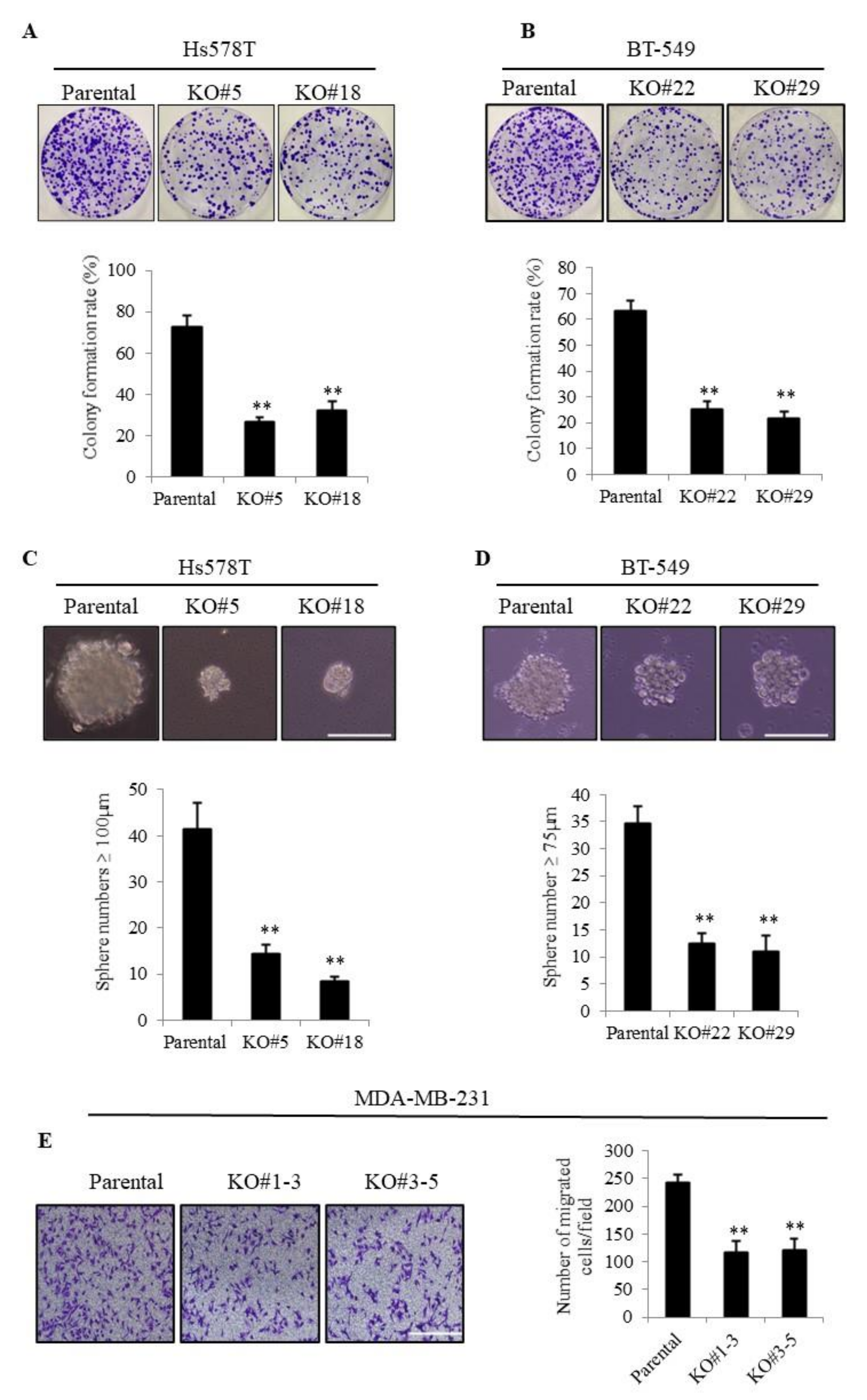
3.1. TMEPAI Promotes Colony Formation, Tumor Sphere Formation, and Cell Migration in TNBC Cells
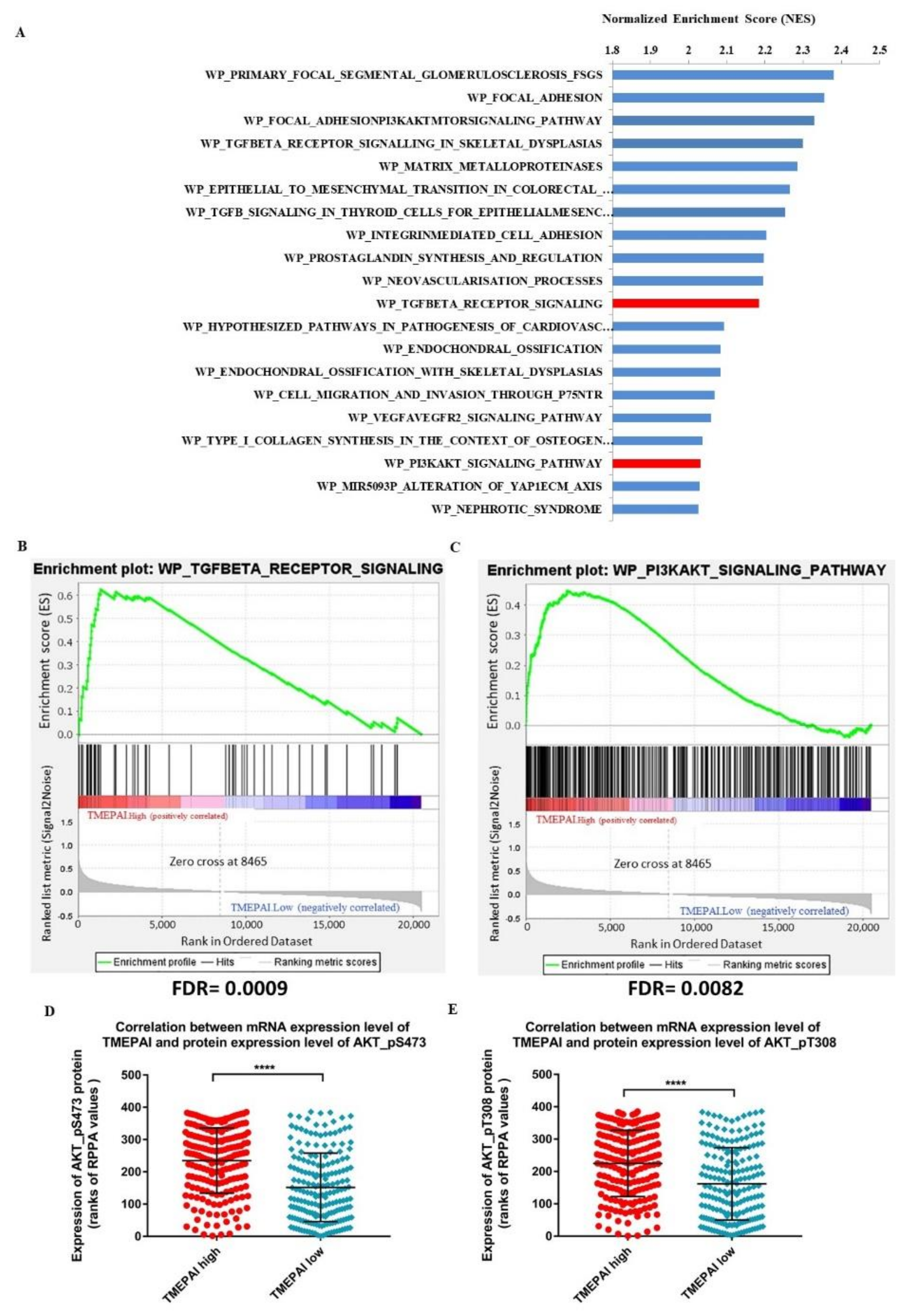
3.2. Correlation of High Expression of TMEPAI and High Levels of AKT Ser473 Phosphorylation among TCGA Invasive Breast Cancer Cases (BRCA)
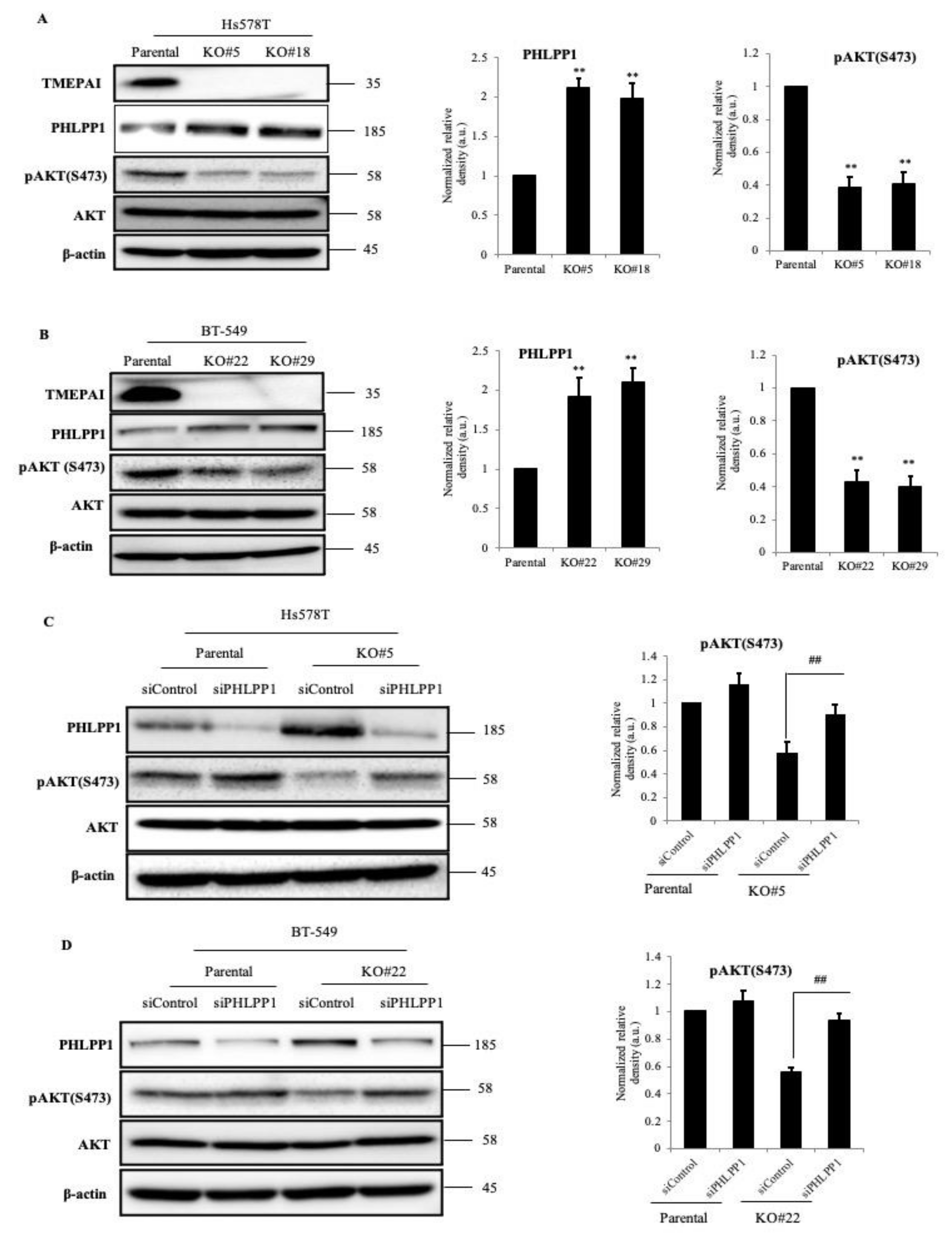
3.3. TMEPAI Promotes PI3K/AKT Signaling through Phosphorylation of Ser473 of AKT by Suppressing PHLPP1
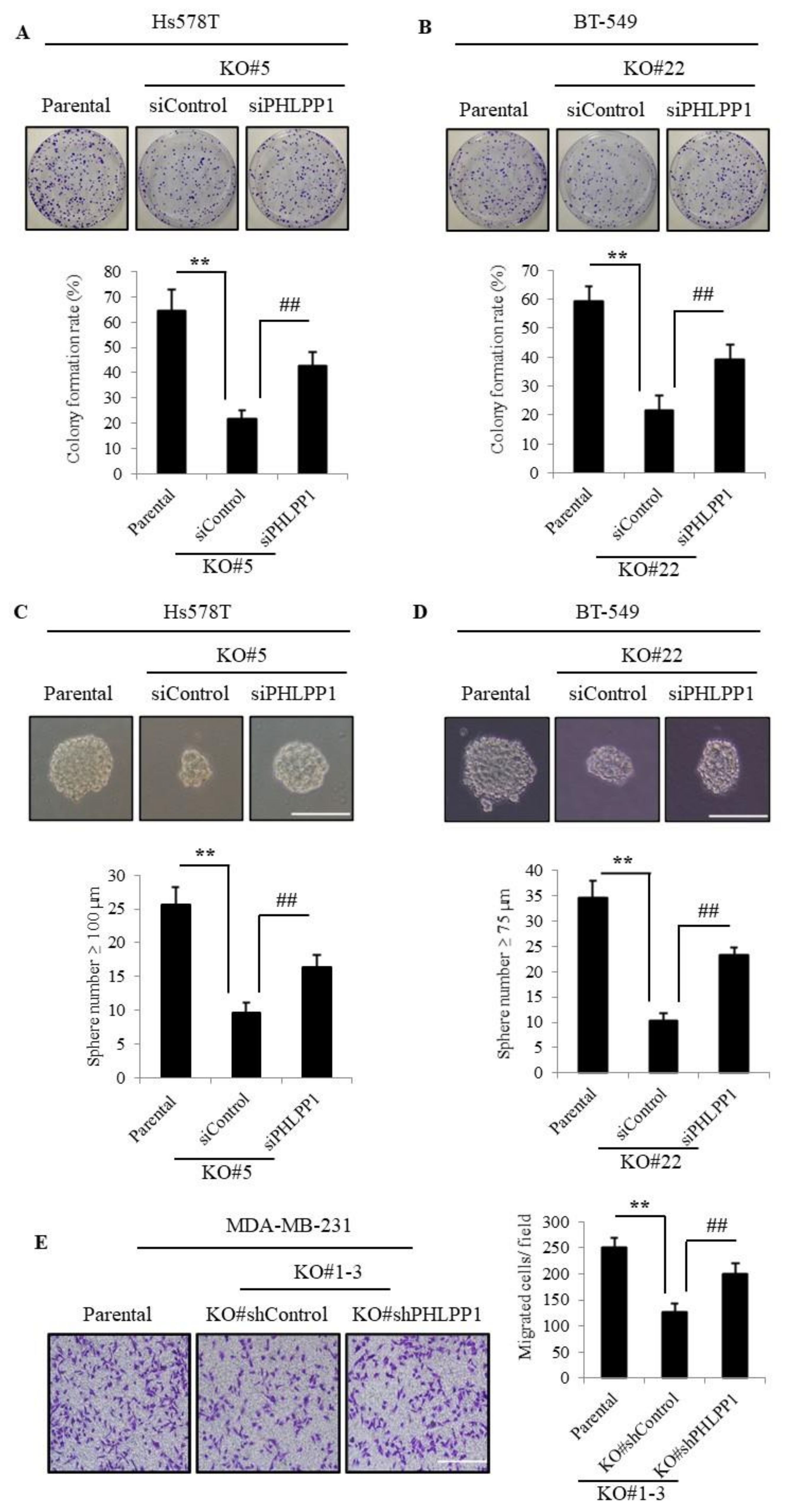
3.4. Knockdown of PHLPP1 Rescues Colony Formation, Tumor Sphere Formation, and Cell Migration in TMEPAI-KO Cells
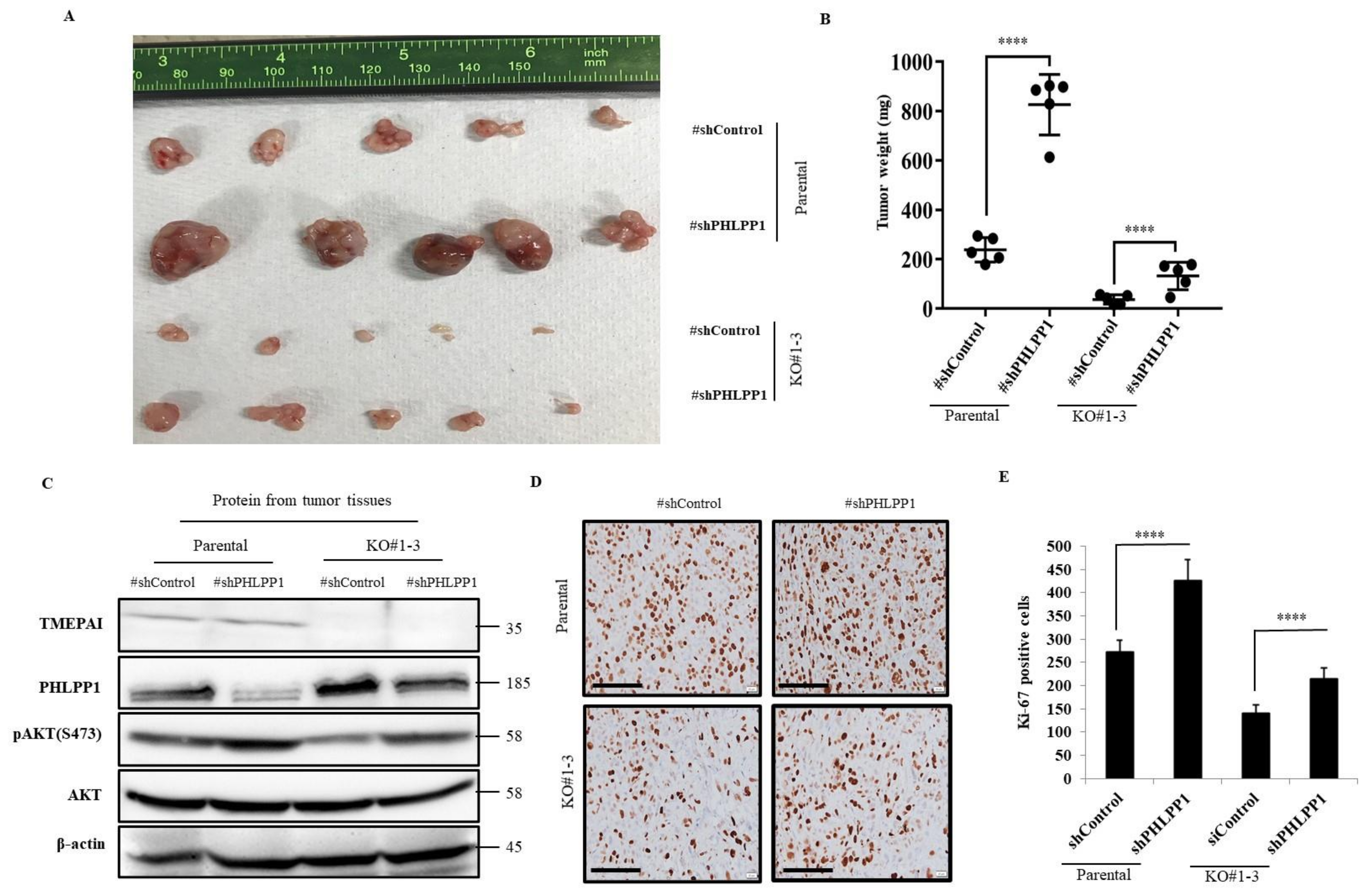
3.5. Knockdown of PHLPP1 Promotes In Vivo Tumor Formation in a Mouse Xenograft Model
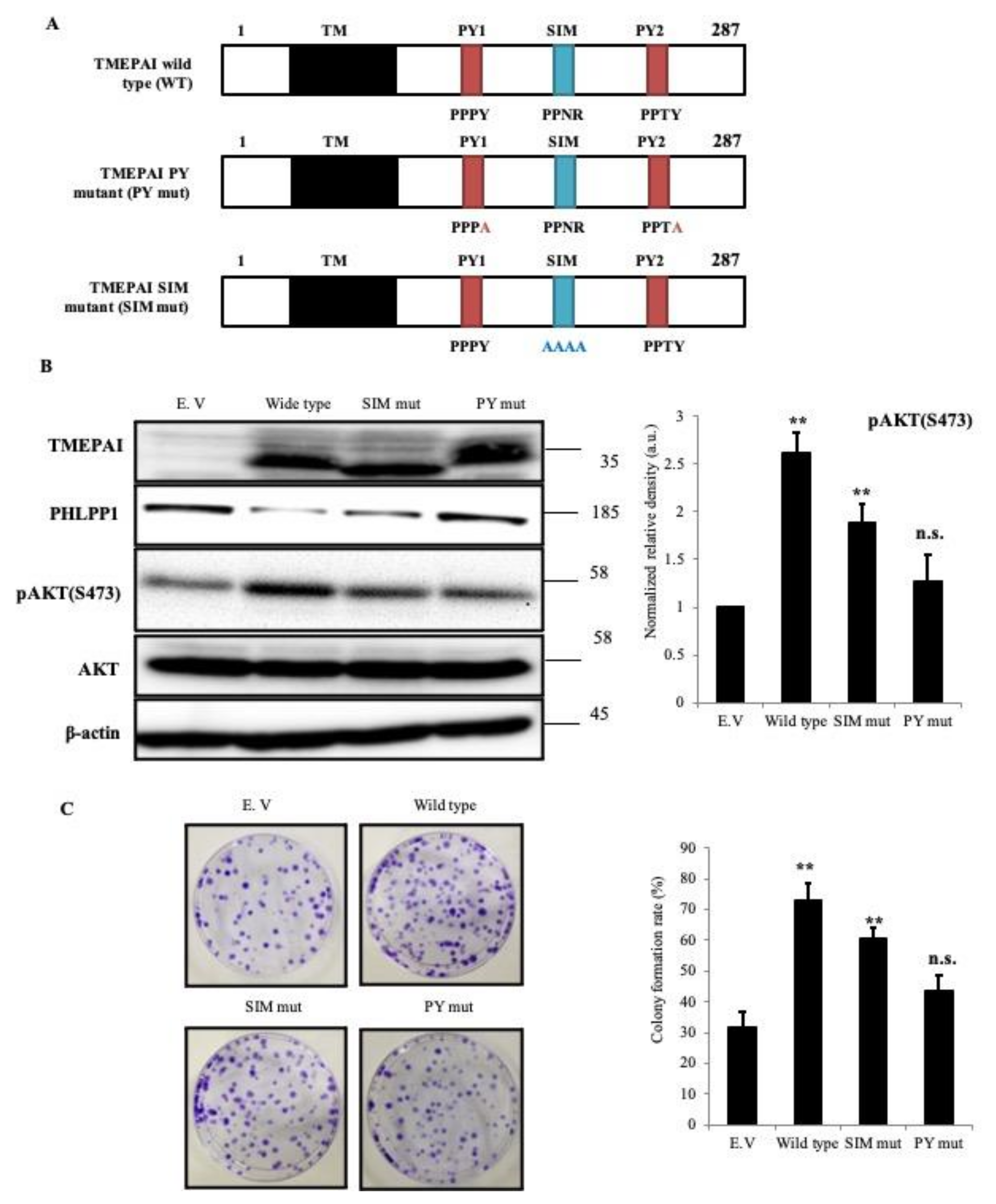
3.6. PY Motifs of TMEPAI Mediate Downregulation of PHLPP1 to Facilitate AKT Ser473 Phosphorylation
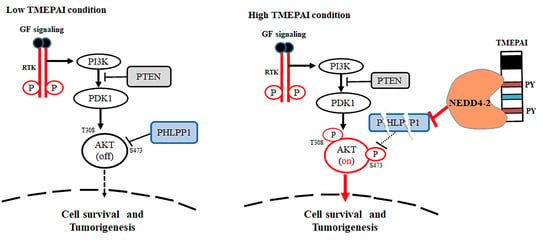
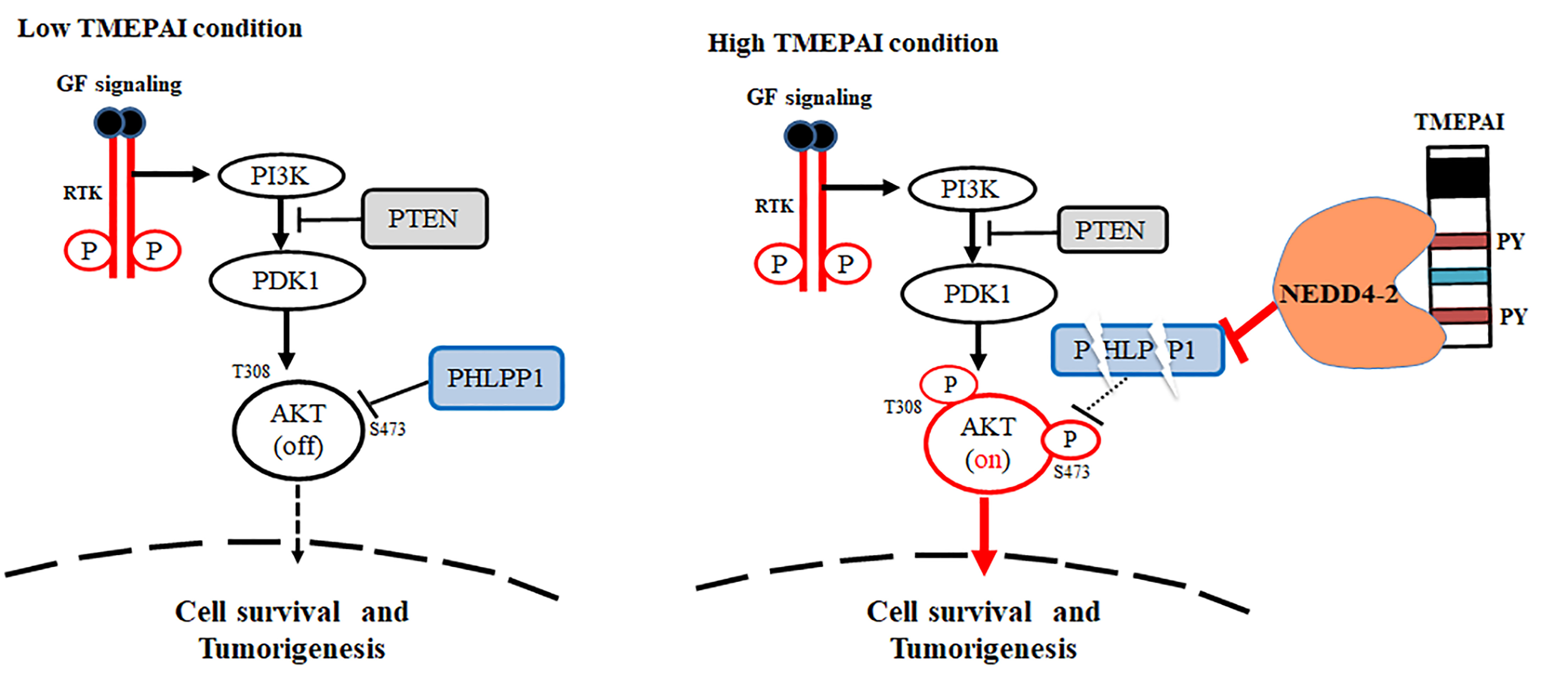
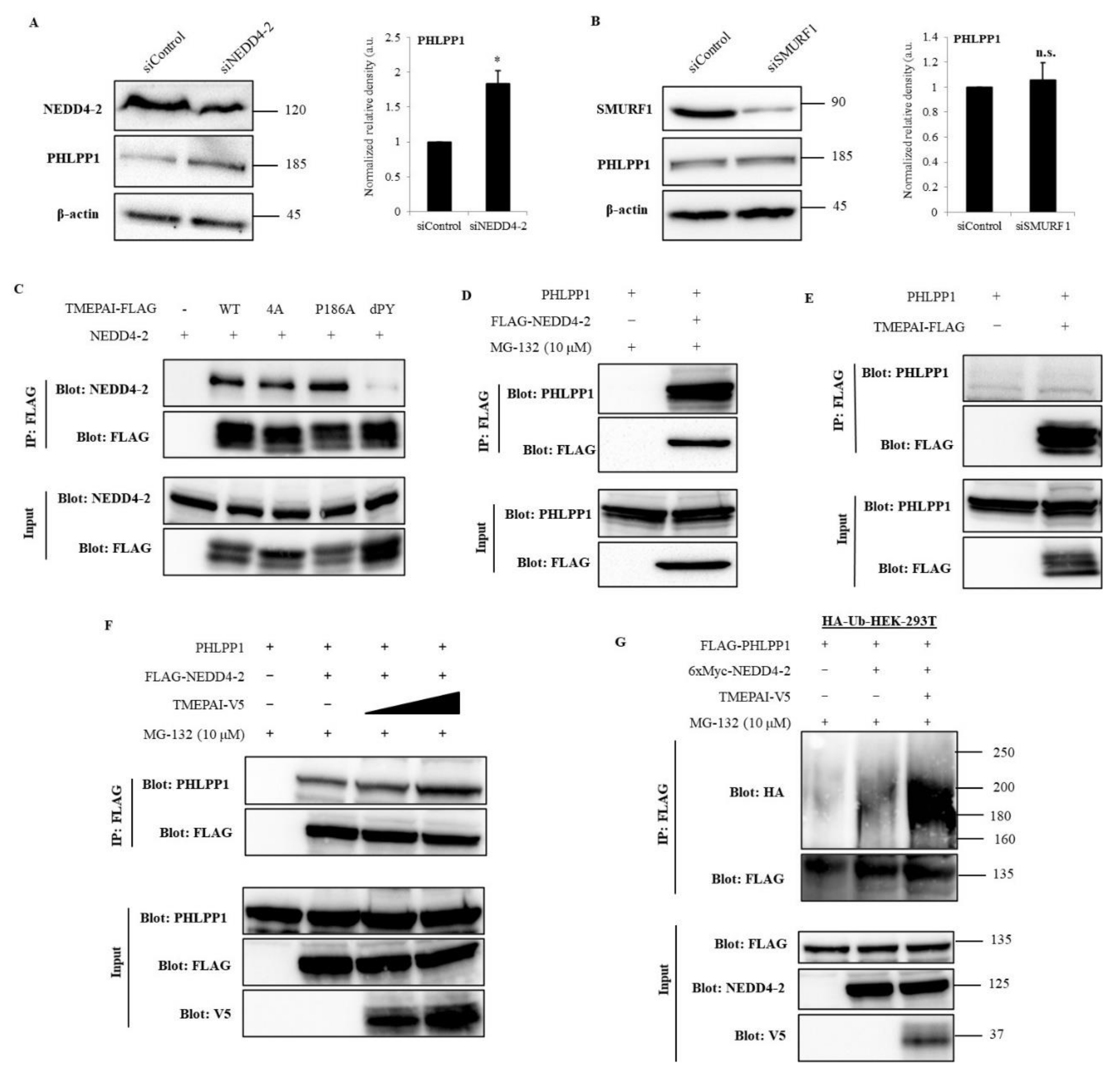
3.7. NEDD4-2 Mediates TMEPAI-Induced Proteasomal Degradation of PHLPP1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortman, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Wu, L.; Yang, X. Targeting the Hippo Pathway for Breast Cancer Therapy. Cancers 2018, 10, 422. