Small Ones to Fight a Big Problem—Intervention of Cancer Metastasis by Small Molecules

 , and
, and
Abstract
:1. Introduction
1.1. The Demanding Clinical Need for Metastasis Intervention
1.2. Exploiting the Metastatic Cascade to Find Vulnerabilities for Metastasis Intervention
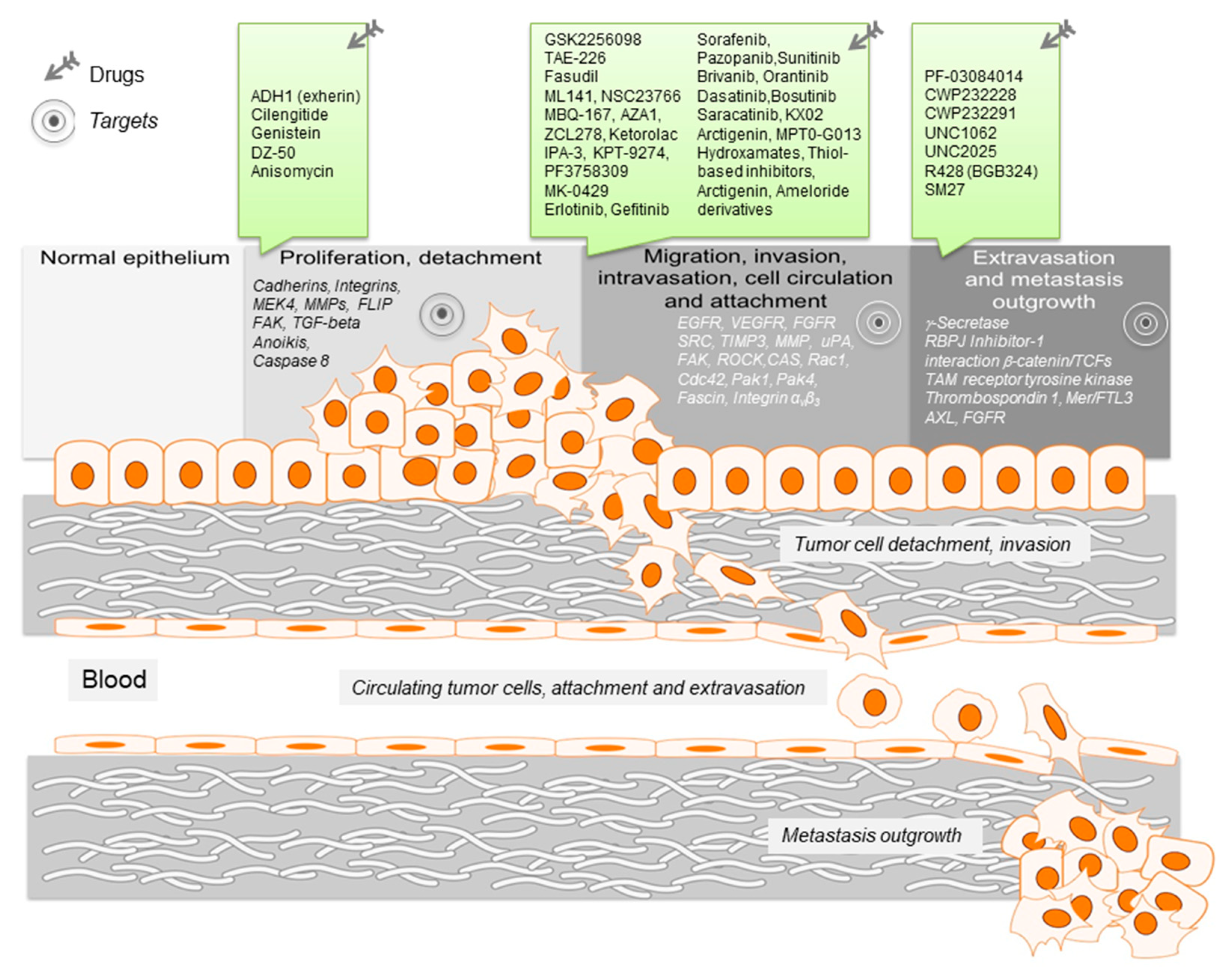
2. Targets for Therapeutic Intervention during Metastasis Formation
2.1. Tumor Cell Detachment—Principiis Obsta
2.2. Migration of Tumor Cells—Stop Moving
2.3. Invasion Intervention—Stop the Invaders
2.4. Metastasis Outgrowth—Intervention of Settlement
2.5. Metastasis Suppressors—Natural Borne Inhibitors
3. Conclusion and Future Prospects of Anti-Metastatic Therapy
Funding
Conflicts of Interest
References
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; A Weinberg, R. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Budczies, J.; Kluck, K.; Walther, W.; Stein, U. Decoding and targeting the molecular basis of MACC1-driven metastatic spread: Lessons from big data mining and clinical-experimental approaches. Semin. Cancer Boil. 2020, 60, 365–379. [Google Scholar] [CrossRef]
- Zijlstra, A.; The Board Members of the Metastasis Research Society; Von Lersner, A.; Yu, D.; Borrello, L.; Oudin, M.; Kang, Y.; Sahai, E.; Fingleton, B.; Stein, U. The importance of developing therapies targeting the biological spectrum of metastatic disease. Clin. Exp. Metastasis 2019, 36, 305–309. [Google Scholar] [CrossRef]
- Valastyan, S.; A Weinberg, R. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef]
- Fidler, I.J.; Poste, G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008, 9, 808. [Google Scholar] [CrossRef]
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Berim, L.D.; Kos, B.M.; Evande, R.; Meza, J.L.; Shostrom, V.; Schwarz, J.K.; Grem, J.L. A phase I study of ADH-1 with cisplatin (Cisp) and gemcitabine (Gem) in patients (Pts) with unresectable or metastatic pancreatic and biliary tract cancers. J. Clin. Oncol. 2017, 35, 306. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Haddad, T.; Qin, R.; Lupu, R.; Satele, D.; Eadens, M.; Goetz, M.P.; Erlichman, C.; Molina, J.R. A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 1221–1227. [Google Scholar] [CrossRef]
- Manegold, C.; Vansteenkiste, J.; Cardenal, F.; Schuette, W.; Woll, P.J.; Ulsperger, E.; Kerber, A.; Eckmayr, J.; Von Pawel, J. Randomized phase II study of three doses of the integrin inhibitor cilengitide versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer. Investig. New Drugs 2012, 31, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Pavese, J.M.; Krishna, S.N.; Bergan, R.C. Genistein inhibits human prostate cancer cell detachment, invasion, and metastasis. Am. J. Clin. Nutr. 2014, 100, S431–S436. [Google Scholar] [CrossRef]
- Frey, R.S.; Li, J.; Singletary, K. Effects of genistein on cell proliferation and cell cycle arrest in nonneoplastic human mammary epithelial cells: Involvement of Cdc2, p21waf/cip1, p27kip1, and Cdc25C expression11Abbreviations: PTK, protein tyrosine kinase; cdk, cyclin-dependent kinase; DMEM, Dulbecco’s Modified Eagle’s Medium; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; and ER, estrogen receptor. Biochem. Pharmacol. 2001, 61, 979–989. [Google Scholar] [CrossRef]
- Hemaiswarya, S.; Doble, M. Potential synergism of natural products in the treatment of cancer. Phytother. Res. 2006, 20, 239–249. [Google Scholar] [CrossRef]
- Cui, S.; Wang, J.; Wu, Q.; Qian, J.; Yang, C.; Bo, P. Genistein inhibits the growth and regulates the migration and invasion abilities of melanoma cells via the FAK/paxillin and MAPK pathways. Oncotarget 2017, 8, 21674–21691. [Google Scholar] [CrossRef]
- Garrison, J.B.; Shaw, Y.-J.; Chen, C.-S.; Kyprianou, N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007, 67, 11344–11352. [Google Scholar] [CrossRef] [Green Version]
- Mawji, I.A.; Simpson, C.D.; Gronda, M.; Williams, M.A.; Hurren, R.; Henderson, C.J.; Datti, A.; Wrana, J.L.; Schimmer, A.D. A Chemical Screen Identifies Anisomycin as an Anoikis Sensitizer That Functions by Decreasing FLIP Protein Synthesis. Cancer Res. 2007, 67, 8307–8315. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; He, D.-H.; Zajac-Kaye, M.; Hochwald, S.N. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014, 13, 3143–3149. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, W.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [CrossRef]
- Negoro, N.; Hoshiga, M.; Seto, M.; Kohbayashi, E.; Ii, M.; Fukui, R.; Shibata, N.; Nakakoji, T.; Nishiguchi, F.; Sasaki, Y.; et al. The Kinase Inhibitor Fasudil (HA-1077) Reduces Intimal Hyperplasia through Inhibiting Migration and Enhancing Cell Loss of Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 1999, 262, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Nöth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Boil. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onesto, C.; Shutes, A.; Picard, V.; Schweighoffer, F.; Der, C.J. Characterization of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. Methods Enzymol. 2008, 439, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [Green Version]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; De La Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, Novel Small Molecule Inhibitor of Rac GTPase. J. Boil. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef] [Green Version]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef] [Green Version]
- Zins, K.; Lucas, T.; Reichl, P.; Abraham, D.; Aharinejad, S. A Rac1/Cdc42 GTPase-Specific Small Molecule Inhibitor Suppresses Growth of Primary Human Prostate Cancer Xenografts and Prolongs Survival in Mice. PLoS ONE 2013, 8, e74924. [Google Scholar] [CrossRef] [Green Version]
- Zins, K.; Gunawardhana, S.; Lucas, T.; Abraham, D.; Aharinejad, S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of PAK1 activity. J. Transl. Med. 2013, 11, 295. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesland, A.; Zhao, Y.; Chen, Y.-H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42–intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Viaud, J.; Peterson, J. An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol. Cancer Ther. 2009, 8, 2559–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Aboud, O.; Chen, C.-H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, B.J.; Lee, H.; Kim, S.-H.; Heo, J.-N.; Choi, S.-W.; Yeon, J.-T.; Lee, J.; Lee, J.; Cho, J.Y.; Kim, S.H.; et al. PF-3758309, p21-activated kinase 4 inhibitor, suppresses migration and invasion of A549 human lung cancer cells via regulation of CREB, NF-κB, and β-catenin signalings. Mol. Cell. Biochem. 2013, 389, 69–77. [Google Scholar] [CrossRef]
