Epigenetic Research in Stem Cell Bioengineering—Anti-Cancer Therapy, Regenerative and Reconstructive Medicine in Human Clinical Trials

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Overview of Epigenetic Modification of Genome
2.1. DNA Methylation
2.2. Histone Modification
2.3. Non-Coding RNA
3. Advanced Epigenetic Research in Human Stem Cells—A Novel Bioengineering Tool
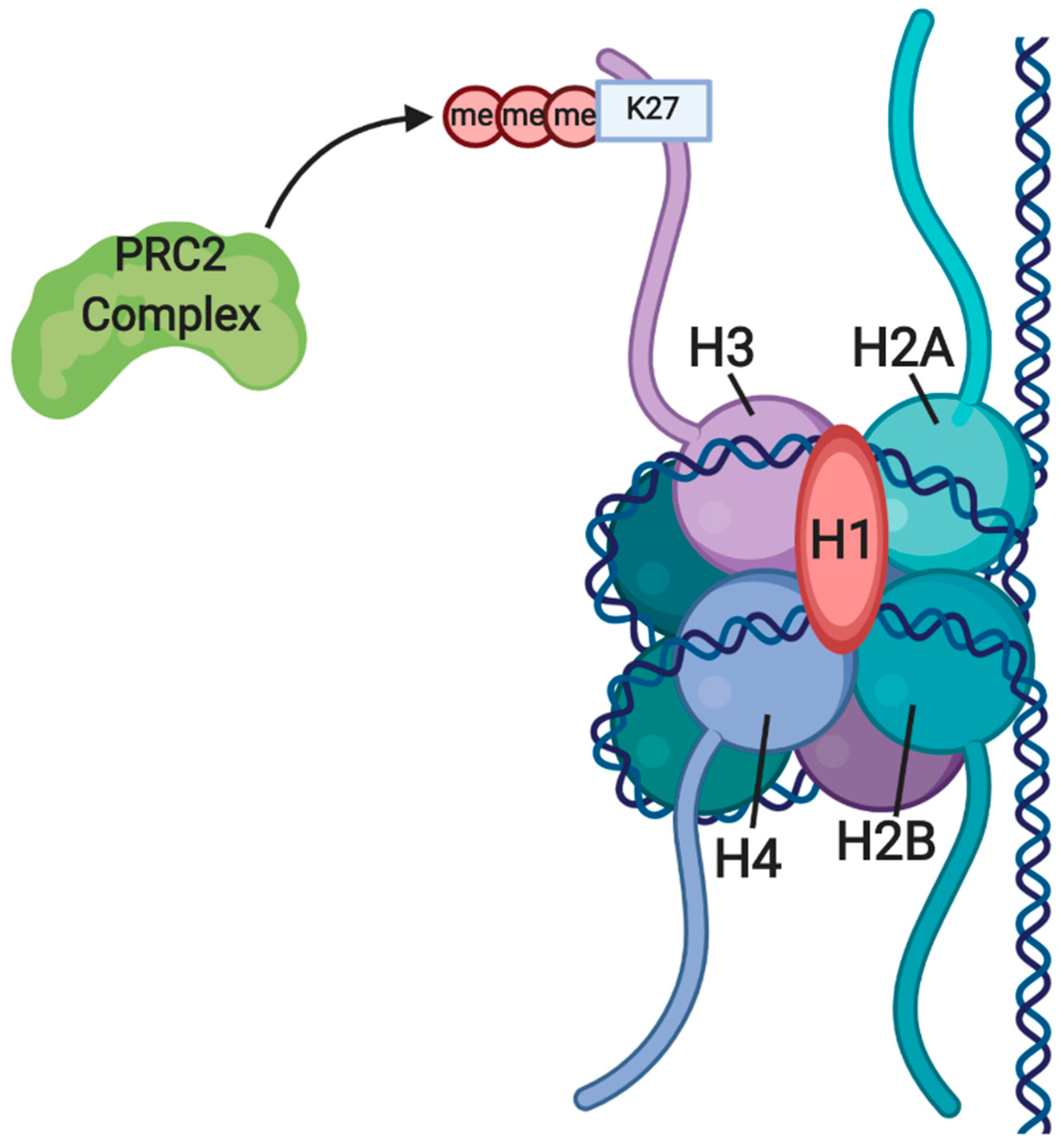
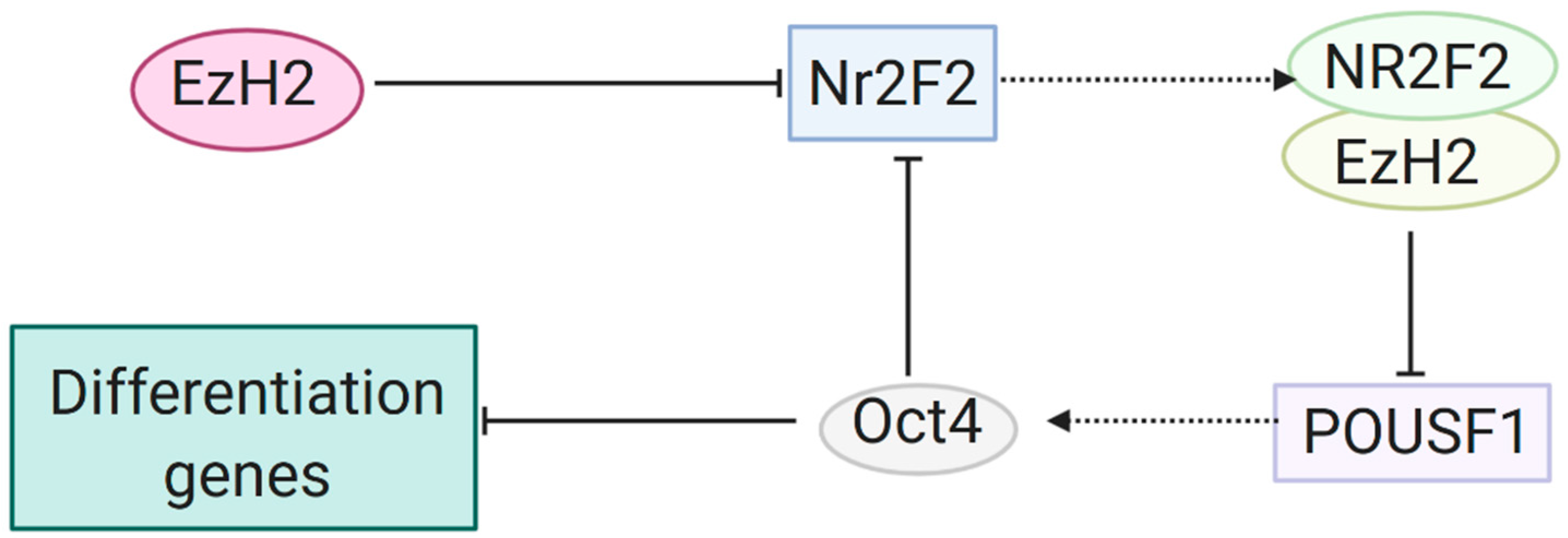
3.1. Gene Silencing through H3K27me by EZH2 and the PRC2 Complex
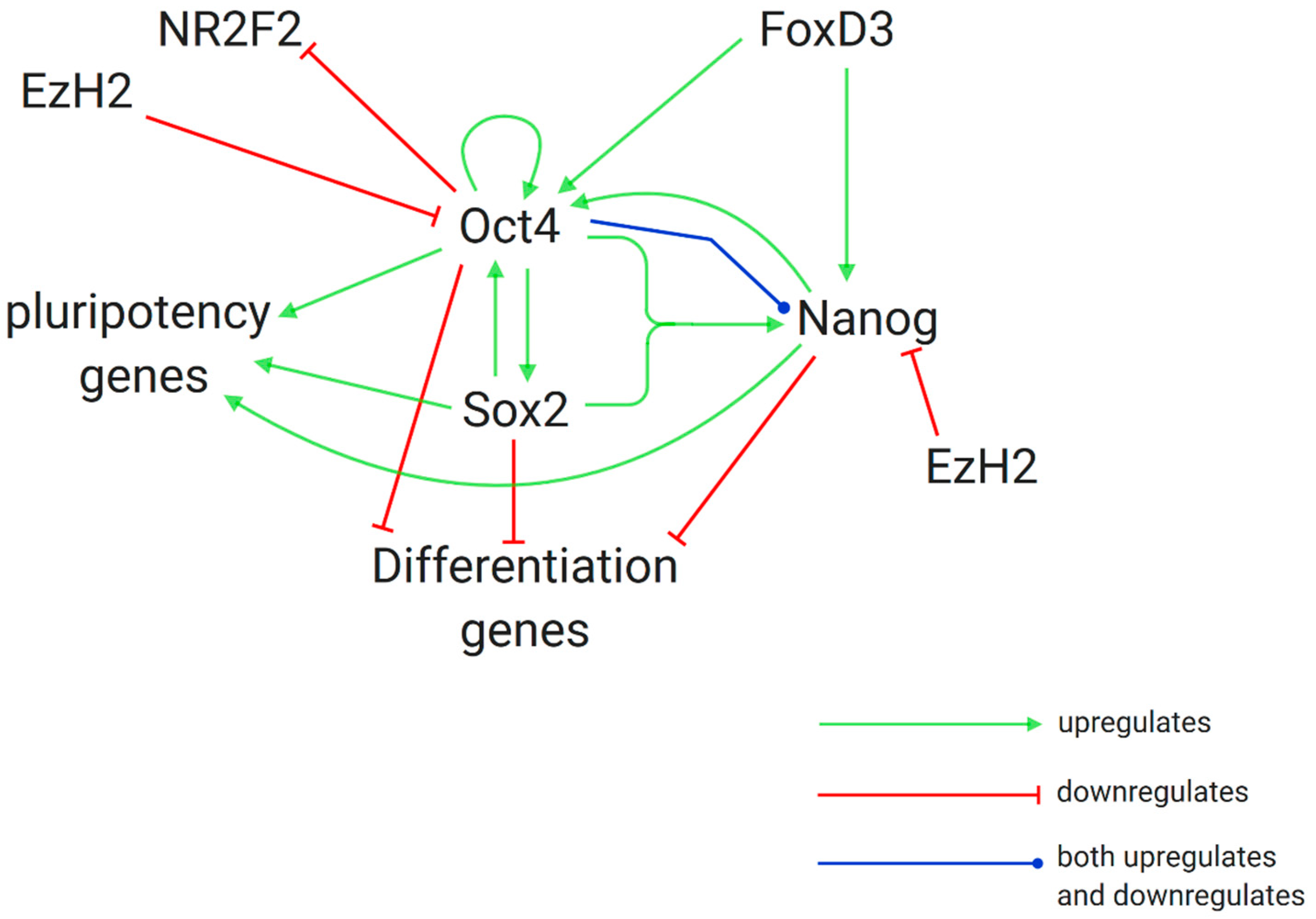
3.2. Further Research on Epigenetic Modifications and Epigenetic Inheritance
3.3. Cancer Research and Anti-Cancer Therapy—Living in the Shadow of Epigenetic Genome Modification
4. Epigenetic Genome Modification and Regenerative Medicine
5. Epigenome and Human Clinical Trials
5.1. Epigenetic Drugs in Cancer
5.2. Mechanisms in Cancer
5.3. Regenerative Medicine
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Lopomo, A.; Burgio, E.; Migliore, L. Epigenetics of Obesity. Prog. Mol. Biol. Transl. Sci. 2016, 140, 151–184. [Google Scholar] [CrossRef] [PubMed]
- Muka, T.; Koromani, F.; Portilla, E.; O’Connor, A.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Franco, O.H. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int. J. Cardiol. 2016, 212, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovrei, L.; Maver, A.; Zadel, M.; Peterli, B. The Role of Epigenetics in Neurodegenerative Diseases. Neurodegener. Dis. 2013, 345, 345. [Google Scholar] [CrossRef] [Green Version]
- Carson, C.; Lawson, H.A. Epigenetics of metabolic syndrome. Physiol. Genom. 2018, 50, 947–955. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Peng, X.; Li, Z.; Zhou, Q.; Huang, S.; Wang, Y.; Li, J.; Song, Y. Epigenetics and bone diseases. Genet. Res. (Camb.) 2018, 100, e6. [Google Scholar] [CrossRef] [Green Version]
- Del Real, A.; Riancho-Zarrabeitia, L.; López-Delgado, L.; Riancho, J.A. Epigenetics of Skeletal Diseases. Curr. Osteoporos. Rep. 2018, 16, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Tabolacci, E.; Pietrobono, R.; Moscato, U.; Oostra, B.A.; Chiurazzi, P.; Neri, G. Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 2005, 13, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Adenocarcinoma, D.; Sato, N.; Maitra, A.; Fukushima, N.; Heek, N.T.; Van Matsubayashi, H.; Iacobuzio-Donahue, C.A.; Rosty, C.; Goggins, M.; van Heek, N.T. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003, 63, 4158–4166. [Google Scholar]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar]
- Schneider, E.; Pliushch, G.; Hajj, N.; El Galetzka, D.; Puhl, A.; Schorsch, M.; Frauenknecht, K.; Riepert, T.; Tresch, A.; Müller, A.M.; et al. Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic Acids Res. 2010, 38, 3880–3890. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Chen, C.; Yang, M.C.K.; Yang, T.P. Evidence that silencing of the HPRT promoter by DNA methylation is mediated by critical CpG sites. J. Biol. Chem. 2001, 276, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, M.; Hermann, A.; Gowher, H.; Jeltsch, A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002, 269, 4981–4984. [Google Scholar] [CrossRef]
- Siegfried, Z.; Eden, S.; Mendelsohn, M.; Feng, X.; Tsuberi, B.Z.; Cedar, H. DNA methylation represses transcription in vivo. Nat. Genet. 1999, 22, 203–206. [Google Scholar] [CrossRef]
- Wolf, S.F.; Jolly, D.J.; Lunnen, K.D.; Friedmann, T.; Migeon, B.R. Methylation of the hypoxanthine phosphoribosyltransferase locus on the human X chromosome: Implications for X chromosome inactivation. Proc. Natl. Acad. Sci. USA 1984, 81, 2806–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.S.; Estécio, M.R.H.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.-P.J. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennartsson, A.; Ekwall, K. Histone modification patterns and epigenetic codes. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 863–868. [Google Scholar] [CrossRef]