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Huang, L.; Gest, C.; Xi, X.; Janin, A.; Soria, C.; Li, H.; Lu, H. Opposite regulation by PI3K/Akt and MAPK/ERK pathways of tissue factor expression, cell-associated procoagulant activity and invasiveness in MDA-MB-231 cells. J. Hematol. Oncol. 2012, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Fukushima, H.; Inuzuka, H.; Wan, L.; Liu, P.; Gao, D.; Sarkar, F.H.; Wei, W. Skp2 is a promising therapeutic target in breast cancer. Front. Oncol. 2012, 1, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002. [Google Scholar] [CrossRef] [Green Version]
- Majerus, P.W.; Kisseleva, M.V.; Norris, F.A. The role of phosphatases in inositol signaling reactions. J. Biol. Chem. 1999, 274, 10669–10672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Itoh, T.; Takenawa, T. Phosphatidylinositol 4-phosphate 5-kinase type I is regulated through phosphorylation response by extracellular stimuli. J. Biol. Chem. 2001, 276, 4781–4787. [Google Scholar] [CrossRef] [Green Version]
- Bozulic, L.; Hemmings, B.A. PIKKing on PKB: Regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol. 2009, 21, 256–261. [Google Scholar] [CrossRef]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, A.K.; Niederst, M.J.; Newton, A.C. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 2013, 280, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Singha, P.K.; Pandeswara, S.; Geng, H.; Lan, R.; Venkatachalam, M.A.; Saikumar, P. TGF-β induced TMEPAI/PMEPA1 inhibits canonical Smad signaling through R-Smad sequestration and promotes non-canonical PI3K/Akt signaling by reducing PTEN in triple negative breast cancer. Genes Cancer 2014, 5, 320–336. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Newton, A.C. Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): A new player in cell signaling. J. Biol. Chem. 2012, 287, 3610–3616. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, C.; Edderkaoui, M.; Moore, R.M.; Eibl, G.; Kasahara, N.; Treger, J.; Grippo, P.J.; Mayerle, J.; Lerch, M.M.; Gukovskaya, A.S. The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology 2012, 142, 377–387.e875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Niederst, M.; Fecteau, J.F.; Nguyen, V.M.; Kipps, T.J.; Messmer, D.; Newton, A.C.; Handel, T.M. Mechanisms and consequences of the loss of PHLPP1 phosphatase in chronic lymphocytic leukemia (CLL). Leukemia 2012, 26, 1689–1692. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.L.; Wang, Z.C.; De Nicolo, A.; Lu, X.; Brown, M.; Miron, A.; Liao, X.; Iglehart, J.D.; Livingston, D.M.; Ganesan, S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006, 9, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.L.; Shi, Y.; Petrovics, G.; Sun, C.; Makarem, M.; Zhang, W.; Sesterhenn, I.A.; McLeod, D.G.; Sun, L.; Moul, J.W.; et