- Murray, B.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.-M.; Zager, M.; et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef] [Green Version]
- Pickarski, M.; Gleason, A.; Bednar, B.; Duong, L.T. Orally active αvβ3 integrin inhibitor MK-0429 reduces melanoma metastasis. Oncol. Rep. 2015, 33, 2737–2745. [Google Scholar] [CrossRef] [Green Version]
- Karp, D.; Ferrante, K.; Tensfeldt, T.; Thurer, R.; LoCicero, J.; Huberman, M.; Wirth, F.; Hellman, R.; Poulin, P.; Silberman, S.; et al. A phase I dose escalation study of epidermal growth factor receptor (EGFR) tyrosine kinase (TK) inhibitor CP-358,774 in patients (pts) with advanced solid tumors. Lung Cancer 2000, 29, 65. [Google Scholar] [CrossRef]
- Baselga, J.; Rischin, D.; Ranson, M.; Calvert, H.; Raymond, E.; Kieback, D.; Kaye, S.; Gianni, L.; Harris, A.L.; Bjork, T.; et al. Phase I Safety, Pharmacokinetic, and Pharmacodynamic Trial of ZD1839, a Selective Oral Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, in Patients With Five Selected Solid Tumor Types. J. Clin. Oncol. 2002, 20, 4292–4302. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marti, A.; Navarro, A.; Felip, E. Epidermal growth factor receptor first generation tyrosine-kinase inhibitors. Transl. Lung Cancer Res. 2019, 8, S235–S246. [Google Scholar] [CrossRef]
- Lacal, P.M.; Graziani, G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; Rosen, L.S.; Sawyer, M.; Wilding, G.; Noberasco, C.; Jayson, G.; Rustin, G.; McArthur, G.; Velasquez, L.; Galbraith, S. A phase I study of BMS-582664 (brivanib alaninate), an oral dual inhibitor of VEGFR and FGFR tyrosine kinases, in patients (pts) with advanced/metastatic solid tumors: Safety, pharmacokinetic (PK), and pharmacodynamic (PD) findings. J. Clin. Oncol. 2007, 25, 3559. [Google Scholar] [CrossRef]
- Laird, A.D.; Vajkoczy, P.; Shawver, L.K.; Thurnher, A.; Liang, C.; Mohammadi, M.; Schlessinger, J.; Ullrich, A.; Hubbard, S.R.; A Blake, R.; et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000, 60, 4152–4160. [Google Scholar]
- Creedon, H.; Brunton, V.G. Src Kinase Inhibitors: Promising Cancer Therapeutics? Crit. Rev. Oncog. 2012, 17, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.C.; Logothetis, C.J. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.I.; Zhang, J.; Phillips, K.A.; Araujo, J.C.; Najjar, A.M.; Volgin, A.Y.; Gelovani, J.G.; Kim, S.-J.; Wang, Z.; Gallick, G.E. Targeting Src Family Kinases Inhibits Growth and Lymph Node Metastases of Prostate Cancer in an Orthotopic Nude Mouse Model. Cancer Res. 2008, 68, 3323–3333. [Google Scholar] [CrossRef] [Green Version]
- Ammer, A.G.; Kelley, L.C.; Hayes, K.E.; Evans, J.V.; Lopez-Skinner, L.A.; Martin, K.H.; Frederick, B.; Rothschild, B.L.; Raben, D.; Elvin, P.; et al. Saracatinib Impairs Head and Neck Squamous Cell Carcinoma Invasion by Disrupting Invadopodia Function. J. Cancer Sci. Ther. 2009, 1, 052–061. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Friedl, P. Mapping proteolytic cancer cell-extracellular matrix interfaces. Clin. Exp. Metastasis 2008, 26, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Tube Travel: The Role of Proteases in Individual and Collective Cancer Cell Invasion. Cancer Res. 2008, 68, 7247–7249. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Christofori, G. Mechanisms of Motility in Metastasizing Cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-W.; Lin, C.-W.; Yang, W.-E.; Yang, S. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919864247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towle, M.J.; Lee, A.; Maduakor, E.C.; E Schwartz, C.; Bridges, A.J.; A Littlefield, B. Inhibition of urokinase by 4-substituted benzo[b]thiophene-2-carboxamidines: An important new class of selective synthetic urokinase inhibitor. Cancer Res. 1993, 53, 2553–2559. [Google Scholar]
- Meyer, J.E.; Brocks, C.; Graefe, H.; Mala, C.; Thäns, N.; Bürgle, M.; Rempel, A.; Rotter, N.; Wollenberg, B.; Lang, S. The Oral Serine Protease Inhibitor WX-671 – First Experience in Patients with Advanced Head and Neck Carcinoma. Breast Care 2008, 3, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.; Harbeck, N.; Brünner, N.; Jänicke, F.; Meisner, C.; Mühlenweg, B.; Jansen, H.; Dorn, J.; Nitz, U.; Kantelhardt, E.J.; et al. Cancer therapy trials employing level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev. Mol. Diagn. 2011, 11, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Dai, J.; Keller, J.M.; Mizokami, A.; Xia, S.; Keller, E.T. Notch Pathway Inhibition Using PF-03084014, a γ-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Clin. Cancer Res. 2015, 21, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of Selective -Secretase Inhibitor PF-03084014 for Its Antitumor Efficacy and Gastrointestinal Safety to Guide Optimal Clinical Trial Design. Mol. Cancer Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2012, 11, 1565–1575. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST Domain Mutations in Notch Receptors Comprise an Oncogenic Driver Segment in Triple-Negative Breast Cancer Sensitive to a -Secretase Inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef] [Green Version]
- Kummar, S.; Coyne, G.O.; Do, K.T.; Turkbey, B.; Meltzer, P.S.; Polley, E.; Choyke, P.L.; Meehan, R.; Vilimas, R.; Horneffer, Y.; et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis). J. Clin. Oncol. 2017, 35, 1561–1569. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, G.; Krishnan, M.; Ha, E.; Chun, K.-S. Selective Wnt/β-catenin Small-molecule Inhibitor CWP232228 Impairs Tumor Growth of Colon Cancer. Anticancer. Res. 2019, 39, 3661–3667. [Google Scholar] [CrossRef]