- Zhao, J. Sumoylation regulates diverse biological processes. Cell. Mol. Life Sci. 2007, 64, 3017–3033. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Turner, B.M. Defining an epigenetic code. Nat. Cell Biol. 2007, 9, 2–6. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Li, F.; Huarte, M.; Zaratiegui, M.; Vaughn, M.W.; Shi, Y.; Martienssen, R.; Cande, W.Z. Lid2 Is Required for Coordinating H3K4 and H3K9 Methylation of Heterochromatin and Euchromatin. Cell 2008, 135, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Shukla, A.; Schneider, J.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Bhaumik, S.R.; Shilatifard, A. Histone Crosstalk between H2B Monoubiquitination and H3 Methylation Mediated by COMPASS. Cell 2007, 131, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, E.M.; Muratore-Schroeder, T.L.; Cook, R.G.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D. Cathepsin L Proteolytically Processes Histone H3 During Mouse Embryonic Stem Cell Differentiation. Cell 2008, 135, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, J.L.; Kingston, R.E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998, 67, 545–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Hansen, K.H.; Christensen, J.; Agger, K.; Cloos, P.A.C.; Helin, K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008, 22, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [Green Version]
- Feingold, E.A.; Good, P.J.; Guyer, M.S.; Kamholz, S.; Liefer, L.; Wetterstrand, K.; Collins, F.S.; Gingeras, T.R.; Kampa, D.; Sekinger, E.A.; et al. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, S. Small noncoding RNAs: Biogenesis, function, and emerging significance in toxicology. J. Biochem. Mol. Toxicol. 2010, 24, 195–216. [Google Scholar] [CrossRef]
- Uchida, S.; Dimmeler, S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015, 116, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson-Smith, A.C.; Sasaki, H.; Cattanach, B.M.; Surani, M.A. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 1993, 362, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Herzing, L.B.K.; Romer, J.T.; Horn, J.M.; Ashworth, A. Xist has properties of the X-chromosome inactivation centre. Nature 1997, 386, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Astori, G.; Vignati, F.; Bardelli, S.; Tubio, M.; Gola, M.; Albertini, V.; Bambi, F.; Scali, G.; Castelli, D.; Rasini, V.; et al. “In vitro” and multicolor phenotypic characterization of cell subpopulations identified in fresh human adipose tissue stromal vascular fraction and in the derived mesenchymal stem cells. J. Transl. Med. 2007, 5. [Google Scholar] [CrossRef] [Green Version]
- Boyer, L.A.; Tong, I.L.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Rumman, M.; Dhawan, J.; Kassem, M. Concise Review: Quiescence in Adult Stem Cells: Biological Significance and Relevance to Tissue Regeneration. Stem Cells 2015, 33, 2903–2912. [Google Scholar] [CrossRef]
- Saldaña, L.; Bensiamar, F.; Vallés, G.; Mancebo, F.J.; García-Rey, E.; Vilaboa, N. Immunoregulatory potential of mesenchymal stem cells following activation by macrophage-derived soluble factors. Stem Cell Res. Ther. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Raghunath, M.; Nurcombe, V.; Hui, J.H.; van Wijnen, A.J.; Cool, S.M. Concise Review: Multifaceted Characterization of Human Mesenchymal Stem Cells for Use in Regenerative Medicine. Stem Cells Transl. Med. 2017, 6, 2173–2785. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Van Seuningen, I. Epigenetics, stem cells and epithelial cell fate. Differentiation 2009, 78, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Bates, S.E. Epigenetic Modifiers: Basic Understanding and Clinical Development. Clin. Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Chudin, E.; Wu, B.; Zhou, L.; Garcia, E.W.; Liu, Y.; Shin, S.; Plaia, T.W.; Auerbach, J.M.; Arking, D.E.; et al. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006, 16, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzanska, L. Human Neural Stem Cells: From Generation to Differentiation and Application; Springer Publishing: New York, NY, USA, 2018. [Google Scholar]
