1. Introduction
Neuroblastoma is the most common extracranial solid tumor in children, accounting for about 8% of all childhood cancers, and is diagnosed using a combination of laboratory tests, radiologic imaging and pathology assessment. Elevated urine catecholamine metabolites and imaging findings of meta-iodo-benzyl-guanidine (MIBG) radiotracer avidity (occurring each in approximately 90% of patients) are tested early in the diagnostic process and are sufficient for provisional diagnosis. On the other hand, histological classification,
MYCN amplification (MNA), segmental chromosome aberrations (SCA) and
ALK activating mutations detected via tissue biopsy, are the current molecular foundations of risk-group stratification and targeted therapeutics [
1]. Increase in demand for tumor genetic testing for neuroblastoma diagnosis is posing a challenge to current practice, as the small size of the core needle biopsies obtained are required for multiple molecular tests: single nucleotide polymorphism (SNP) chromosome microarray analysis (CMA) or fluorescence in situ hybridization (FISH) for SCA and/or MNA, and DNA sequencing for
ALK somatic variants [
2].
Sampling body fluids such as peripheral blood, also known as “liquid biopsy”, offers an alternative biological source material for analysis of tumor-specific biomarkers within cell free circulating tumor DNA (ctDNA). Liquid biopsy is rapidly emerging as a revolutionizing approach for cancer diagnosis and treatment through interrogation of tumor-shed biomolecules [
3]. The unique challenges in the pediatric setting, and neuroblastoma diagnosis, make such a minimally invasive approach to disease diagnosis highly attractive.
Over the past decade, there has been growing interest in the utility of ctDNA in neuroblastoma. Early efforts toward the noninvasive identification of recurrent prognostic copy number aberrations showed high sensitivity and specificity for MNA tumors (91% and 98%, respectively) [
4], but only low-to-moderate rates for detection of 17q-gain (51–59% and 71–94%, respectively) [
5] and 11q-loss (26% and 100%, respectively) [
6]. These different rates are likely due to the masking effect of healthy circulating-free DNA (cfDNA) on moderate copy number changes, or potentially these aberrations being present at sub-clonal fractions. More recently, advances in genomic technologies made it possible to perform genome-wide assessment of genomic abnormalities from cfDNA, thereby capturing all neuroblastoma subtypes with 68–100% overall sensitivity (Reviewed in [
7,
8,
9,
10]). Chromosomal microarray is a key tool used for diagnosis of SCA (and MNA) in patients with neuroblastoma. Importantly, Chicard and colleagues [
11] reported the use of a microarray for cfDNA analysis, reaching a sensitivity of 23–75%, which was dependent on disease stage with accurate copy number detection highly compromised due to: (1) background cfDNA shed from normal blood cells, and (2) the highly degraded nature of ctDNA, which may have reflected collecting blood under suboptimal conditions and preforming subsequent analysis. This may further hinder the utility of liquid biopsy in clinical practice.
Lately, the efficacy of ALK inhibitors had shown potential in
ALK-driven refractory or relapsed neuroblastoma [
12] and is currently being investigated as upfront treatment for newly diagnosed patients with high-risk disease and confirmed
ALK mutated tumors (COG ANBL1531, ClinicalTrails.gov identifier NCT03126916). However, access to adequate core needle tissue to determine
ALK somatic variants is challenging, with only 10% of samples reported to be successfully genotyped (either mutated or wild type) (COG personal communications). This is disappointing as the prevalence of
ALK somatic variants in this population has been reported to be relatively high (14%) [
13].
ALK somatic variants identified in cfDNA using digital-droplet PCR (ddPCR) in a series of small-cohort studies [
14,
15] highlight the feasibility of liquid biopsy in this setting.
The aim of our study was to evaluate and optimize pre-analytical processes for detecting neuroblastoma biomarkers in the circulation. We studied a series of 10 cfDNA samples obtained from patients with neuroblastoma and assessed whether SCA and MNA could be identified using various modified protocol conditions. We also evaluated the dynamics of detectable MNA and ALK variants throughout the course of treatment of 6 patients, 3 of which treated with the ALK inhibitor Lorlatinib, to assess ctDNA utility monitor therapeutic biomarkers and treatment efficacy in real-time.
2. Materials and Methods
2.1. Patients and Samples
Eligibility criteria for this prospective observational study included diagnosis of neuroblastoma and confirmed MNA status. Accordingly, sixteen patients enrolled: 13 at initial diagnosis, and 3 at disease relapse. The study was conducted under research protocols approved by the Sydney Children’s Hospitals Network Human Research Ethics Committee, (reference number: HREC/17/SCHN/302). Forty-four samples (from thirteen patients) prospectively collected from patients treated at the Children’s Hospital at Westmead (CHW), with all patients’ parents consented to the study. Additional thirteen samples collected from children in long-term remission from solid cancers (
n = 3 neuroblastoma,
n = 5 hepatoblastoma, and
n = 5 sarcomas) who are otherwise healthy. Additional two and one retrospective samples obtained from Children’s Cancer Institute Tumour Bank (CCI-TB) (NSW) and from Zero Childhood Cancer (ZCC) PRISM Program (NSW), respectively. Additional clinical, pathological, and radiological information is found in
Supplementary Materials: Table S1.
Blood samples collected in Streck (Cell-Free DNA BCT®, STRECK, La Vista, NE, USA, catalog No. 218997) or EDTA tubes and processed for plasma. Samples from CHW/ZCC collected in Streck and processed at ambient temperature in a double-centrifugation protocol: first centrifugation at 1600× g for 10 min followed by plasma supernatant aspiration into new tubes without disturbing the buffy coat layer, then a second centrifugation of the plasma supernatant at 15,500× g for 10 min, followed by aspirating the top of phase into new tubes without disturbing the pellet, and storing at −80 °C until DNA isolation. Samples collected at CCI-TB (in EDTA) centrifuged at approximately 1000× g for 10 min before plasma supernatant isolation and storage at −80 °C. An additional high-speed centrifugation step (15,000× g for 10 min at 4 °C) after thawing of stored samples was preformed prior to DNA extraction.