- Cortes, J.E.; Faderl, S.; Pagel, J.; Jung, C.W.; Yoon, S.-S.; Koh, Y.; Pardanani, A.D.; Hauptschein, R.S.; Lee, K.-J.; Lee, J.-H. Phase 1 study of CWP232291 in relapsed/refractory acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). J. Clin. Oncol. 2015, 33, 7044. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W.; Stashko, M.A.; DeRyckere, D.; Cummings, C.T.; Hunter, D.; Yang, C.; Jayakody, C.N.; Cheng, N.; Simpson, C.; et al. UNC1062, a new and potent Mer inhibitor. Eur. J. Med. Chem. 2013, 65, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a Potent and Orally Bioavailable MER/FLT3 Dual Inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, K.; Torella, R.; Foglieni, C.; Bugatti, A.; Tomaselli, S.; Zetta, L.; Presta, M.; Rusnati, M.; Taraboletti, G.; Colombo, G.; et al. Direct and allosteric inhibition of the FGF2/HSPGs/FGFR1 ternary complex formation by an antiangiogenic, thrombospondin-1-mimic small molecule. PLoS ONE 2012, 7, e36990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, G.; Margosio, B.; Ragona, L.; Neves, M.; Bonifacio, S.; Annis, D.S.; Stravalaci, M.; Tomaselli, S.; Giavazzi, R.; Rusnati, M.; et al. Non-peptidic thrombospondin-1 mimics as fibroblast growth factor-2 inhibitors: An integrated strategy for the development of new antiangiogenic compounds. J. Biol. Chem. 2010, 285, 8733–8742. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Tsujino, Y.; Moriguchi, K.; Tatematsu, M.; Ushijima, T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2’-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci. 2006, 97, 64–71. [Google Scholar] [CrossRef]
- Avan, A.; Crea, F.; Paolicchi, E.; Funel, N.; Galvani, E.; E Marquez, V.; Honeywell, R.J.; Danesi, R.; Peters, G.J.; Elisa, G. Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-deazaneplanocin A with gemcitabine in pancreatic cancer cells. Mol. Cancer Ther. 2012, 11, 1735–1746. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.D.; Liu, Y.; Nagji, A.S.; Theodosakis, N.; Jones, D.R. Combined proteasome and histone deacetylase inhibition attenuates epithelial-mesenchymal transition through E-cadherin in esophageal cancer cells. J. Thorac. Cardiovasc. Surg. 2010, 139, 1224–1232. [Google Scholar] [CrossRef] [Green Version]
- Labbozzetta, M.; Poma, P.; Vivona, N.; Gulino, A.; D’Alessandro, N.; Notarbartolo, M. Epigenetic changes and nuclear factor-κB activation, but not microRNA-224, downregulate Raf-1 kinase inhibitor protein in triple-negative breast cancer SUM 159 cells. Oncol. Lett. 2015, 10, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.-C.; Collins, J.; Marino, N.; Steeg, P. The Nm23-H1 metastasis suppressor as a translational target. Eur. J. Cancer 2010, 46, 1278–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujii, M.; Kawano, S.; Dubois, R.N. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc. Natl. Acad. Sci. USA 1997, 94, 3336–3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, L.; Ward, P.M. Cell detachment and metastasis. Cancer Metastasis Rev. 1983, 2, 111–127. [Google Scholar] [CrossRef]
- Li, D.-M.; Feng, Y.-M. Signaling mechanism of cell adhesion molecules in breast cancer metastasis: Potential therapeutic targets. Breast Cancer Res. Treat. 2011, 128, 7–21. [Google Scholar] [CrossRef]
- Albelda, S.M. Role of integrins and other cell adhesion molecules in tumor progression and metastasis. Lab. Investig. 1993, 68, 4–17. [Google Scholar] [PubMed]
- Aplin, A.E.; Howe, A.; Alahari, S.K.; Juliano, R.L. Signal transduction and signal modulation by cell adhesion receptors: The role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol. Rev. 1998, 50, 197–263. [Google Scholar]
- Cavallaro, U.; Christofori, G. Cell adhesion in tumor invasion and metastasis: Loss of the glue is not enough. Biochim. Biophys. Acta (BBA) Bioenerg. 2001, 1552, 39–45. [Google Scholar] [CrossRef]
- Alizadeh, A.M.; Shiri, S.; Farsinejad, S. Metastasis review: From bench to bedside. Tumor Boil. 2014, 35, 8483–8523. [Google Scholar] [CrossRef]
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous Expression of N-Cadherin in Breast Cancer Cells Induces Cell Migration, Invasion, and Metastasis. J. Cell Boil. 2000, 148, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-Cadherin Promotes Motility in Human Breast Cancer Cells Regardless of Their E-Cadherin Expression. J. Cell Boil. 1999, 147, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, H.; Pietilä, M.; Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 2016, 115, 1017–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, S.; Kyprianou, N. Targeting anoikis resistance in prostate cancer metastasis. Mol. Asp. Med. 2010, 31, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- I Deryugina, E.; A Bourdon, M.; Luo, G.X.; A Reisfeld, R.; Strongin, A. Matrix metalloproteinase-2 activation modulates glioma cell migration. J. Cell Sci. 1997, 110, 110. [Google Scholar]
- Brooks, P.C.; Stromblad, S.; Sanders, L.C.; Von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; A Cheresh, D. Localization of Matrix Metalloproteinase MMP-2 to the Surface of Invasive Cells by Interaction with Integrin αvβ3. Cell 1996, 85, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Charles, J.M. Matrix metalloproteinases (MMPs) in health and disease: An overview. Front. Biosci. 2006, 11, 1696. [Google Scholar] [CrossRef]
- John, A.; Tuszynski, G. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol. Oncol. Res. 2001, 7, 14–23. [Google Scholar] [CrossRef]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Boil. 2015, 44, 200–206. [Google Scholar] [CrossRef]
- Frisch, S.M.; Screaton, R. Anoikis mechanisms. Curr. Opin. Cell Boil. 2001, 13, 555–562. [Google Scholar] [CrossRef]
- Gilmore, A.P. Anoikis. Cell Death Differ 2005, 12 (Suppl. 2), 1473–1477. [Google Scholar] [CrossRef]
- Duxbury, M.; Ito, H.; Zinner, M.; Ashley, S.; Whang, E. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery 2004, 135, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-N.; Koo, K.H.; Sung, J.Y.; Yun, U.-J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Boil. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, C.A.; Fox, J.; Majkut, J.; Fiedler, G.E.; Roberts, J.; Humphreys, L.; Boffey, R.J.; Perrior, T.R.; Harrison, T.; Longley, D.B. Abstract 382: Development and pre-clinical assessment of a first-in-class small molecule inhibitor of FLIP. Exp. Mol. Ther. 2019, 79, 382. [Google Scholar] [CrossRef]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adhes. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Rammal, H.; Saby, C.; Magnien, K.; Van Gulick, L.; Garnotel, R.; Buache, E.; El Btaouri, H.; Jeannesson, P.; Morjani, H. Discoidin Domain Receptors: Potential Actors and Targets in Cancer. Front. Pharmacol. 2016, 7, 1321. [Google Scholar] [CrossRef]
- Sieg, D.J.; Hauck, C.R.; Schlaepfer, D.D. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999, 112, 2677–2691. [Google Scholar]