- Aguilar-Gallardo, C.; Simón, C. Cells, stem cells, and cancer stem cells. Semin. Reprod. Med. 2013, 31, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Alexanian, A.R. Epigenetic modifiers promote efficient generation of neural-like cells from bone marrow-derived mesenchymal cells grown in neural environment. J. Cell. Biochem. 2007, 100, 362–371. [Google Scholar] [CrossRef]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate decision of mesenchymal stem cells: Adipocytes or osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Hemming, S.; Cakouros, D.; Isenmann, S.; Cooper, L.; Menicanin, D.; Zannettino, A.; Gronthos, S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2014, 32, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Al Hezaimi, K.; Zhou, X.; Park, N.H.; Wang, C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell 2012, 11, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yu, B.; Hong, C.; Wang, C.Y. KDM6B epigenetically regulates odontogenic differentiation of dental mesenchymal stem cells. Int. J. Oral Sci. 2013, 5, 200–205. [Google Scholar] [CrossRef] [Green Version]
- De Haan, G.; Gerrits, A. Epigenetic control of hematopoietic stem cell aging: The case of Ezh2. Ann. N. Y. Acad. Sci. 2007, 1106, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.H.; Yu, Y.L.; Hung, M.C. The roles of EZH2 in cell lineage commitment. Am. J. Transl. Res. 2011, 3, 243–250. [Google Scholar] [PubMed]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef] [Green Version]
- Sher, F.; Rößler, R.; Brouwer, N.; Balasubramaniyan, V.; Boddeke, E.; Copray, S. Differentiation of Neural Stem Cells into Oligodendrocytes: Involvement of the Polycomb Group Protein Ezh2. Stem Cells 2008, 26, 2875–2883. [Google Scholar] [CrossRef]
- Rosa, A.; Brivanlou, A.H. A regulatory circuitry comprised of miR-302 and the transcription factors OCT4 and NR2F2 regulates human embryonic stem cell differentiation. EMBO J. 2011, 30, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Pursani, V.; Pethe, P.; Bashir, M.; Sampath, P.; Tanavde, V.; Bhartiya, D. Genetic and Epigenetic Profiling Reveals EZH2-mediated Down Regulation of OCT-4 Involves NR2F2 during Cardiac Differentiation of Human Embryonic Stem Cells. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Villasante, A.; Piazzolla, D.; Li, H.; Gomez-Lopez, G.; Djabali, M.; Serrano, M. Epigenetic regulation of Nanog expression by Ezh2 in pluripotent stem cells. Cell Cycle 2011, 10, 1488–1498. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. Regulation of Stem cell pluripotency and differentiation involves a mutual regulatory circuit of the Nanog, OCT4, and SOX2 pluripotency transcription factors with polycomb Repressive Complexes and Stem Cell microRNAs. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef]
- Pan, G.; Li, J.; Zhou, Y.; Zheng, H.; Pei, D. A negative feedback loop of transcription factors that controls stem cell pluripotency and self-renewal. FASEB J. 2006, 20, 1730–1732. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.; Tam, W.; Yeap, L.; Li, P.; Ang, Y.; Lim, B.; Robson, P.; Ng, H. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol. Cell. Biol. 2005, 25, 6031–6046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandy, R.A.; Whitfield, T.W.; Wu, H.; Fitzgerald, M.P.; VanOudenhove, J.J.; Zaidi, S.K.; Montecino, M.A.; Lian, J.B.; van Wijnen, A.J.; Stein, J.L.; et al. Genome-Wide Studies Reveal that H3K4me3 Modification in Bivalent Genes Is Dynamically Regulated during the Pluripotent Cell Cycle and Stabilized upon Differentiation. Mol. Cell. Biol. 2016, 36, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Xu, M.; Zhu, B. Epigenetic inheritance mediated by histone lysine methylation: Maintaining transcriptional states without the precise restoration of marks? Philos. Trans. R. Soc. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [Green Version]