For
Section 3.2.2, blood samples collected in Streck, PAXgene (PAXgene
® Blood ccfDNA Tube, product reference 768165, BD Biosciences Macquarie Park, NSW, Australia), and EDTA tubes. Samples processed in a double-centrifugation protocol as described above, with Streck and PAXgene tubes processed at ambient temperature and samples in EDTA processed at cooled 4 °C conditions. Gentle agitation to mimic sample transfer conditions performed using horizontal orbital rotation at a low speed of 3.75 RPM.
2.2. DNA Isolation
Cell free circulating DNA was extracted from 0.25–3 mL of frozen plasma samples using the QIAamp Circulating Nucleic Acid kit (Qiagen, catalog No. 55114, Chadstone, VIC, Australia) according to the manufacturer’s instructions, except for increasing the proteinase digest step to 60 min for plasma samples collected in Streck and PAXgene tubes, as recommended by Streck and PAXgene tube product literature. In cases of starting volume of plasma lower than 3 mL, volumes adjusted to 3 mL with PBS. DNA was eluted in 50 µL buffer provided with the kit and stored at −80 °C until PCR analysis. DNA amount was evaluated using Qubit dsDNA High Sensitivity Assay Kit (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA).
Genomic/constitutional DNA was extracted from cell lines and whole blood samples using AllPrep DNA/RNA/Protein kit (Qiagen; catalog No. 80004) and QIAGEN DNeasy Blood & Tissue kit (Qiagen; catalog No. 69504), respectively, according to the manufacturer’s instructions. Cell lines used in this study: neuroblastoma cell lines-Kelly, BE2C, IMR32, SK-N-SH purchased from ECACC (European Collection of Authenticated Cell Cultures, Salisbury, UK), and sarcoma cell lines–RH30, A673, and ES8 kindly provided by Dr Belinda Kramer (Advanced Cellular Therapeutics, Children’s Cancer Research Unit, Kid’s Research, The Children’s Hospital at Westmead, NSW, Australia). All cell lines subjected to Short Tandem Repeat (STR) profiling by CellBank Australia (Westmead, Australia) to confirm identity.
2.3. cfDNA Treatment
Purified cfDNA was treated with either with the Infinium® HD FFPE DNA Restore Kit (Illumina, San Diego, CA, USA) or SureTag Purification Columns (Agilent, Santa Clara, CA, USA), according to the manufacturers’ instructions.
2.4. SNP Chromosomal Microarray Analysis (SNP CMA)
SNP CMA was performed with the Infinium CytoSNP−850K v1.2 Beadchip (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions. Microarray data was analyzed using BlueFuse Multi v4.5 software (Illumina, San Diego, CA, USA) based on the reference human genome (hg19/GRCh37) according to laboratory protocols within the CHW Cytogenetics Department.
Satisfactory SNP microarray data quality control (QC) metrics are usually defined by Median Log R Deviation (Log R Dev) and Median B-Allele Frequency Deviation (BAF Dev) and SNP call rate. For DNA samples with high molecular weight and high purity, satisfactory QC metrics are <0.2, <0.03 and >0.98, respectively. It was anticipated that SNP CMA QC metrics using ctDNA samples would not meet these typically values.
The mean effective resolution for detecting copy number abnormalities is ~20–50 kb and SCAs were defined as being >5 Mb in size. Smaller abnormalities were defined as focal gain or loss.
2.5. ddPCR
(1) MNA: We designed a ddPCR assay to evaluate
MYCN copy number levels compared with the reference gene
THNSL2.
THNSL2 was chosen as it is positioned near the centromere of the same chromosome arm as
MYCN, to control for a false positive result due to numerical change of whole chromosome 2. We also included evaluation of ALK gene levels as: (a)
ALK amplification is known to occur in 3–4% of neuroblastoma sporadic cases [
16] almost exclusively in the context of MNA due to their close proximity on chromosome 2p23–24 [
17], and (b) to increase the robustness for normalization by using both
ALK and
THNSL2 as reference genes. Simultaneous detection of the three target genes using only two fluorophores included strategies to optimize primer/probe-sets concentration that allow a clean segregation of the droplets by levels of fluorescence in the data output [
18] (
Figure S1).
Samples analyzed for MYCN, ALK, and THNSL2 genomic regions using PrimePCR Copy Number Assays: dHsaCP2506554 (MYCN, HEX), dHsaCP1000588 (ALK, 6-FAM), and dHsaCP2506297 (THNSL2, 6-FAM), respectively (Bio-Rad Laboratories, Gladesville, NSW, Australia). Duplex MYCN/THNSL2 ddPCR reaction consisted of 10 μL ddPCR™ Supermix for Probes (No dUTP) (Bio-Rad Laboratories), 1 μL of MYCN and THNSL2 PrimePCR assay (final concentrations of primers and probes of 900 nM and 250 nM, respectively), 4 units of HaeIII restriction enzyme (New England BioLabs, Notting Hill, VIC, Australia), and cfDNA/water for a final volume of 20 μL. For triplex MYCN/ALK/THNSL2 ddPCR, additional 1 μL of ALK PrimePCR assay was included, while THNSL2 PrimePCR assay was reduced to 0.4 μL. In duplex ALK/THNSL2 dHsaCP2506297 (THNSL2, HEX) was used. NTCs contained purified water instead of cfDNA. The ddPCR reaction mixture was used for droplet generation, and amplification was carried out in a C1000 Touch Thermal Cycler (Bio-Rad Laboratories) under the following conditions: 95 °C for 10 min, 40 cycles of 94 °C for 30 s, 60 °C for 1 min; then 98 °C for 10 min. ddPCR was performed using the QX200 ddPCR system according to manufacturer’s instructions (Bio-Rad Laboratories). QuantaSoft™ Analysis Pro v1.0 software (Bio-Rad Laboratories) used for data analysis. Target and reference/s copies were within the dynamic range of the instrument to ensure accurate detection level. Each sample was tested in 2 to 3 replicates preformed in at least 2 different experiments.