- McLean, G.W.; Carragher, N.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Serrels, B.; Sandilands, E.; Serrels, A.; Baillie, G.; Houslay, M.; Brunton, V.G.; Canel, M.; Machesky, L.M.; Anderson, K.; Frame, M. A Complex between FAK, RACK1, and PDE4D5 Controls Spreading Initiation and Cancer Cell Polarity. Curr. Boil. 2010, 20, 1086–1092. [Google Scholar] [CrossRef] [Green Version]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Boil. 2004, 265, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Durand-Onaylı, V.; Haslauer, T.; Härzschel, A.; Hartmann, T.N. Rac GTPases in Hematological Malignancies. Int. J. Mol. Sci. 2018, 19, 4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009, 28, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef]
- Wilkinson, S.; Paterson, H.F.; Marshall, C.J. Cdc42–MRCK and Rho–ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nature 2005, 7, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Nicholas, N.S.; Wells, C.M. Role of p-21-Activated Kinases in Cancer Progression. Int. Rev. Cell Mol. Biol. 2014, 309, 347–387. [Google Scholar] [CrossRef]
- Schoumacher, M.; El-Marjou, F.; Laé, M.; Kambou, N.; Louvard, D.; Robine, S.; Vignjevic, D. Conditional expression of fascin increases tumor progression in a mouse model of intestinal cancer. Eur. J. Cell Boil. 2014, 93, 388–395. [Google Scholar] [CrossRef]
- Vignjevic, D.; Schoumacher, M.; Gavert, N.; Janssen, K.-P.; Jih, G.; Laé, M.; Louvard, D.; Ben-Ze’Ev, A.; Robine, S. Fascin, a Novel Target of β-Catenin-TCF Signaling, Is Expressed at the Invasive Front of Human Colon Cancer. Cancer Res. 2007, 67, 6844–6853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claessens, M.M.A.E.; Bathe, M.; Frey, E.; Bausch, A.R. Actin-binding proteins sensitively mediate F-actin bundle stiffness. Nat. Mater. 2006, 5, 748–753. [Google Scholar] [CrossRef]
- Jawhari, A.U.; Buda, A.; Jenkins, M.; Shehzad, K.; Sarraf, C.; Noda, M.; Farthing, M.J.G.; Pignatelli, M.; Adams, J.C. Fascin, an Actin-Bundling Protein, Modulates Colonic Epithelial Cell Invasiveness and Differentiation in Vitro. Am. J. Pathol. 2003, 162, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Morton, J.P.; Ma, Y.; Karim, S.A.; Zhou, Y.; Faller, W.; Woodham, E.F.; Morris, H.T.; Stevenson, R.P.; Juin, A.; et al. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology 2014, 146, 1386–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, V.; Lewis, S.; Adams, J.C.; Martin, R.M. Association of fascin-1 with mortality, disease progression and metastasis in carcinomas: A systematic review and meta-analysis. BMC Med. 2013, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, U.; Arlt, F.; Walther, W.; Smith, J.; Waldman, T.; Harris, E.D.; Mertins, S.D.; Heizmann, C.W.; Allard, D.; Birchmeier, W.; et al. The Metastasis-Associated Gene S100A4 Is a Novel Target of β-catenin/T-cell Factor Signaling in Colon Cancer. Gastroenterology 2006, 131, 1486–1500. [Google Scholar] [CrossRef]
- Takenaga, K.; Nakamura, Y.; Sakiyama, S.; Hasegawa, Y.; Sato, K.; Endo, H. Binding of pEL98 protein, an S100-related calcium-binding protein, to nonmuscle tropomyosin. J. Cell Boil. 1994, 124, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Roy-Luzarraga, M.; Hodivala-Dilke, K. Molecular Pathways: Endothelial Cell FAK-A Target for Cancer Treatment. Clin. Cancer Res. 2016, 22, 3718–3724. [Google Scholar] [CrossRef] [Green Version]
- Mak, G.; Soria, J.-C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- Fukami, S.; Tomioka, D.; Murakami, Y.; Honda, T.; Hatakeyama, S. Pharmacological profiling of a dual FAK/IGF-1R kinase inhibitor TAE226 in cellular and in vivo tumor models. BMC Res. Notes 2019, 12, 347. [Google Scholar] [CrossRef]
- Carboni, S.S.C.M.; Lima, N.A.R.; Pinheiro, N.M.; Murta, B.M.T.; Crema, V.O. HA-1077 inhibits cell migration/invasion of oral squamous cell carcinoma. Anti Cancer Drugs 2015, 26, 1–930. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Zong, Z.; Tian, D. The Rho kinase inhibitor fasudil inhibits the migratory behaviour of 95-D lung carcinoma cells. Biomed. Pharmacother. 2010, 64, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Biroc, S.L.; Li, W.-W.; Alicke, B.; Xuan, J.-A.; Pagila, R.; Ohashi, Y.; Okada, T.; Kamata, Y.; Dinter, H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol. Cancer Ther. 2006, 5, 2158–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutting, S.; Heidenreich, J.; Cherpokova, D.; Amin, E.; Zhang, S.-C.; Ahmadian, M.R.; Brakebusch, C.; Nieswandt, B. Critical off-target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J. Thromb. Haemost. 2015, 13, 827–838. [Google Scholar] [CrossRef]
- Castoria, G.; D’Amato, L.; Ciociola, A.; Giovannelli, P.; Giraldi, T.; Sepe, L.; Paolella, G.; Barone, M.V.; Migliaccio, A.; Auricchio, F. Androgen-Induced Cell Migration: Role of Androgen Receptor/Filamin A Association. PLoS ONE 2011, 6, e17218. [Google Scholar] [CrossRef] [Green Version]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and Mechanism of Action of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. J. Boil. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.-Q.; Chen, Q.-Y.; Jiao, D.-M.; Yao, Q.-H.; Wang, Y.-Y.; Hu, H.-Z.; Wu, Y.-Q.; Song, J.; Yan, J. Silencing of Rac1 modifies lung cancer cell migration, invasion and actin cytoskeleton rearrangements and enhances chemosensitivity to antitumor drugs. Int. J. Mol. Med. 2011, 28, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Van Eygen, S.; Krysko, D.V.; Vandenabeele, P.; Nys, K.; Rillaerts, K.; Garg, A.D.; Verfaillie, T.; Agostinis, P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014, 5, e1127. [Google Scholar] [CrossRef] [Green Version]
- Arnst, J.L.; Hein, A.L.; Taylor, M.A.; Palermo, N.Y.; Contreras, J.I.; Sonawane, Y.A.; Wahl, A.O.; Ouellette, M.M.; Natarajan, A.; Yan, Y. Discovery and characterization of small molecule Rac1 inhibitors. Oncotarget 2017, 8, 34586–34600. [Google Scholar] [CrossRef] [Green Version]
- Rane, C.K.; Minden, A. P21 activated kinase signaling in cancer. Semin. Cancer Boil. 2019, 54, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.E.; Myers, C.; Chernoff, J.; Peterson, J. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Boil. 2008, 15, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Bi, S.; Hou, J.; Zhao, Z.; Wang, C.; Xie, S. Targeting p21-activated kinase 1 inhibits growth and metastasis via Raf1/MEK1/ERK signaling in esophageal squamous cell carcinoma cells. Cell Commun. Signal. 2019, 17, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Suzuki, K. Membrane transport of WAVE2 and lamellipodia formation require Pak1 that mediates phosphorylation and recruitment of stathmin/Op18 to Pak1–WAVE2–kinesin complex. Cell. Signal. 2009, 21, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Huang, J.; Liu, B.; Xing, B.; Bordeleau, F.; Reinhart-King, C.A.; Li, W.; Zhang, J.J.; Huang, X.-Y. Improving fascin inhibitors to block tumor cell migration and metastasis. Mol. Oncol. 2016, 10, 966–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.-K.; Han, S.; Xing, B.; Huang, J.; Liu, B.; Bordeleau, F.; Reinhart-King, C.A.; Zhang, J.J.; Huang, X.-Y. Targeted inhibition of fascin function blocks tumour invasion and metastatic colonization. Nat. Commun. 2015, 6, 7465. [Google Scholar] [CrossRef] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Raab-Westphal, S.; Marshall, J.; Goodman, S.L. Integrins as Therapeutic Targets: Successes and Cancers. Cancers 2017, 9, 110. [Google Scholar] [CrossRef]