- Reverón-Gómez, N.; González-Aguilera, C.; Stewart-Morgan, K.R.; Petryk, N.; Flury, V.; Graziano, S.; Johansen, J.V.; Jakobsen, J.S.; Alabert, C.; Groth, A. Accurate Recycling of Parental Histones Reproduces the Histone Modification Landscape during DNA Replication. Mol. Cell 2018, 72, 239–249.e5. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, C.J.; Hyland, E.M. Yeast epigenetics: The inheritance of histone modification states. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Park, J.W.; Han, J.W. Targeting epigenetics for cancer therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.I.; Matsuno, Y.; Hyodo, M.; Fujimori, H. Genomic-destabilization-associated mutagenesis and clonal evolution of cells with mutations in tumor-suppressor genes. Cancers (Basel) 2019, 11, 1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constâncio, V.; Nunes, S.P.; Moreira-Barbosa, C.; Freitas, R.; Oliveira, J.; Pousa, I.; Oliveira, J.; Soares, M.; Dias, C.G.; Dias, T.; et al. Early detection of the major male cancer types in blood-based liquid biopsies using a DNA methylation panel. Clin. Epigenet. 2019, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.P.; Moreira-Barbosa, C.; Salta, S.; de Sousa, S.P.; Pousa, I.; Oliveira, J.; Soares, M.; Rego, L.; Dias, T.; Rodrigues, J.; et al. Cell-free DNA methylation of selected genes allows for early detection of the major cancers in women. Cancers (Basel) 2018, 10, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.X.; Sheng, D.Q.; Cheng, L.; Song, X.Y. Current Landscape of Epigenetics in Lung Cancer: Focus on the Mechanism and Application. J. Oncol. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Chen, J.; Li, L.-Y.; Wu, M. Epigenetic plasticity of enhancers in cancer. Transcription 2020, 1–11. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63. [Google Scholar] [CrossRef]
- Tortorella, S.M.; Hung, A.; Karagiannis, T.C. The Implication of Cancer Progenitor Cells and the Role of Epigenetics in the Development of Novel Therapeutic Strategies for Chronic Myeloid Leukemia. Antioxid. Redox Signal. 2015, 22, 1425–1462. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.S.; Schmezer, P.; Breuer, R.; Haas, S.F.; Essers, M.A.; Krammer, P.H.; Li-Weber, M. The traditional Chinese medical compound Rocaglamide protects nonmalignant primary cells from DNA damage-induced toxicity by inhibition of p53 expression. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.; Ghosh, S.; Maitra, A.; Biswas, N.K.; Panda, C.K.; Roy, B.; Sarin, R.; Majumder, P.P. Epigenomic dysregulation-mediated alterations of key biological pathways and tumor immune evasion are hallmarks of gingivo-buccal oral cancer. Clin. Epigenet. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zayas, J.; Qin, B.; Wang, L. Targeting DNA methylation for treating triple-negative breast cancer. Pharmacogenomics 2019, 20, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Liu, H.; Wan, L.; Zhang, W.; Wang, Q.; Zhang, S.; Shang, S.; Zhang, Y.; Pang, D. The MS-lincRNA landscape reveals a novel lincRNA BCLIN25 that contributes to tumorigenesis by upregulating ERBB2 expression via epigenetic modification and RNA–RNA interactions in breast cancer. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.P.; Diniz, F.; Moreira-Barbosa, C.; Constâncio, V.; Silva, A.V.; Oliveira, J.; Soares, M.; Paulino, S.; Cunha, A.L.; Rodrigues, J.; et al. Subtyping Lung Cancer Using DNA Methylation in Liquid Biopsies. J. Clin. Med. 2019, 8, 1500. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Barbosa, C.; Barros-Silva, D.; Costa-Pinheiro, P.; Torres-Ferreira, J.; Constâncio, V.; Freitas, R.; Oliveira, J.; Antunes, L.; Henrique, R.; Jerónimo, C. Comparing diagnostic and prognostic performance of two-gene promoter methylation panels in tissue biopsies and urines of prostate cancer patients. Clin. Epigenet. 2018, 10. [Google Scholar] [CrossRef]