(2) ALK variants: NM_004304.5 (ALK):c.3522C > A (p.F1174L) [ALK p.F1174L] analyzed using PrimePCR Mutation Detection Assays: dHsaCP2000084 (p.F1174L c.3522C > A, HEX) and dHsaCP2000083 (ALK wild type, 6-FAM) as described above, with Tm 55 °C.
NM_004304.5 (ALK):c.3733T > A (p.F1245I) [
ALK p.F1245I] analyzed using primers: 5′-TGGCTGTCAGTATTTGGAGG-3′ and 5′ CAGGAAGAGCACAGTCACTT 3′, wild type probe: 5′-[HEX] A+ACCACTTCATCCACCGGTGA-[IABkFQ]-3′, and
ALK c.3733T > A probe: 5′-[6-FAM] AACCACATCATCCACCGGTGAGT [IABkFQ]-3′, as described above (A+-LNA nucleotide). Wild type and variant
ALK sequences were used following the dMIQE guidelines [
19].
ALK assays tested positive on genomic DNA from the primary tumors of Dx1, Dx9, and Rec3 to demonstrate VAF of 19.13%, 35.15%, and 29.51%, respectively. Tm optimizations conducted on tumor DNA. All cell lines used in this study tested negative for either p.F1174L or p.F1245I, with the exception of Kelly and SK-N-SH which tested positive for ALK p.F1174L, confirming the specificity of the assays.
2.6. qPCR
Samples analyzed for
MYCN, and
THNSL2 genomic regions in the Rotor Gene Q (QIAGEN) real-time PCR, similarly to ddPCR: qPCR reactions consisted of 10 μL ddPCR™ Supermix for Probes (No dUTP) (Bio-Rad Laboratories), 0.8 μL of
MYCN and
THNSL2 PrimePCR Copy Number Assays (dHsaCP2506554, and dHsaCP2506297, respectively; final concentrations of primers and probes of 500 nM and 200 nM, respectively), 4 units of HaeIII restriction enzyme (New England BioLabs), and cfDNA/water for a final volume of 20 μL. NTCs contained purified water instead of cfDNA. Amplification was carried out in a Rotor Gene Q (QIAGEN) under the following conditions: 95 °C for 10 min, 40 cycles of 94 °C for 30 s, 60 °C for 1 min; then 98 °C for 10 min. Each sample was tested in 3 replicates. Data collected from the yellow (HEX) and green (FAM) channels. Rotor Gene Q Series Software 2.3.1 (QIAGEN) was used for data output, with thresholds adjusted manually to generate equal copy number with minimal variance of
MYCN and
THNSL2 in cell lines samples A673 and ES8. Primers’ efficiencies calculated from serial dilution of Kelly cell line DNA generating matched
MYCN and
THNSL2 calling in appropriate dynamic range to ensure accurate detection level. Pfaffl method [
20] used to calculate
MYCN copy number:
95% CI calculated with t statistics for 3 degrees of freedom.
2.7. Statistical Analysis
Mann–Whitney nonparametric
t test, paired
t test, Pearson correlation test, and Bland-Altham method comparison test were performed using GraphPad Prism version 9.1.1 for macOS, GraphPad Software, San Diego, CA, USA,
www.graphpad.com (accessed on 15 April 2021).
4. Discussion
Over the past decade, there has been growing interest in the utility of circulating biomarkers such as ctDNA and liquid biopsy of cancers. This approach can provide a global picture of the cancer in real time, at multiple time points, and with minimal invasiveness. A number of studies were able to accurately detect MNA and SCA in serum and plasma from patients with neuroblastoma as a tumor surrogate with varied diagnostic accuracies, which depended on disease stage [
7]. In more recent years it became clear that mutations of the
ALK tyrosine kinase domain constitute an important potential therapeutic target in neuroblastoma [
12] and can be detected in ctDNA [
14,
15]. In the current clinical setting, surgical or core biopsies are carried out to provide histological diagnosis. However, these techniques do not always provide enough material for the analysis of biological markers. Here, we examined cfDNA isolated from plasma samples collected from patients with neuroblastoma and found that ctDNA is the predominant DNA type in plasma of metastatic patients and thus represents a relevant model for ctDNA analysis. We present strategies for optimizing plasma collection and ctDNA testing ensuring the accuracy and reproducibility of those analytes, which is required for clinical application of circulating biomarkers.