- Goubran, H.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of Tumor Growth and Metastasis: The Role of Tumor Microenvironment. Cancer Growth Metastasis 2014, 7, CGM–S11285. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Boil. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2013, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J.S. Spatial regulation of tumor cell protrusions by RhoC. Cell Adhes. Migr. 2014, 8, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, M.A.; Deryugina, E.I.; Niessen, S.; Cravatt, B.F.; Quigley, J.P. Activity-based Protein Profiling Implicates Urokinase Activation as a Key Step in Human Fibrosarcoma Intravasation. J. Boil. Chem. 2006, 281, 15997–16005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, L.A.S.; Jensen-Taubman, S.; Stetler-Stevenson, W.G. Matrix Metalloproteinases. Am. J. Pathol. 2012, 181, 1895–1899. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Wyckoff, J.; Liang, F.; Sidani, M.; Violini, S.; Tsai, K.-L.; Zhang, Z.Y.; Sahai, E.; Condeelis, J.S.; E Segall, J. Epidermal Growth Factor Receptor Overexpression Results in Increased Tumor Cell MotilityIn vivoCoordinately with Enhanced Intravasation and Metastasis. Cancer Res. 2006, 66, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325. [Google Scholar] [CrossRef]
- Kedrin, D.; Wyckoff, J.; Boimel, P.; Coniglio, S.J.; Hynes, N.E.; Arteaga, C.L.; E Segall, J. ERBB1 and ERBB2 have distinct functions in tumor cell invasion and intravasation. Clin. Cancer Res. 2009, 15, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- McMahon, B.J.; Kwaan, H.C. Components of the Plasminogen-Plasmin System as Biologic Markers for Cancer. Adv. Exp. Med. Biol. 2015, 867, 145–156. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nature 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Naumov, G.N.; Townson, J.L.; Macdonald, I.C.; Wilson, S.M.; Bramwell, V.H.; Groom, A.C.; Chambers, A. Ineffectiveness of Doxorubicin Treatment on Solitary Dormant Mammary Carcinoma Cells or Late-developing Metastases. Breast Cancer Res. Treat. 2003, 82, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Van Tetering, G.; Van Diest, P.; Verlaan, I.; Van Der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 Is Required for Notch1 Site 2 Cleavage*. J. Boil. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, M.J.; Selkoe, D.J. The Notch Ligands, Jagged and Delta, Are Sequentially Processed by α-Secretase and Presenilin/γ-Secretase and Release Signaling Fragments. J. Boil. Chem. 2003, 278, 34427–34437. [Google Scholar] [CrossRef] [Green Version]
- Giuli, M.V.; Giuliani, E.; Screpanti, I.; Bellavia, D.; Checquolo, S. Notch Signaling Activation as a Hallmark for Triple-Negative Breast Cancer Subtype. J. Oncol. 2019, 2019, 8707053-15. [Google Scholar] [CrossRef]
- Wu, C.X.; Xu, A.; Zhang, C.C.; Olson, P.; Chen, L.; Lee, T.K.W.; Cheung, T.T.; Lo, C.-M.; Wang, X.Q. Notch Inhibitor PF-03084014 Inhibits Hepatocellular Carcinoma Growth and Metastasis via Suppression of Cancer Stemness due to Reduced Activation of Notch1–Stat3. Mol. Cancer Ther. 2017, 16, 1531–1543. [Google Scholar] [CrossRef] [Green Version]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Mollen, E.W.J.; Ient, J.; Tjan-Heijnen, V.C.G.; Boersma, L.J.; Miele, L.; Smidt, M.L.; Vooijs, M. Moving Breast Cancer Therapy up a Notch. Front. Oncol. 2018, 8, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, C.; Safarova, A.; Smith, M.; Chung, R.; Bruyneel, A.A.N.; Gomez-Galeno, J.; Oswald, F.; Larson, C.J.; Cashman, J.R.; Ruiz-Lozano, P.; et al. Disruption of NOTCH signaling by a small molecule inhibitor of the transcription factor RBPJ. Sci. Rep. 2019, 9, 10811–10819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozawa, Y.; Havens, A.M.; Pienta, K.J.; Taichman, R.S. The bone marrow niche: Habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia 2008, 22, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altundağ, M.K.; Altundag, O.; Elkiran, E.T.; Cengiz, M.; Ozişik, Y. Addition of granulocyte-colony stimulating factor (G-CSF) to adjuvant treatment may increase survival in patients with operable breast cancer: Interaction of G-CSF with dormant micrometastatic breast cancer cells. Med. Hypotheses 2004, 63, 56–58. [Google Scholar] [CrossRef]
- Saito, Y.; Uchida, N.; Tanaka, S.; Suzuki, N.; Tomizawa-Murasawa, M.; Sone, A.; Najima, Y.; Takagi, S.; Aoki, Y.; Wake, A.; et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 2010, 28, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Tran, F.H.; Zheng, J.J. Modulating the wnt signaling pathway with small molecules. Protein Sci. 2017, 26, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Seto, K.; Sakabe, T.; Itaba, N.; Azumi, J.; Oka, H.; Morimoto, M.; Umekita, Y.; Shiota, G. A Novel Small-molecule WNT Inhibitor, IC-2, Has the Potential to Suppress Liver Cancer Stem Cells. Anticancer. Res. 2017, 37, 3569–3579. [Google Scholar] [CrossRef] [Green Version]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Boil. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Ma, Z.; Cheng, Y.; Hu, W.; Deng, C.; Jiang, S.; Li, T.; Chen, F.; Yang, Y. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol. Cancer 2018, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Ma, Z.; Hu, W.; Wang, N.; Gong, B.; Fan, C.; Jiang, S.; Li, T.; Gao, J.; Yang, Y. Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 2017, 8, e2700. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, M.; Kim, S. Gas6 induces cancer cell migration and epithelial–mesenchymal transition through upregulation of MAPK and Slug. Biochem. Biophys. Res. Commun. 2013, 434, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL Axis Regulates Prostate Cancer Invasion, Proliferation, and Survival in the Bone Marrow Niche. Neoplasia 2010, 12, 116–IN4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. 2011, 17, 6130–6139. [Google Scholar] [CrossRef] [Green Version]