al. PMEPA1, an androgen-regulated NEDD4-binding protein, exhibits cell growth inhibitory function and decreased expression during prostate cancer progression. Cancer Res. 2003, 63, 4299–4304. [Google Scholar] [PubMed]
- Watanabe, Y.; Itoh, S.; Goto, T.; Ohnishi, E.; Inamitsu, M.; Itoh, F.; Satoh, K.; Wiercinska, E.; Yang, W.; Shi, L.; et al. TMEPAI, a transmembrane TGF-β-inducible protein, sequesters Smad proteins from active participation in TGF-β signaling. Mol. Cell 2010, 37, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Giannini, G.; Ambrosini, M.I.; Di Marcotullio, L.; Cerignoli, F.; Zani, M.; MacKay, A.R.; Screpanti, I.; Frati, L.; Gulino, A. EGF- and cell-cycle-regulated STAG1/PMEPA1/ERG1.2 belongs to a conserved gene family and is overexpressed and amplified in breast and ovarian cancer. Mol. Carcinog. 2003, 38, 188–200. [Google Scholar] [CrossRef]
- Li, H.; Xu, L.L.; Masuda, K.; Raymundo, E.; McLeod, D.G.; Dobi, A.; Srivastava, S. A feedback loop between the androgen receptor and a NEDD4-binding protein, PMEPA1, in prostate cancer cells. J. Biol. Chem. 2008, 283, 28988–28995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rae, F.K.; Hooper, J.D.; Nicol, D.L.; Clements, J.A. Characterization of a novel gene, STAG1/PMEPA1, upregulated in renal cell carcinoma and other solid tumors. Mol. Carcinog. 2001, 32, 44–53. [Google Scholar] [CrossRef]