The combination of chromosomal microarray and FISH analyses is the current standard of practice for clinical genomic investigations for the detection of SCA and MNA, respectively, in neuroblastoma molecular risk-stratification. In the present study, we examined the utility of ctDNA to provide SCA and MNA profiles using SNP array, with orthogonal conformation of MNA status via ddPCR. SNP CMA is a key technique already incorporated into routine clinical diagnostic workflows. A major challenge associated with applying SNP CMA for ctDNA is providing adequate DNA input quantities which are not readily available from most cfDNA cancer samples. Moreover, neuroblastoma is diagnosed mostly in infants and very young children (90% younger than 5 years, median age 18 months) [
1] which therefore able to provide only limited amounts of blood for a liquid biopsy test (typically 0.5–1 mL plasma). Hence, 200 ng input DNA required according to manufacturer’s recommendation for the Infinium CytoSNP-850K Beadchip array is not feasible for many patients with neuroblastoma, including all the patients in our cohort with localized disease. Here, the Infinium
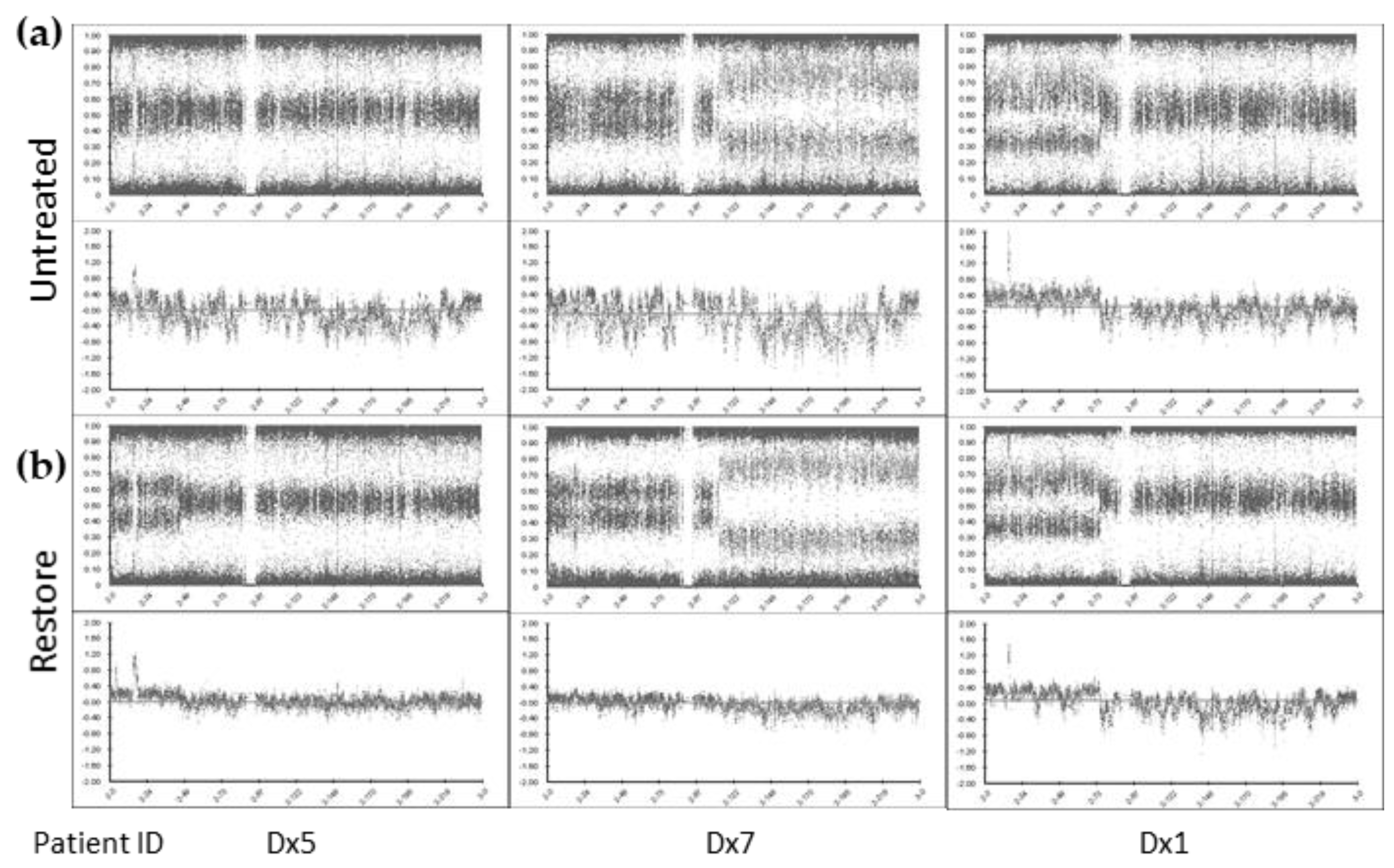
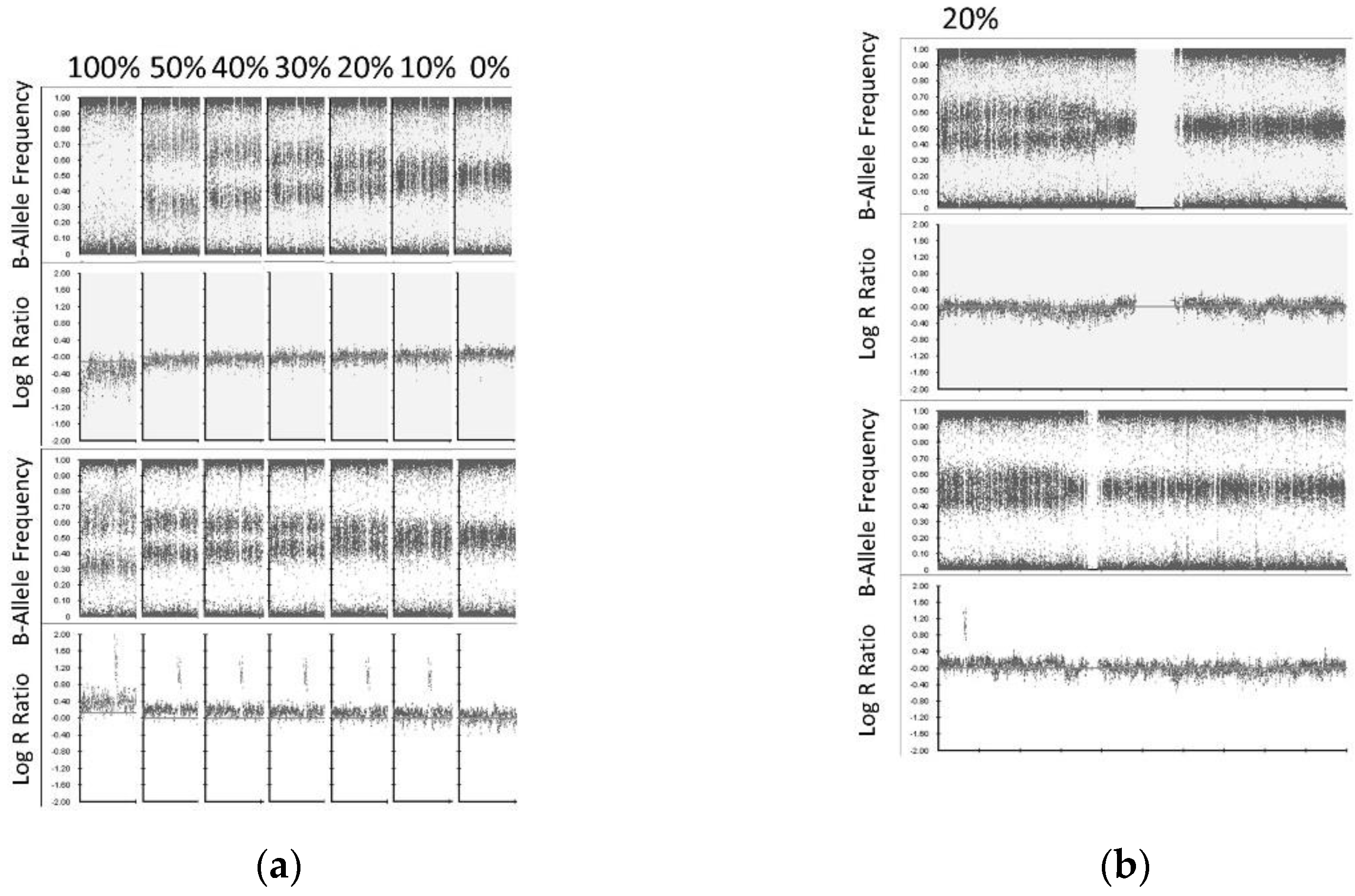
® FFPE DNA Restoration kit has been employed for cfDNA repair and increased the detectability of ctDNA by SNP CMA. We successfully analyze 10/10 (100%) cfDNA samples by SNP CMA with cfDNA input amount as low as 30 ng. We also demonstrated the limit of detection for this approach with SCA and MNA detected at the lower limit of 20% and 10% of the sample, respectively.