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galoway, A.; Rifkin, D.B.; Mignatti, P. Fibroblast Growth Factor-2 (FGF-2) Induces Vascular Endothelial Growth Factor (VEGF) Expression in the Endothelial Cells of Forming Capillaries: An Autocrine Mechanism Contributing to Angiogenesis. J. Cell Boil. 1998, 141, 1659–1673. [Google Scholar] [CrossRef]
- Zhao, M.; Li, Z.; Qu, H. An evidence-based knowledgebase of metastasis suppressors to identify key pathways relevant to cancer metastasis. Sci. Rep. 2015, 5, 15478. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Steeg, P.S. Metastasis suppressors: Functional pathways. Lab. Investig. 2017, 98, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a Novel Gene Associated With Low Tumor Metastatic Potential. J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef]
- Steeg, P.S.; Bevilacqua, G.; Sobel, M.E.; Liotta, L.A. Identification and Characterization of Differentially Expressed Genes in Tumor Metastasis: The nm23 Gene. Boundaries Promot. Progress. Carcinog. 1991, 57, 355–361. [Google Scholar] [CrossRef]
- A Stahl, J.; Leone, A.; Rosengard, A.M.; Porter, L.; King, C.R.; Steeg, P.S. Identification of a second human nm23 gene, nm23-H2. Cancer Res. 1991, 51, 445–449. [Google Scholar]
- Hartsough, M.T. Nm23-H1 Metastasis Suppressor Phosphorylation of Kinase Suppressor of Ras via a Histidine Protein Kinase Pathway. J. Boil. Chem. 2002, 277, 32389–32399. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Smith, P.C.; Zhang, L.; Rubin, M.A.; Dunn, R.L.; Yao, Z.; Keller, E.T. Effects of Raf Kinase Inhibitor Protein Expression on Suppression of Prostate Cancer Metastasis. J. Natl. Cancer Inst. 2003, 95, 878–889. [Google Scholar] [CrossRef]
- Yoshida, B.A.; Dubauskas, Z.; Chekmareva, M.A.; Christiano, T.R.; Stadler, W.M.; Rinker-Schaeffer, C.W. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 17. Cancer Res. 1999, 59, 5483–5487. [Google Scholar]
- Nakano, T.; Tani, M.; Ishibashi, Y.; Kimura, K.; Park, Y.; Imaizumi, N.; Tsuda, H.; Aoyagi, K.; Sasaki, H.; Ohwada, S.; et al. Biological properties and gene expression associated with metastatic potential of human osteosarcoma. Clin. Exp. Metastasis 2003, 20, 665–674. [Google Scholar] [CrossRef]
- Griend, D.V.; Kocherginsky, M.; Hickson, J.A.; Stadler, W.M.; Lin, A.; Rinker-Schaeffer, C. Suppression of Metastatic Colonization by the Context-Dependent Activation of the c-Jun NH2-Terminal Kinase Kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005, 65, 10984–10991. [Google Scholar] [CrossRef] [Green Version]
- Ohtaki, T.; Shintani, Y.; Matsumoto, H.; Hori, A.; Kanehashi, K.; Terao, Y.; Kumano, S.; Takatsu, Y.; Masuda, Y.; Ishibashi, Y.; et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature 2001, 411, 613–617. [Google Scholar] [CrossRef]
- Gelman, I. Suppression of tumor and metastasis progression through the scaffolding functions of SSeCKS/Gravin/AKAP12. Cancer Metastasis Rev. 2012, 31, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, M.; Ihara, Y.; Matsuzawa, Y.; Taniguchi, N. Aberrant Glycosylation of E-cadherin Enhances Cell-Cell Binding to Suppress Metastasis. J. Boil. Chem. 1996, 271, 13811–13815. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.C.; Lou, W.; Sleeman, J.P.; Isaacs, J.T. Metastasis suppression by the standard CD44 isoform does not require the binding of prostate cancer cells to hyaluronate. Cancer Res. 1998, 58, 2350–2352. [Google Scholar]
- Jackson, P.; Marreiros, A.; Russell, P.J. KAI1 tetraspanin and metastasis suppressor. Int. J. Biochem. Cell Boil. 2005, 37, 530–534. [Google Scholar] [CrossRef]
- Durkin, M.E.; Yuan, B.-Z.; Zhou, X.; Zimonjic, A.B.; Lowy, U.R.; Thorgeirsson, S.S.; Popescu, N.C. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J. Cell. Mol. Med. 2007, 11, 1185–1207. [Google Scholar] [CrossRef] [Green Version]
- Said, N.; Theodorescu, D. Pathways of metastasis suppression in bladder cancer. Cancer Metastasis Rev. 2009, 28, 327–333. [Google Scholar] [CrossRef]
- Kim, T.Y.; Vigil, D.; Der, C.J.; Juliano, R.L. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev. 2009, 28, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Seraj, M.J.; Samant, R.S.; Verderame, M.F.; Welch, D.R. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000, 60, 2764–2769. [Google Scholar]
- Meehan, W.J.; Samant, R.S.; Hopper, J.E.; Carrozza, M.J.; Shevde, L.A.; Workman, J.L.; Eckert, K.A.; Verderame, M.F.; Welch, D.R. Breast Cancer Metastasis Suppressor 1 (BRMS1) Forms Complexes with Retinoblastoma-binding Protein 1 (RBP1) and the mSin3 Histone Deacetylase Complex and Represses Transcription. J. Boil. Chem. 2003, 279, 1562–1569. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Radisky, D.C.; Yang, D.; Xu, R.; Radisky, E.S.; Bissell, M.J.; Bishop, J.M. MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat. Cell Biol. 2012, 14, 567–574. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Szmulewitz, R.Z.; Rinker-Schaeffer, C.W. Metastasis-suppressor genes in clinical practice: Lost in translation? Nat. Rev. Clin. Oncol. 2011, 8, 333–342. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Wang, X.; Ci, H.; Zhou, L.; Zhu, B.; Wu, S.; Wang, D. Evaluation of the correlation of vasculogenic mimicry, Notch4, DLL4, and KAI1/CD82 in the prediction of metastasis and prognosis in non-small cell lung cancer. Med. 2018, 97, e13817. [Google Scholar] [CrossRef]
- Ferguson, A.W.; Flatow, U.; Macdonald, N.J.; Larminat, F.; A Bohr, V.; Steeg, P.S. Increased sensitivity to cisplatin by nm23-transfected tumor cell lines. Cancer Res. 1996, 56, 2931–2935. [Google Scholar]
- Scambia, G.; Ferrandina, G.; Marone, M.; Panici, P.B.; Giannitelli, C.; Piantelli, M.; Leone, A.; Mancuso, S. nm23 in ovarian cancer: Correlation with clinical outcome and other clinicopathologic and biochemical prognostic parameters. J. Clin. Oncol. 1996, 14, 334–342. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Kuo, C.-C.; Wu, C.-C.; Hsiao, C.-W.; Hu, J.-M.; Hsu, C.-H.; Chou, Y.-C.; Shih, Y.-L.; Lin, Y.-W. NKX6.1 hypermethylation predicts the outcome of stage II colorectal cancer patients undergoing chemotherapy. Genes Chromosom. Cancer 2018, 57, 268–277. [Google Scholar] [CrossRef]