- Hu, B.B.; Wang, X.Y.; Gu, X.Y.; Zou, C.; Gao, Z.J.; Zhang, H.; Fan, Y. N6-methyladenosine (m6A) RNA modification in gastrointestinal tract cancers: Roles, mechanisms, and applications. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, C.; Sun, Y.; He, X.; Xue, D. m 6 A RNA modification modulates gene expression and cancer-related pathways in clear cell renal cell carcinoma. Epigenomics 2019, 12, 87–99. [Google Scholar] [CrossRef]
- Orouji, E.; Peitsch, W.K.; Orouji, A.; Houben, R.; Utikal, J. Oncogenic Role of an Epigenetic Reader of m6A RNA Modification: YTHDF1 in Merkel Cell Carcinoma. Cancers (Basel) 2020, 12, 202. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Chen, Z.; Zheng, Y.; Liu, Y.; Gao, J.; Lin, S.; Chen, S. MiR-506 Targets UHRF1 to Inhibit Colorectal Cancer Proliferation and Invasion via the KISS1/PI3K/NF-κB Signaling Axis. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wei, J.; Chen, Z.; Yuan, Y.; Li, X.; Zhang, Y.; Meng, Y.; Hu, Y.; Du, H. Integrative Analysis Reveals Comprehensive Altered Metabolic Genes Linking with Tumor Epigenetics Modification in Pan-Cancer. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Pu, L.; Cui, H. Mitoepigenetics and Its Emerging Roles in Cancer. Front. Cell Dev. Biol. 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, B.; Pappa, S.; Díez-Villanueva, A.; Mallona, I.; Custodio, J.; Barrero, M.J.; Peinado, M.A.; Jordà, M. Tissue and cancer-specific expression of DIEXF is epigenetically mediated by an Alu repeat. Epigenetics 2020, 1–15. [Google Scholar] [CrossRef]
- Yang, L.; Lei, Q.; Li, L.; Yang, J.; Dong, Z.; Cui, H. Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c-Myc expression in colorectal cancer. Clin. Epigenet. 2019, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, K.; Zhang, Y.; Barnes, S.D.; Jaichander, P.; Zheng, Y.; Hassan, M.; Malladi, V.S.; Skapek, S.X.; Xu, L.; et al. Twist2 amplification in rhabdomyosarcoma represses myogenesis and promotes oncogenesis by redirecting MyoD DNA binding. Genes Dev. 2019, 33, 626–640. [Google Scholar] [CrossRef] [Green Version]
- Fachal, L.; Aschard, H.; Beesley, J.; Barnes, D.R.; Allen, J.; Kar, S.; Pooley, K.A.; Dennis, J.; Michailidou, K.; Turman, C.; et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat. Genet. 2020, 52, 56–73. [Google Scholar] [CrossRef]
- Aloia, L.; McKie, M.A.; Vernaz, G.; Cordero-Espinoza, L.; Aleksieva, N.; van den Ameele, J.; Antonica, F.; Font-Cunill, B.; Raven, A.; Aiese Cigliano, R.; et al. Epigenetic remodelling licences adult cholangiocytes for organoid formation and liver regeneration. Nat. Cell Biol. 2019, 21, 1321–1333. [Google Scholar] [CrossRef]
- Robertson, F.L.; Marqués-Torrejón, M.A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. DMM Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Abbas, M.N.; Kausar, S.; Cui, H. Therapeutic potential of natural products in glioblastoma treatment: Targeting key glioblastoma signaling pathways and epigenetic alterations. Clin. Transl. Oncol. 2019. [Google Scholar] [CrossRef]
- Schötterl, S.; Hübner, M.; Armento, A.; Veninga, V.; Wirsik, N.M.; Bernatz, S.; Lentzen, H.; Mittelbronn, M.; Naumann, U. Viscumins functionally modulate cell motility-associated gene expression. Int. J. Oncol. 2017, 50, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Quan Dean, J. Reprogramming the genome to totipotency in mouse embryos. Trends Cell Biol. 2015, 25, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stueve, T.R.; Marconett, C.N.; Zhou, B.; Borok, Z.; Laird-Offringa, I.A. The importance of detailed epigenomic profiling of different cell types within organs. Epigenomics 2016, 8, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilja, T.; Wallenborg, K.; Björkman, K.; Albåge, M.; Eriksson, M.; Lagercrantz, H.; Rohdin, M.; Hermanson, O. Novel alterations in the epigenetic signature of MeCP2-targeted promoters in lymphocytes of Rett syndrome patients. Epigenetics 2013, 8, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Santiago, R.; Merkel, A.; Castellano, G.; Heath, S.; Raya, Á.; Tolosa, E.; Martí, M.J.; Consiglio, A.; Ezquerra, M. Whole-genome DNA hyper-methylation in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. Clin. Epigenet. 2019, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Kang, Y.; Wang, M.; Li, Y.; Xu, T.; Yang, W.; Song, H.; Wu, H.; Shu, Q.; Jin, P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 2018, 27, 3936–3950. [Google Scholar] [CrossRef]