The diagnostic accuracy of cfDNA for the determination of MNA status in advanced stage neuroblastoma was investigated since the beginning of the century (even before the concept of “liquid biopsy” emerged) using PCR techniques, with quantitative real-time PCR (qPCR) MNA detection reported in large cohorts of patients [
4]. Here, we compared results between ddPCR and qPCR for MNA detection and found them both to reliably detect normal copy levels,
MYCN gain, and MNA, with ddPCR demonstrating a superior sensitivity to detect CNAs.
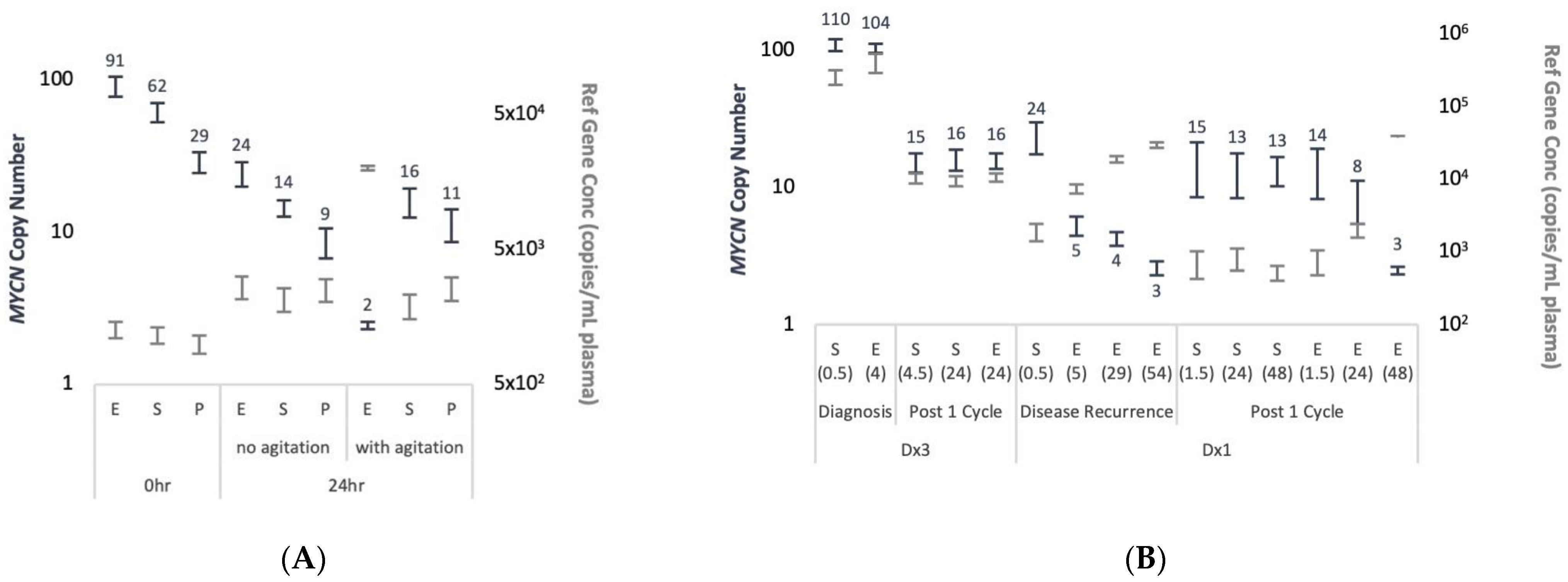
The detectability of ctDNA from cfDNA is largely influenced by pre-analytical factors, as evaluated from data on SNVs, but to our knowledge was not assessed for CNAs. We evaluated the impact of pre-analytical procedures for plasma processing on the detectability of MNA in ddPCR. We found that EDTA blood collection tubes do not prevent the release of background cfDNA and so eliminate MNA detection. This was observed in conditions mimicking routine sample transfer in a hospital environment, both in artificial spiking experiments and in real-life patient’s samples scenarios.
There is great potential for ctDNA molecular diagnosis in neuroblastoma. In 2005, a case report by Combaret et al. highlighted the utility of MYCN evaluation in cfDNA, where provisional diagnosis was made but tumor biopsy was not possible. A 13-day-old infant presented with left adrenal tumor and elevated urinary catecholamine metabolites, however, thrombocytopenia and hypofibrinogenemia confounded the collection of tumor tissue. Evaluation of serum MYCN by qPCR revealed high-level MNA, and the child was assigned with high-intensity chemotherapy. Three weeks after a significant response, blood coagulation parameters had returned to normal and biopsy tissue was obtained, which showed greater than 50 MYCN copies by qPCR.