- Patel, S.A.; Vanharanta, S. Epigenetic determinants of metastasis. Mol. Oncol. 2017, 11, 79–96. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Baba, T.; Mandai, M.; Matsumura, N.; Hamanishi, J.; Kharma, B.; Kondoh, E.; Yoshioka, Y.; Oishi, S.; Fujii, N.; et al. GPR54 Is a Target for Suppression of Metastasis in Endometrial Cancer. Mol. Cancer Ther. 2011, 10, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Hartsough, M.T.; E Clare, S.; Mair, M.; Elkahloun, A.G.; Sgroi, D.; Osborne, C.; Clark, G.; Steeg, P.S. Elevation of breast carcinoma Nm23-H1 metastasis suppressor gene expression and reduced motility by DNA methylation inhibition. Cancer Res. 2001, 61, 2320–2327. [Google Scholar]
- Kong, B.; Lv, Z.-D.; Wang, Y.; Jin, L.-Y.; Ding, L.; Yang, Z.-C. Down-regulation of BRMS1 by DNA hypermethylation and its association with metastatic progression in triple-negative breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11076–11083. [Google Scholar]
- Au, S.L.-K.; Wong, C.C.-L.; Lee, J.M.-F.; Wong, C.-M.; Ng, I.O.-L. EZH2-Mediated H3K27me3 Is Involved in Epigenetic Repression of Deleted in Liver Cancer 1 in Human Cancers. PLoS ONE 2013, 8, e68226. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Baritaki, S.; Marathe, H.; Feng, J.; Park, S.; Beach, S.; Bazeley, P.S.; Beshir, A.B.; Fenteany, G.; Mehra, R.; et al. Polycomb Protein EZH2 Regulates Tumor Invasion via the Transcriptional Repression of the Metastasis Suppressor RKIP in Breast and Prostate Cancer. Cancer Res. 2012, 72, 3091–3104. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Farquhar, K.S.; Yun, J.; Frankenberger, C.A.; Bevilacqua, E.; Yeung, K.; Kim, E.-J.; Balazsi, G.; Rosner, M.R. Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc. Natl. Acad. Sci. USA 2014, 111, E364–E373. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotoh, K.; Asaoka, T.; Wada, H.; et al. A Histone Deacetylase Inhibitor Suppresses Epithelial-Mesenchymal Transition and Attenuates Chemoresistance in Biliary Tract Cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef] [Green Version]
- Bu, Y.; Gelman, I.H. v-Src-mediated Down-regulation of SSeCKS Metastasis Suppressor Gene Promoter by the Recruitment of HDAC1 into a USF1-Sp1-Sp3 Complex. J. Boil. Chem. 2007, 282, 26725–26739. [Google Scholar] [CrossRef] [Green Version]
- Yasui, W.; Oue, N.; Ono, S.; Mitani, Y.; Ito, R.; Nakayama, H. Histone acetylation and gastrointestinal carcinogenesis. Ann. N. Y. Acad. Sci. 2003, 983, 220–231. [Google Scholar] [CrossRef]
- Li, G.-F.; Qian, T.-L.; Li, G.-S.; Yang, C.-X.; Qin, M.; Huang, J.; Sun, M.; Han, Y.-Q. Sodium valproate inhibits MDA-MB-231 breast cancer cell migration by upregulating NM23H1 expression. Genet. Mol. Res. 2012, 11, 77–86. [Google Scholar] [CrossRef]
- Chen, M.; Nie, J.; Liu, Y.; Li, X.; Zhang, Y.; Brock, M.V.; Feng, K.; Wu, Z.; Li, X.; Shi, L.; et al. Phase Ib/II study of safety and efficacy of low-dose decitabine-primed chemoimmunotherapy in patients with drug-resistant relapsed/refractory alimentary tract cancer. Int. J. Cancer 2018, 143, 1530–1540. [Google Scholar] [CrossRef]
- Goncalves, P.H.; Heilbrun, L.K.; Barrett, M.T.; Kummar, S.; Hansen, A.R.; Siu, L.L.; Piekarz, R.L.; Sukari, A.W.; Chao, J.; Pilat, M.J.; et al. A phase 2 study of vorinostat in locally advanced, recurrent, or metastatic adenoid cystic carcinoma. Oncotarget 2017, 8, 32918–32929. [Google Scholar] [CrossRef] [Green Version]
- Caponigro, F.; Di Gennaro, E.; Ionna, F.; Longo, F.; Aversa, C.; Pavone, E.; Maglione, M.G.; Di Marzo, M.; Muto, P.; Cavalcanti, E.; et al. Phase II clinical study of valproic acid plus cisplatin and cetuximab in recurrent and/or metastatic squamous cell carcinoma of Head and Neck-V-CHANCE trial. BMC Cancer 2016, 16, 918. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Xu, Z.; Li, H. NSAIDs Use and Reduced Metastasis in Cancer Patients: Results from a meta-analysis. Sci. Rep. 2017, 7, 1875. [Google Scholar] [CrossRef]
- Lundholm, K.; Axelsson, H.; Lönnroth, C.; Andersson, M. Mechanisms behind COX-1 and COX-2 inhibition of tumor growth in vivo. Int. J. Oncol. 2010, 37, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-G.; Huang, J.-A.; Yang, Y.-N.; Huang, H.; Luo, H.-S.; Yu, J.-P.; Meier, J.J.; Schrader, H.; Bastian, A.; Schmidt, W.E.; et al. The effects of acetylsalicylic acid on proliferation, apoptosis, and invasion of cyclooxygenase-2 negative colon cancer cells. Eur. J. Clin. Investig. 2002, 32, 838–846. [Google Scholar] [CrossRef]
- Natarajan, K.; Mori, N.; Artemov, D.; Bhujwalla, Z.M. Exposure of Human Breast Cancer Cells to the Anti-inflammatory Agent Indomethacin Alters Choline Phospholipid Metabolites and Nm23 Expression1. Neoplasia 2002, 4, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, R.; Imanishi, Y.; Shibata, K.; Sakai, N.; Sakamoto, K.; Shigetomi, S.; Habu, N.; Otsuka, K.; Sato, Y.; Watanabe, Y.; et al. Restoration of E-cadherin expression by selective Cox-2 inhibition and the clinical relevance of the epithelial-to-mesenchymal transition in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.M.; Song, J.; Saini, V.; Wong, Y.H. Small Molecules as Drugs to Upregulate Metastasis Suppressors in Cancer Cells. Curr. Med. Chem. 2019, 26, 5876–5899. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Shibata, T.; Nakamura, M.; Yamashita, H.; Yoshioka, D.; Okubo, M.; Maruyama, N.; Kamano, T.; Kamiya, Y.; Fujita, H.; et al. Chronic Aspirin Use Suppresses CDH1 Methylation in Human Gastric Mucosa. Dig. Dis. Sci. 2009, 55, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Iiizumi-Gairani, M.; Okuda, H.; Kobayashi, A.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Xing, F.; Fukuda, K.; Modur, V.; et al. KAI1 gene is engaged in NDRG1 gene-mediated metastasis suppression through the ATF3-NFkappaB complex in human prostate cancer. J. Biol. Chem. 2011, 286, 18949–18959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, T.; Miki, T.; Nishimura, N.; Nokihara, H.; Hamada, H.; Mukaida, N.; Sone, S. Nuclear factor-kappaB-dependent expression of metastasis suppressor KAI1/CD82 gene in lung cancer cell lines expressing mutant p53. Cancer Res. 2001, 61, 673–678. [Google Scholar]
- Xu, J.; Hua, X.; Jin, H.; Zhu, J.; Li, Y.; Li, J.; Huang, C. NFκB2 p52 stabilizes rhogdiβ mRNA by inhibiting AUF1 protein degradation via a miR-145/Sp1/USP8-dependent axis. Mol. Carcinog. 2019, 58, 777–793. [Google Scholar] [CrossRef]