- Schnerch, A.; Rampalii, S.; Bhatia, M. Histone modification profiling in normal and transformed human embryonic stem cells using micro chromatin immunoprecipitation, scalable to genome-wide microarray analyses. Methods Mol. Biol. 2013, 1029, 149–161. [Google Scholar] [CrossRef]
- A Phase II Study of Epigenetic Therapy to Overcome Chemotherapy Resistance in Refractory Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT00404508 (accessed on 6 April 2020).
- Gene Expression Variation and Implant Wound Healing Among Smokers and Diabetics. Available online: https://clinicaltrials.gov/ct2/show/NCT01663298?cond=Gene+Expression+Variation+and+Implant+Wound+Healing+Among+Smokers+and+Diabetics&draw=2&rank=1 (accessed on 6 April 2020).
- Tao, H.; Li, H.; Su, Y.; Feng, D.; Wang, X.; Zhang, C.; Ma, H.; Hu, Q. Histone methyltransferase G9a and H3K9 dimethylation inhibit the self-renewal of glioma cancer stem cells. Mol. Cell. Biochem. 2014, 394, 23–30. [Google Scholar] [CrossRef]
- Hydralazine and Valproate Plus Cisplatin Chemoradiation in Cervical Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00404326?cond=Hydralazine+and+Valproate+Plus+Cisplatin+Chemoradiation+in+Cervical+Cancer&draw=2&rank=1 (accessed on 6 April 2020).
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; De Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3177–3183. [Google Scholar] [CrossRef] [Green Version]
- Van den Boom, V.; Maat, H.; Geugien, M.; Rodríguez López, A.; Sotoca, A.M.; Jaques, J.; Brouwers-Vos, A.Z.; Fusetti, F.; Groen, R.W.J.; Yuan, H.; et al. Non-canonical PRC1.1 Targets Active Genes Independent of H3K27me3 and Is Essential for Leukemogenesis. Cell Rep. 2016, 14, 332–346. [Google Scholar] [CrossRef] [Green Version]
- Study of Azacitidine in Adult Taiwanese Subjects With Higher-Risk Myelodysplastic Syndromes (MDS). Available online: https://clinicaltrials.gov/ct2/show/NCT01201811?cond=Study+of+Azacitidine+in+Adult+Taiwanese+Subjects+With+Higher-Risk+Myelodysplastic+Syndromes+%28MDS%29&draw=2&rank=1 (accessed on 6 April 2020).
- Torres, C.M.; Biran, A.; Burney, M.J.; Patel, H.; Henser-Brownhill, T.; Cohen, A.H.S.; Li, Y.; Ben-Hamo, R.; Nye, E.; Spencer-Dene, B.; et al. The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidaza to Restore Hormone Thx Prostate. Available online: https://clinicaltrials.gov/ct2/show/NCT00384839 (accessed on 6 April 2020).
- A Study of Venetoclax in Combination With Azacitidine Versus Azacitidine in Treatment Naïve Subjects With Acute Myeloid Leukemia Who Are Ineligible for Standard Induction Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT02993523 (accessed on 6 April 2020).
- A Trial of Epigenetic Priming in Patients With Newly Diagnosed Acute Myeloid Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT03164057 (accessed on 6 April 2020).
- Azacytidine Prior to in Vivo T-cell Depleted Allo Stem Cell Transplant for Patients With Myeloid Malignancies in CR. Available online: https://clinicaltrials.gov/ct2/show/NCT02497404?cond=Azacytidine+Prior+to+in+Vivo+T-cell+Depleted+Allo+Stem+Cell+Transplant+for+Patients+With+Myeloid+Malignancies+in+CR&draw=2&rank=1 (accessed on 6 April 2020).
- Diagnosis of RSTS: Identification of the Acetylation Profiles as Epigenetic Markers for Assessing Causality of CREBBP Variants. Available online: https://clinicaltrials.gov/ct2/show/NCT04122742?cond=Diagnosis+of+RSTS%3A+Identification+of+the+Acetylation+Profiles+as+Epigenetic+Markers+for+Assessing+Causality+of+CREBBP+Variants&draw=2&rank=1 (accessed on 6 April 2020).