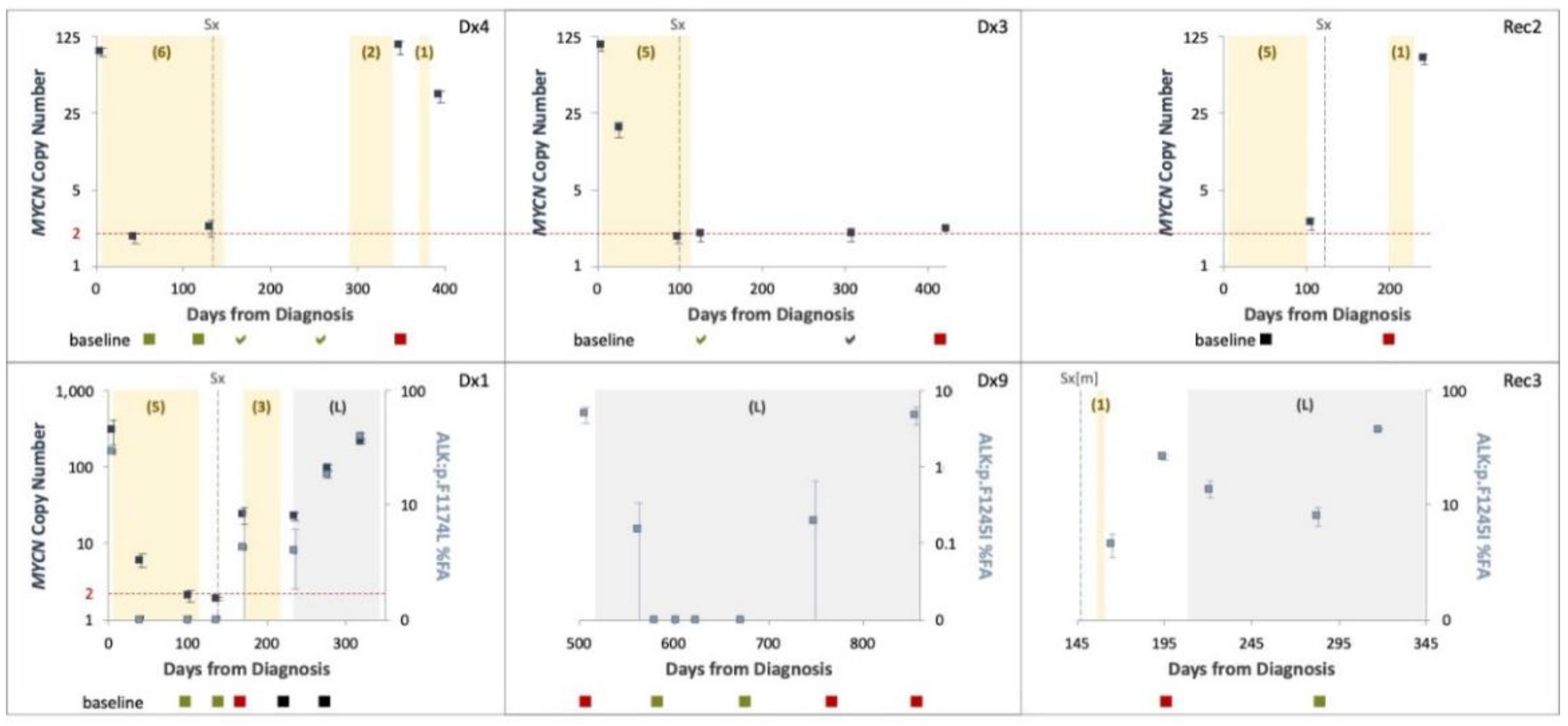
In our study, for patient ID Dx1 the initial biopsy suffered crush artefact and was difficult to interpret. In addition, biopsy at relapse was done neurosurgically (brain metastases) and so carried inherent clinical risks. On both occasions, cfDNA analysis demonstrated MNA and the ALK p.F1174L variant, and MIBG showed positive avidity. Thus, alternative approaches that overcome technically difficulties or high-risk surgical biopsy of the primary and/or recurrent tumors such as combining imaging and liquid biopsy can be very valuable.
Low-risk neuroblastoma represents 25% of cases, is classified in infants younger than 18 months, with either localized or metastatic (to the liver, skin, and bone marrow sites only) non-MNA disease. Clinically the disease is monitored without treatment as it usually spontaneously regress [
24]. Molecular cytogenetics studies of low-risk tumors show hyperdiploidy (having extra chromosomes) rather than SCA. In this very young population, a surrogate marker to detect tumor-specific alterations in a non-invasive setting and avoiding tissue biopsy would be very desirable since accessibility of the tumor, morbidities associated with surgical risks and exposure to anesthetics (during MIBG imaging) can present key technical and practical challenges. While we show that localized neuroblastoma shed higher-than-normal cfDNA amounts, we were not able to analyze any of the four localized cases using SNP array due to low-input cfDNA levels. In addition, although several localized pediatric diseases were found to shed high ctDNA VAF in some cases (e.g., [
25]), there was not enough to proceed with SNP CMA, given the assay sensitivity or LOD for identify low levels of MNA (10%) and SCA (20%). Next generation sequencing (NGS) is used routinely to detect DNA somatic variants when VAF ≥ 5% and might offer a better solution in this setting. However, NGS has its own limitations as benchmarking copy number variant callers tend to be challenging [
26,