- Teixeira, L.F.S.; Peron, J.P.S.; Bellini, M.H. Silencing of nuclear factor kappa b 1 gene expression inhibits colony formation, cell migration and invasion via the downregulation of interleukin 1 beta and matrix metallopeptidase 9 in renal cell carcinoma. Mol. Boil. Rep. 2019, 47, 1143–1151. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, D.L.; Jiao, X.L.; Dong, Q. S100A4 regulates migration and invasion in hepatocellular carcinoma HepG2 cells via NF-kappaB-dependent MMP-9 signal. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2372–2382. [Google Scholar]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Thomas, H.; Gruenert, S. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicek, M.; Fukuyama, R.; Welch, D.R.; Sizemore, N.; Casey, G.; Cıcek, M.; Hagiwara, T.; Kono, S.; Yin, G.; Toyomura, K.; et al. Breast Cancer Metastasis Suppressor 1 Inhibits Gene Expression by Targeting Nuclear Factor-κB Activity. Cancer Res. 2005, 65, 3586–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samant, R.S.; Clark, D.W.; A Fillmore, R.; Cicek, M.; Metge, B.J.; Chandramouli, K.H.; Chambers, A.; Casey, G.; Welch, D.R.; A Shevde, L. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-κB activation. Mol. Cancer 2007, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Mayo, M.W.; Xiao, A.; Hall, E.H.; Amin, E.B.; Kadota, K.; Adusumilli, P.S.; Jones, D.R. Loss of BRMS1 Promotes a Mesenchymal Phenotype through NF-κB-Dependent Regulation ofTwist1. Mol. Cell Boil. 2014, 35, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Wang, H.; Boyd, D.D. KiSS-1Represses 92-kDa Type IV Collagenase Expression by Down-regulating NF-κB Binding to the Promoter as a Consequence of IκBα-induced Block of p65/p50 Nuclear Translocation. J. Boil. Chem. 2000, 276, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Liu, M.; Cowell, J.K. Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. Int. J. Cancer 2011, 129, 2825–2835. [Google Scholar] [CrossRef]
- Cho, S.G.; Li, D.; Stafford, L.J.; Luo, J.; Rodriguez-Villanueva, M.; Wang, Y.; Liu, M. KiSS1 suppresses TNFalpha-induced breast cancer cell invasion via an inhibition of RhoA-mediated NF-kappaB activation. J. Cell Biochem. 2009, 107, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Althouse, S.K.; Nabell, L.; Rugo, H.; Carey, L.; Kimmick, G.; Jones, D.R.; Merino, M.J.; Steeg, P.S. A phase II study of medroxyprogesterone acetate in patients with hormone receptor negative metastatic breast cancer: Translational breast cancer research consortium trial 007. Breast Cancer Res. Treat. 2014, 148, 99–106. [Google Scholar] [CrossRef]


{kind=link}
| Biologic Process, Pathway or Biological Function | Direct Target | Drug | Ref. |
|---|---|---|---|
| Proliferation and detachment | |||
| Cadherin signaling | N-cadherin | ADH1 (exherin) | [11] |
| Integrin signaling | αvβ3 and αvβ5 integrin | Cilengitide | [12,13,14] |
| FAK/TGF-beta signaling, MMPs | MEK4 | Genistein | [15,16,17,18] |
| Integrin signaling, anoikis | Integrin β1 | DZ-50 | [19] |
| Anoikis, caspase 8 activity | FLIP | Anisomycin | [20] |
| Migration, invasion, intravasation, circulation and attachment | |||
| Focal Adhesions, cytoskeletal remodeling | FAK | GSK2256098, TAE-226 | [21,22] |
| Stress fiber formation | ROCK | Fasudil | [23] |
| Actomyosin contraction | Cdc42 | ML-141/CID2950007, CID44216842 | [24] |
| Actomyosin contraction | Rac, Cdc42 | EHT-1864, NSC23766, Ehop-016, MBQ-167, AZA1, AZA197, Ketorolac | [25,26,27,28,29,30,31] |
| Actomyosin contraction | Rho | ZCL278 | [32] |
| cytoskeleton turnover | PAK | IPA3, KPT-9274, PF3758309 | [33,34,35] |
| extravasation of circulating tumor cells | αvβ3 integrin, fascin | MK-0429, NP-G2-029, NP-G2-044 | [35,36,37] |
| EGFR mediated signaling | EGFR | Erlotinib, Gefitinib | [38,39,40] |
| VEGFR signaling | VEGFR | Sorafenib, Pazopanib, Sunitinib | [41] |
| FGFR and VEGFR signaling; ATP competitor | FGFR, VEGFR | Brivanib | [42] |
| PDGFR and FGFR1 signaling; ATP competitor | FGFR | Orantinib | [43] |
| SRC signaling | SRC | Dasatinib, Bosutinib | [44,45,46] |
| SRC signaling, invadopodia formation | FAK, CAS | Saracatinib | [44,47] |
| SRC signaling, tubulin polymerization | SRC/tubulin | KX02, KX2-391 | [44] |
| MMP activity, invasion | MMP | Hydroxamates, thiol-based inhibitors, S3304, cis-ACCP, Ro-28-2653, B-428, WX-671 | [48,49,50] |
| TIMP expression for MMP interference | TIMP3 | Arctigenin, diallyl-disulfide, MPT0-G013 | [51] |
| uPA mediated growth factor activation | uPA | ameloride derivatives | [52,53,54] |
| Extravasation and metastasis outgrowth | |||
| Notch signaling | γ-secretase | PF-03084014 | [55,56,57,58,59] |
| Wnt signaling; transcriptional activity of β-catenin | β-catenin | CWP232228 | [60] |
| Wnt signaling; downstream apoptotic cell death pathway | β-catenin | CWP232291 | [61] |
| TAM signaling; oncogenesis and anti-apoptotic activity | Mer | UNC1062 | [62] |
| TAM signaling; prosurvival and anti-apoptotic pathways | Mer/FTL3 | UNC2025 | [63] |
| TAM signaling; prosurvival, proinflammatory, EMT pathways | AXL | R428 (BGB324) | [64] |
| Angiogenesis; FGF signaling | FGF2-FGFRR1-HSPGs | SM27 | [65,66] |
| Metastasis Suppressors | |||
| epigenetic regulation of gene expression: DNA methylation | DNA methyltransferases | 5-aza-2’-deoxycytidine (decitabine) | [67] |
| epigenetic regulation of gene expression: histone methylation | EZH2 | 3-deazaneplanocin A (DZNep) | [68] |
| epigenetic regulation of gene expression: histone acetylation | histone deacetylases | suberanilohydroxamic acid (SAHA) | [69] |
| NFκB signaling pathway | NFκB | dehydroxymethylepoxyquinomicin | [70] |
| steroid hormone signal transduction | progesterone receptor | aedroxyprogesterone acetate (MPA) | [71] |
| prostaglandin signal transduction | Cox-2 | non-steroidal anti-inflammatory drugs (NSAIDs) | [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobelt, D.; Dahlmann, M.; Dumbani, M.; Güllü, N.; Kortüm, B.; Vílchez, M.E.A.; Stein, U.; Walther, W. Small Ones to Fight a Big Problem—Intervention of Cancer Metastasis by Small Molecules. Cancers 2020, 12, 1454. https://doi.org/10.3390/cancers12061454
Kobelt D, Dahlmann M, Dumbani M, Güllü N, Kortüm B, Vílchez MEA, Stein U, Walther W. Small Ones to Fight a Big Problem—Intervention of Cancer Metastasis by Small Molecules. Cancers. 2020; 12(6):1454. https://doi.org/10.3390/cancers12061454
Chicago/Turabian StyleKobelt, Dennis, Mathias Dahlmann, Malti Dumbani, Nazli Güllü, Benedikt Kortüm, Miguel E. Alberto Vílchez, Ulrike Stein, and Wolfgang Walther. 2020. "Small Ones to Fight a Big Problem—Intervention of Cancer Metastasis by Small Molecules" Cancers 12, no. 6: 1454. https://doi.org/10.3390/cancers12061454