- DNA Methylation in Allogeneic Hematopoietic Stem Cell Transplantation. Available online: https://clinicaltrials.gov/ct2/show/NCT03871296?cond=DNA+Methylation+in+Allogeneic+Hematopoietic+Stem+Cell+Transplantation&draw=2&rank=1 (accessed on 6 April 2020).
- EPIgenetics and in Vivo Resistance of Chronic Myeloid Leukemia Stem Cells to Tyrosine Kinase Inhibitors. Available online: https://clinicaltrials.gov/ct2/show/NCT03481868?cond=EPIgenetics+and+in+Vivo+Resistance+of+Chronic+Myeloid+Leukemia+Stem+Cells+to+Tyrosine+Kinase+Inhibitors&draw=2&rank=1 (accessed on 6 April 2020).
- Genetic and Epigenetic Basis of Chronic Wounds. Available online: https://clinicaltrials.gov/ct2/show/NCT03793062?cond=Genetic+and+Epigenetic+Basis+of+Chronic+Wounds&draw=2&rank=1 (accessed on 6 April 2020).
- Phase II Anti-PD1 Epigenetic Therapy Study in NSCLC. Available online: https://clinicaltrials.gov/ct2/show/NCT01928576 (accessed on 6 April 2020).
- The Efficacy and Safety of Oral Azacitidine Plus Best Supportive Care Versus Placebo and Best Supportive Care in Subjects With Red Blood Cell (RBC) Transfusion-Dependent Anemia and Thrombocytopenia Due to International Prognostic Scoring System (IPSS). Available online: https://clinicaltrials.gov/ct2/show/NCT01566695 (accessed on 6 April 2020).
- Wong, E.; Juneja, S. Acute myeloid leukaemia and myelodysplastic syndromes with 50% or greater erythroblasts: A diagnostic conundrum. Pathology 2015, 47, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Götze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenet. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, A.C.; Döhner, H.; Pleyer, L.; Seymour, J.F.; Fenaux, P.; Dombret, H. Azacitidine in adult patients with acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2017, 116, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Sundar, R.; Cho, B.C.; Brahmer, J.R.; Soo, R.A. Nivolumab in NSCLC: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2015, 7, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Arce, C.; Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Candelaria, M.; Dueñnas-Gonzalez, A. Hydralazine target: From blood vessels to the epigenome. J. Transl. Med. 2006, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Cervera, E.; Candelaria, M.; López-Navarro, O.; Labardini, J.; Gonzalez-Fierro, A.; Taja-Chayeb, L.; Cortes, J.; Gordillo-Bastidas, D.; Dueñas-González, A. Epigenetic therapy with hydralazine and magnesium valproate reverses imatinib resistance in patients with chronic myeloid leukemia. Clin. Lymphoma Myeloma Leuk 2012, 12, 207–212. [Google Scholar] [CrossRef]
- Candelaria, M.; Gallardo-Rincón, D.; Arce, C.; Cetina, L.; Aguilar-Ponce, J.L.; Arrieta, O.; González-Fierro, A.; Chávez-Blanco, A.; de la Cruz-Hernández, E.; Camargo, M.F.; et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2007, 18, 1529–1538. [Google Scholar] [CrossRef]
- De La Cruz-Hernández, E.; Pérez-Cárdenas, E.; Contreras-Paredes, A.; Cantú, D.; Mohar, A.; Lizano, M.; Dueñas-González, A. The effects of DNA methylation and histone deacetylase inhibitors on human papillomavirus early gene expression in cervical cancer, an in vitro and clinical study. Virol. J. 2007, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Hogarth, L.; Hall, A.G.; Skitt, L.; Coulthard, S.A. Epigenetic effects of the thiopurine drugs. Cancer Res. 2005, 65, 647. [Google Scholar]
- Liew, E.; Owen, C. Familial myelodysplastic syndromes: A review of the literature. Haematologica 2011, 96, 1536–1542. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Almeida, A.; Giagounidis, A.; Platzbecker, U.; Garcia, R.; Voso, M.T.; Larsen, S.R.; Valcarcel, D.; Silverman, L.R.; Skikne, B.; et al. Design and rationale of the QUAZAR Lower-Risk MDS (AZA-MDS-003) trial: A randomized phase 3 study of CC-486 (oral azacitidine) plus best supportive care vs placebo plus best supportive care in patients with IPSS lower-risk myelodysplastic syndromes and po. BMC Hematol. 2016, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, W.C.; Yeh, S.P.; Hsiao, L.T.; Lin, S.F.; Chen, Y.C.; Chen, T.Y.; Laille, E.; Galettis, A.; Dong, Q.; Songer, S.; et al. Efficacy, safety, and pharmacokinetics of subcutaneous azacitidine in Taiwanese patients with higher-risk myelodysplastic syndromes. Asia Pac. J. Clin. Oncol. 2017, 13, e430–e439. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Bacalini, M.G.; Gentilini, D.; Boattini, A.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Scurti, M.; Remondini, D.; Capri, M.; et al. Identification of a DNA methylation signature in blood cells from persons with down syndrome. Aging (Albany N.Y.) 2015, 7, 82–96. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Completed Research Studies and Clinical Trials | |||||
|---|---|---|---|---|---|
| No. | Study Title | Condition(s) | Intervention(s) | Reference | ClinicalTrials.gov Identifier |
| 1. | A Phase II Study of Epigenetic Therapy to Overcome Chemotherapy Resistance in Refractory Solid Tumors | Refractory solid tumours | Hydralazine and magnesium valproate | [119] | NCT00404508 |
| 2. | Gene Expression Variation and Implant Wound Healing among Smokers and Diabetics | - Smoking - Diabetes | Dental implant surgery | [120] | NCT01663298 |