27]. Currently, there are no molecular biomarkers for treatment response of high-risk disease. About half of patients with high-risk disease will harbor MNA and so MNA detection using ctDNA could present a potential biomarker for treatment response using either SNP array or ddPCR techniques. However, SNP CMA is limited in detecting MNA at clonal levels <10% and ctDNA input amounts (>30 ng) limit its use to only samples at initial presentation or recurrent disease. Using ddPCR with a detection limit >0.5% VAF for
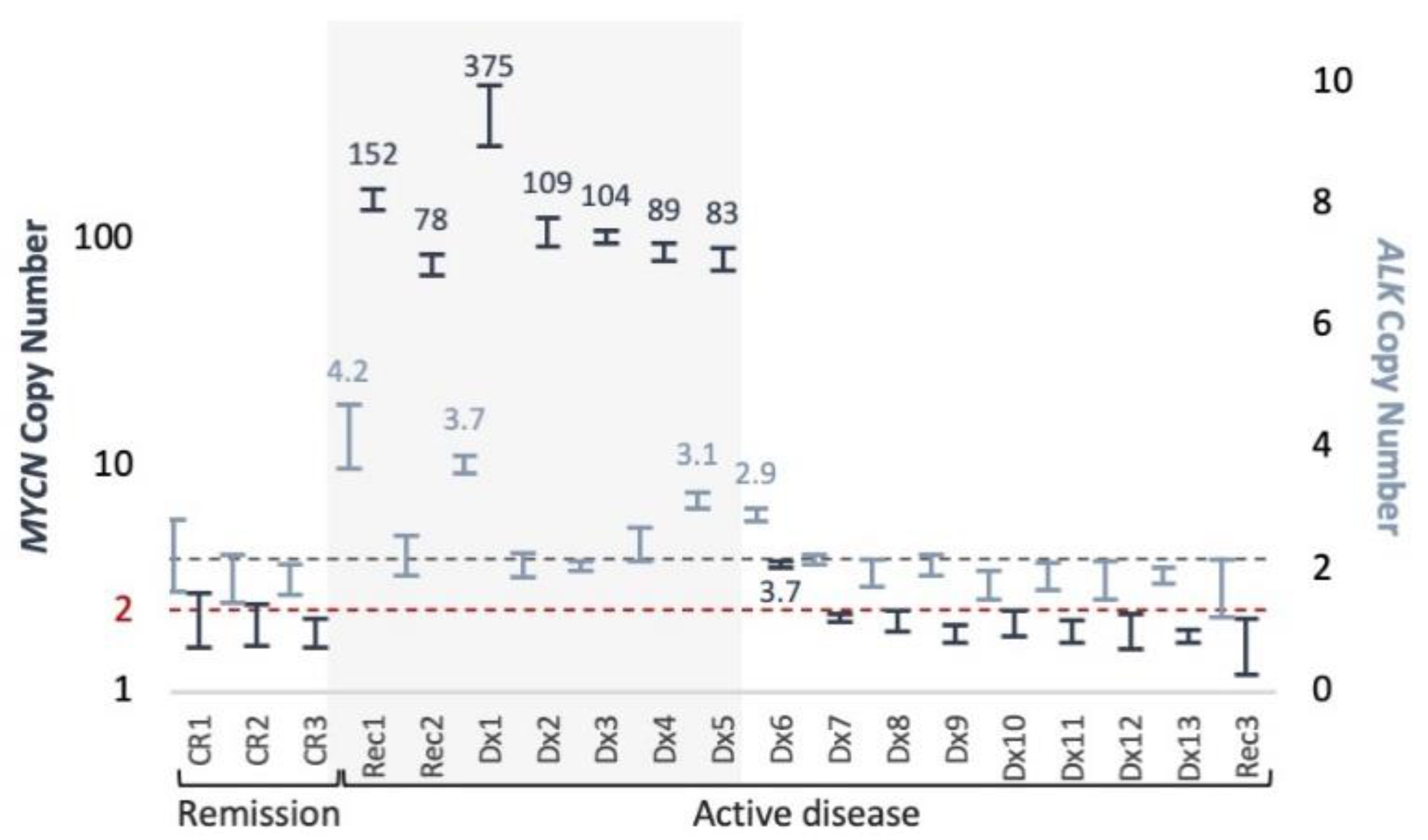
MYCN gain, we were able to monitor treatment response across the course of disease of six patients, testing either MNA (4 cases) or ALK variants (3 cases). We show that longitudinal testing can capture ctDNA dynamics and response to treatment. However, samples taken post neoadjuvant chemotherapy in four cases (Dx4, Dx3, Rec2, Dx1) showed only normal diploid
MYCN levels, although imaging studies performed around the same time demonstrated MIBG avidity. A possible explanation to the difference between ctDNA and MIBG would be that ctDNA is detected in the context of proliferating cells only, while MIBG can detect live neuroblastoma cells, proliferating or static. MNA detected again at disease recurrence in three of these cases (75%). We conclude therefore that our test, having 0.5% VAF LOD, is not sensitive enough for early recurrence or minimal residual disease monitoring.
At the age of precision medicine, the clinical management of high-risk patients harboring hotspots ALK somatic variants is changing. ALK inhibitors are currently being investigated as upfront treatment for newly diagnosed patients with confirmed ALK mutated tumors. However, access to adequate core needle tissue to determine ALK somatic variants at initial diagnosis is challenging, with only 10% of samples reported to be successfully genotyped (either mutated or wild type), despite the prevalence of ALK somatic variants in this population being relatively high (14%). Our results support previous findings in small cohort cases of ALK variants in the circulation of patient with neuroblastoma. Thus, liquid biopsy for ALK variants could potentially overcome current hurdles in this clinical setting.