- Azami, S.; Vo Nguyen, T.; Watanabe, Y.; Kato, M. Cooperative induction of transmembrane prostate androgen induced protein TMEPAI/PMEPA1 by transforming growth factor-β and epidermal growth factor signaling. Biochem. Biophys. Res. Commun. 2015, 456, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, N.; Itoh, S.; Watanabe, Y.; Maeyama, K.; Itoh, F.; Kato, M. Requirement of TCF7L2 for TGF-β dependent transcriptional activation of the TMEPAI gene. J. Biol. Chem. 2010, 285, 38023–38033. [Google Scholar] [CrossRef] [Green Version]
- Nakano, N.; Kato, M.; Itoh, S. Regulation of the TMEPAI promoter by TCF7L2: The C-terminal tail of TCF7L2 is essential to activate the TMEPAI gene. J. Biochem. 2016, 159, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Vo Nguyen, T.; Watanabe, Y.; Shiba, A.; Noguchi, M.; Itoh, S.; Kato, M. PMEPA1/TMEPAI enhances tumorigenic activities in lung cancer cells. Cancer Sci. 2014, 105, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Abdelaziz, M.; Watanabe, Y.; Kato, M. PMEPA1/TMEPAI knockout impairs tumour growth and lung metastasis in MDA-MB-231 cells without changing monolayer culture cell growth. J. Biochem. 2019, 165, 411–414. [Google Scholar] [CrossRef]