| 3. | Histone Methyltransferase G9a and H3K9 Dimethylation Inhibit the Self-Renewal of Glioma Cancer Stem Cells | Glioma | Histone methyltransferase G9a and H3K9 dimethylation | [121] | N/A |
| 4. | Hydralazine and Valproate Plus Cisplatin Chemoradiation in Cervical Cancer | Cervical cancer | Hydralazine and magnesium valproate | [122] | NCT00404326 |
| 5. | Phase I Study of MG98, an Oligonucleotide Antisense Inhibitor of Human DNA Methyltransferase 1, Given as a 7-Day Infusion in Patients with Advanced Solid Tumors | Cancer | MG98, an oligonucleotide to DNA DNMT1 | [123] | N/A |
| 6. | Non-Canonical PRC1.1 Targets Active Genes Independent of H3K27me3 and Is Essential for Leukemogenesis | Acute myeloid leukaemia (AML) | Downregulation of non-canonical PRC1.1 complex delays or prevents both carcinogenesis and its development in mice models | [124] | N/A |
| 7. | Study of Azacitidine in Adult Taiwanese Subjects with Higher-Risk Myelodysplastic Syndromes (MDS) | Myelodysplastic syndromes | Azacitidine | [125] | NCT01201811 |
| 8. | The Linker Histone H1.0 Generates Epigenetic and Functional Intratumor Heterogeneity | - Breast cancer - Glioma and glioblastoma - Melanoma - Kidney renal papillary cell carcinoma Liver cancer | Reversible-silencing of linker histone H1.0 to manipulate tumour proliferation | [126] | N/A |
| 9. | Vidaza to Restore Hormone Thx Prostate | Prostate cancer | Azacitdine for injectable suspension | [127] | NCT00384839 |
| Ongoing Research Studies and Clinical Trials | |||||
|---|---|---|---|---|---|
| No | Study Title | Condition(s) | Intervention(s) | Reference | ClinicalTrials.gov Identifier |
| 1. | A Study of Venetoclax in Combination with Azacitidine versus Azacitidine in Treatment Naïve Subjects with Acute Myeloid Leukemia Who Are Ineligible for Standard Induction Therapy | Acute myeloid leukaemia (AML) | - Azacitidine - Venetoclax - Placebo | [128] | NCT02993523 |
| 2. | A Trial of Epigenetic Priming in Patients with Newly Diagnosed Acute Myeloid Leukemia | - Acute myeloid leukaemia - Myelodysplastic syndromes | - Azacitidine - Decitabine - Cytarabine - Stem cell transplant - etc. | [129] | NCT03164057 |
| 3. | Azacytidine Prior to in Vivo T-cell Depleted Allo Stem Cell Transplant for Patients with Myeloid Malignancies in CR | - Leukaemia - Erythroblastic - Acute myelodysplastic syndromes | - Azacitidine - Fludarabine - Melphalan - Alemtuzumab | [130] | NCT02497404 |
| 4. | Diagnosis of RSTS: Identification of the Acetylation Profiles as Epigenetic Markers for Assessing Causality of CREBBP Variants | Rubinstein–Taybi syndrome | To investigate: - Generation of induced pluripotent Stem cells (iPSC) from fibroblasts obtained by skin biopsy - Histone acetylation profiles of cells of SRT patients with CREBBP mutations - Functional involvement of identified epigenetic alterations - Culture of lymphoblastoid line from blood sample | [131] | NCT04122742 |
| 5. | DNA Methylation in Allogenic Hematopoietic Stem Cell Transplantation | - Aging - Stem cell transplant complications | Investigation comparing DNA methylation of patients | [132] | NCT03871296 |
| 6. | EPIgenetics and in Vivo Resistance of Chronic Myeloid Leukemia Stem Cells to Tyrosine Kinase Inhibitors (EPIK) | - Chronic myeloid leukaemia (CML) - Chronic Phase | Collection of blood and bone marrow | [133] | NCT03481868 |
| 7. | Genetic and Epigenetic Basis of Chronic Wounds | Chronic wounds | Observational | [134] | NCT03793062 |
| 8. | Phase II Anti-PD1 Epigenetic Therapy Study in NSCLC | Non-small-cell lung cancer, epigenetic therapy | - Azacitidine - Entinostat - Nivolumab | [135] | NCT01928576 |
| 9. | The Efficacy and Safety of Oral Azacitidine Plus Best Supportive Care versus Placebo and Best Supportive Care in Subjects with Red Blood Cell (RBC) Transfusion-Dependent Anemia and Thrombocytopenia Due to International Prognostic Scoring System (IPSS) Low Risk Myelodysplastic Syndrome (MDS) | Myelodysplastic syndrome | - Oral azacitidine - Placebo | [136] | NCT01566695 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dompe, C.; Janowicz, K.; Hutchings, G.; Moncrieff, L.; Jankowski, M.; Nawrocki, M.J.; Józkowiak, M.; Mozdziak, P.; Petitte, J.; Shibli, J.A.; et al. Epigenetic Research in Stem Cell Bioengineering—Anti-Cancer Therapy, Regenerative and Reconstructive Medicine in Human Clinical Trials. Cancers 2020, 12, 1016. https://doi.org/10.3390/cancers12041016
Dompe C, Janowicz K, Hutchings G, Moncrieff L, Jankowski M, Nawrocki MJ, Józkowiak M, Mozdziak P, Petitte J, Shibli JA, et al. Epigenetic Research in Stem Cell Bioengineering—Anti-Cancer Therapy, Regenerative and Reconstructive Medicine in Human Clinical Trials. Cancers. 2020; 12(4):1016. https://doi.org/10.3390/cancers12041016
Chicago/Turabian StyleDompe, Claudia, Krzysztof Janowicz, Greg Hutchings, Lisa Moncrieff, Maurycy Jankowski, Mariusz J. Nawrocki, Małgorzata Józkowiak, Paul Mozdziak, Jim Petitte, Jamil A. Shibli, and et al. 2020. "Epigenetic Research in Stem Cell Bioengineering—Anti-Cancer Therapy, Regenerative and Reconstructive Medicine in Human Clinical Trials" Cancers 12, no. 4: 1016. https://doi.org/10.3390/cancers12041016