Several studies have demonstrated the suitability of serial
ALK variants assessment for disease monitoring of ALK-positive non-small-cells lung cancer (NSCLC) [
28]. Madsen and colleagues [
29] reported a total clearance of
ALK variants within two months after ALK inhibitors treatment initiation for patients achieving partial response. In our study
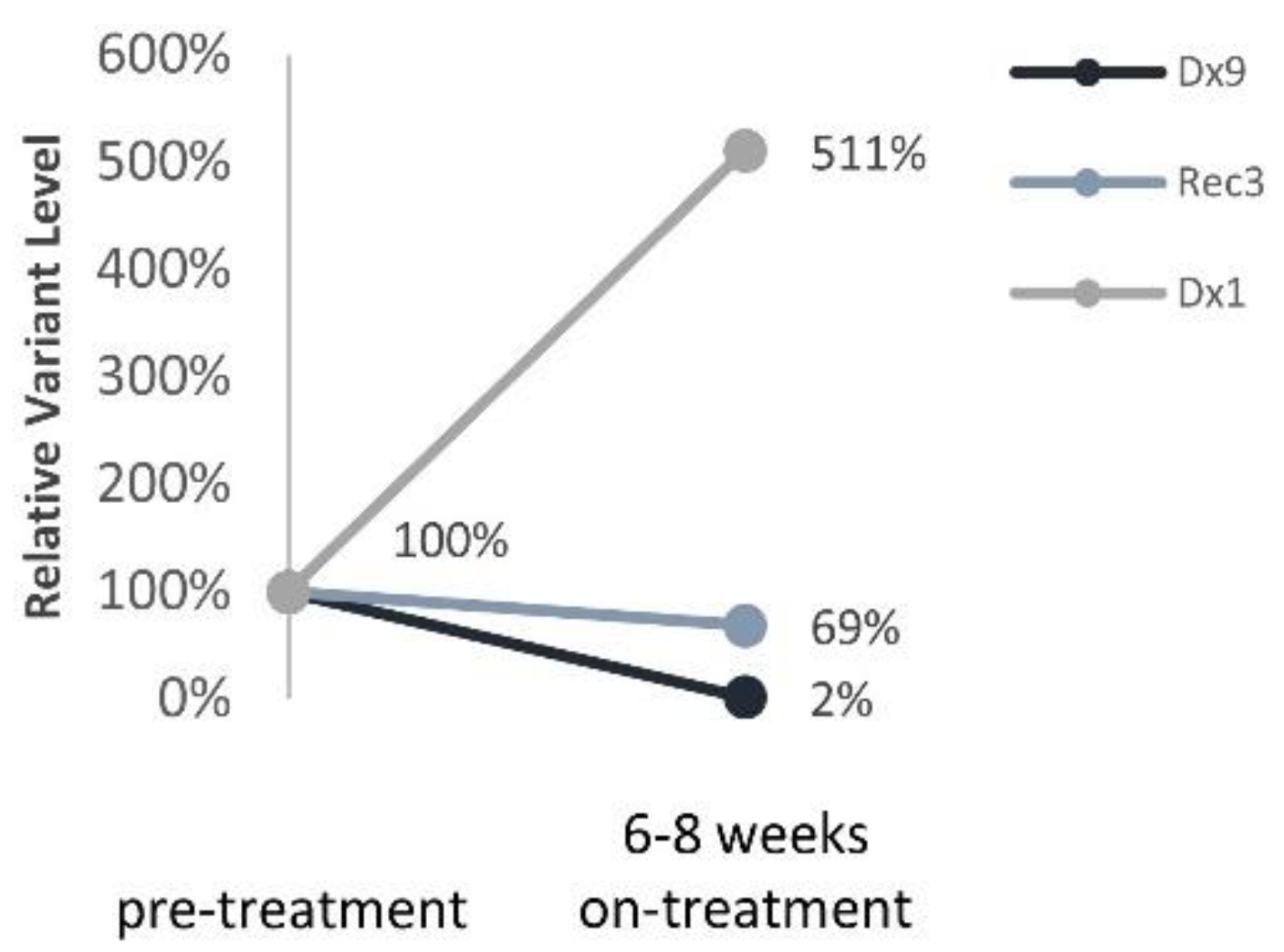
ALK variants were detected in all three patients treated with Lorlatinib 6–8 weeks into the targeted treatment. Nonetheless,
ALK variants’ trajectories reflected response to treatment and thus provide a surrogate biomarker to monitor targeted treatment efficacy in real-time. Sampling ctDNA at shorter intervals might provide earlier identification of treatment failure/success in neuroblastoma.
Unfortunately, it is becoming apparent that therapeutic resistance to ALK inhibition is inevitable for all current compounds. NGS studies demonstrated spatial and temporal evolution of neuroblastoma. Looking into a very narrow lens for characterizing only MYCN and ALK copy numbers, we found in this small study cohort that ALK gain of 4 copies in patient (ID Rec1) with available whole genome sequencing and high-depth targeted sequencing of synchronous tumor samples showed no changes to ALK copy number. However, using NGS for liquid biopsy could offer the opportunity to incorporate additional genomic findings about tumor response and result in earlier detection of somatic variants associated with resistance, and therefore increase treatment precision thereby improving patient survival and quality of life.
The strengths of our study include the use of ctDNA from neuroblastoma patients to identify SNVs and both DNA copy number amplification and SCA by ddPCR and SNP array, which involved optimization of sample collection conditions, novel use of the FFPE DNA restoration kit, evaluating the limit of detection of copy number abnormalities, and lower input ctDNA for SNP microarray analysis. Limitations of our study include the small cohort size, which partly reflects the rarity of neuroblastoma cases and partly the available cfDNA samples at the time of this study. Additional clinical sample are needed to more extensively assess the use of ctDNA, particularly for use of the CytoSNP-850K Beadchip assay. The small DNA fragment size of ctDNA and low input DNA amounts can pose challenges for the assay. In samples with poor quality assay metrics, segmental copy number abnormalities <5 Mb in size or at levels <20% of the sample may go undetected.
To summarize, neuroblastoma offers an attractive disease for ctDNA applications due to the very high tumor burden of most cases at initial presentation, and accordingly, very high ctDNA amounts. Neuroblastoma is one of few pediatric cancers in which biomarkers are routinely used for diagnosis, prognostication and therapeutic actions. Elevated urinary catecholamine metabolites and MIBG avidity, each evident in over 90% of patients, are routinely evaluated for provisional diagnosis and staging, and usually assessed prior to the biopsy procedure. This time window from the provisional diagnosis until the biopsy procedure, allows for cfDNA SCA and MNA profiling. Here, we show how SCA and MNA can be detected by SNP CMA within 1–2 weeks, which is the current diagnostic approach. We also demonstrate how ddPCR can be employed to provide orthogonal confirmation for MNA. With a turnaround time of 1–2 days for ddPCR, both could be achieved at a clinically relevant time frame while the biopsy procedure is been planned. Therefore, reducing core needle tissues could be considered in the context of provisional diagnosis and positive ctDNA-SCA or -MNA findings.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