- Puteri, M.U.; Watanabe, Y.; Wardhani, B.W.K.; Amalia, R.; Abdelaziz, M.; Kato, M. PMEPA1/TMEPAI isoforms function via its PY and Smad-interaction motifs for tumorigenic activities of breast cancer cells. Genes Cells 2020, 25, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, S.L.; Golub, T.R.; Lander, E.S.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MsigDB) 3.0. Bioinfomatics 2011, 27, 1729–1740. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-b (transforming growth factor-b) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-b type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Okita, Y.; Kimura, M.; Xie, R.; Chen, C.; Shen, L.T.; Kojima, Y.; Suzuki, H.; Muratani, M.; Saitoh, M.; Semba, K.; et al. The transcription factor MAFK induces EMT and malignant progression of triple-negative breast cancer cells through its target GPNMB. Sci. Signal. 2017, 10, eaak9397. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Shanmugam, N.; Segawa, T.; Sesterhenn, I.; McLeod, D.G.; Moul, J.W.; Srivastava, S. A novel androgen-regulated gene, PMEPA1, located on chromosome 20q13 exhibits high level expression in prostate. Genomics 2000, 66, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Wang, C.; Zhou, Z.; Chen, C.; Liu, R.; Wang, D. Transforming growth factor-b increases breast cancer stem cell population partially through upregulating PMEPA1 expression. Acta Biochim. Biophys. Sin. 2016, 48, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Huo, X.; Zhao, S.; Yang, J.; Shi, W.; Jing, L.; Li, W.; Li, Y.; Ma, L.; Gao, Y.; et al. Low-density lipoprotein receptor class A domain containing 4 (LDLRAD4) promotes tumorigenesis of hepatic cancer cells. Exp. Cell Res. 2017, 360, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Lai, C.; Zhang, H.; Lai, M. PMEPA1 induces EMT via a non-canonical TGF-β signalling in colorectal cancer. J. Cell. Mol. Med. 2019, 23, 3603–3615. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, W.J.; Lehmann, P.Z.; Li, W. Nuclear PI3K signaling in cell growth and tumorigenesis. Front. Cell Dev. Biol. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Anil Kumar, K.; Reddanna, P. The Paradox of Akt-mTOR Interactions. Front. Oncol. 2013, 3, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Jin, L.; Tseng, H.Y.; Wang, C.Y.; Wilmott, J.S.; Yosufi, B.; Yan, X.G.; Jiang, C.C.; Scolyer, R.A.; Zhang, X.D.; et al. Oncogenic suppression of PHLPP1 in human melanoma. Oncogene 2014, 33, 4756–4766. [Google Scholar] [CrossRef] [Green Version]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful Partners in Cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Ding, K.; Luo, T.; Xu, R.; Zhang, X.; Huang, B.; Chen, A.; Zhang, D.; Miletic, H.; Bjerkvig, R.; et al. PMEPAI isoform a drives progression of glioblastoma by promoting protein degradation of the Hippo pathway kinase LATS1. Oncogene 2020, 39, 1125–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Liu, Y.; Xu, Y.; Juan, J.; Zhang, Z.; Xu, Z.; Cao, B.; Wang, Q.; Zeng, Y.; Mao, X. The transmembrane protein TMEPAI induces myeloma cell apoptosis by promoting degradation of the c-Maf transcription factor. J. Biol. Chem. 2018, 293, 5847–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, M.A.; Abdelaziz, M.; Puteri, M.U.; Vo Nguyen, T.T.; Kudo, K.; Watanabe, Y.; Kato, M. PMEPA1/TMEPAI Is a Unique Tumorigenic Activator of AKT Promoting Proteasomal Degradation of PHLPP1 in Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 4934. https://doi.org/10.3390/cancers13194934
Haque MA, Abdelaziz M, Puteri MU, Vo Nguyen TT, Kudo K, Watanabe Y, Kato M. PMEPA1/TMEPAI Is a Unique Tumorigenic Activator of AKT Promoting Proteasomal Degradation of PHLPP1 in Triple-Negative Breast Cancer Cells. Cancers. 2021; 13(19):4934. https://doi.org/10.3390/cancers13194934
Chicago/Turabian StyleHaque, Md. Anwarul, Mohammed Abdelaziz, Meidi Utami Puteri, Thanh Thao Vo Nguyen, Kosei Kudo, Yukihide Watanabe, and Mitsuyasu Kato. 2021. "PMEPA1/TMEPAI Is a Unique Tumorigenic Activator of AKT Promoting Proteasomal Degradation of PHLPP1 in Triple-Negative Breast Cancer Cells" Cancers 13, no. 19: 4934. https://doi.org/10.3390/cancers13194934