Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Clinico-pathological Features
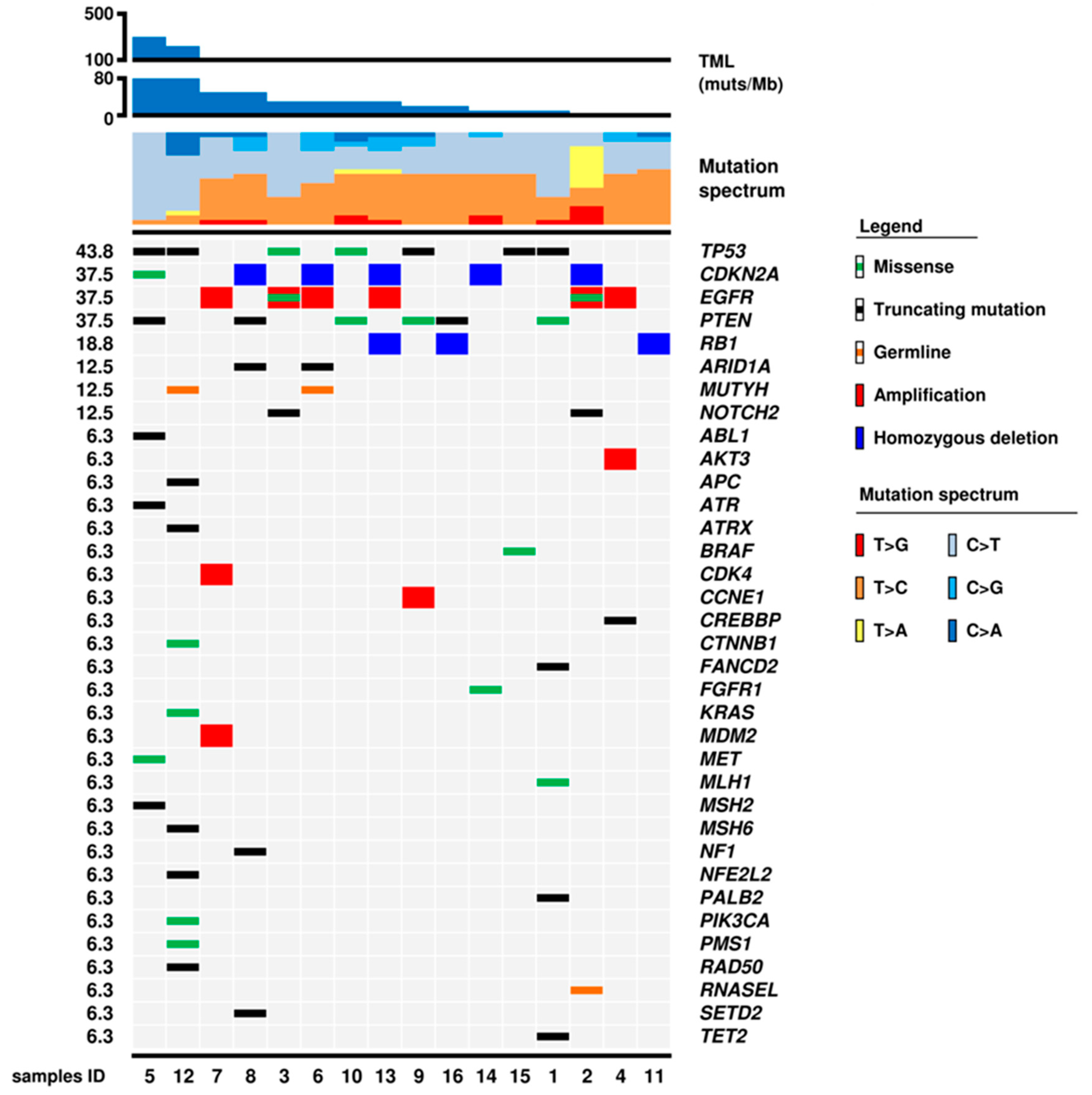
2.2. Mutational Status of 409 Genes
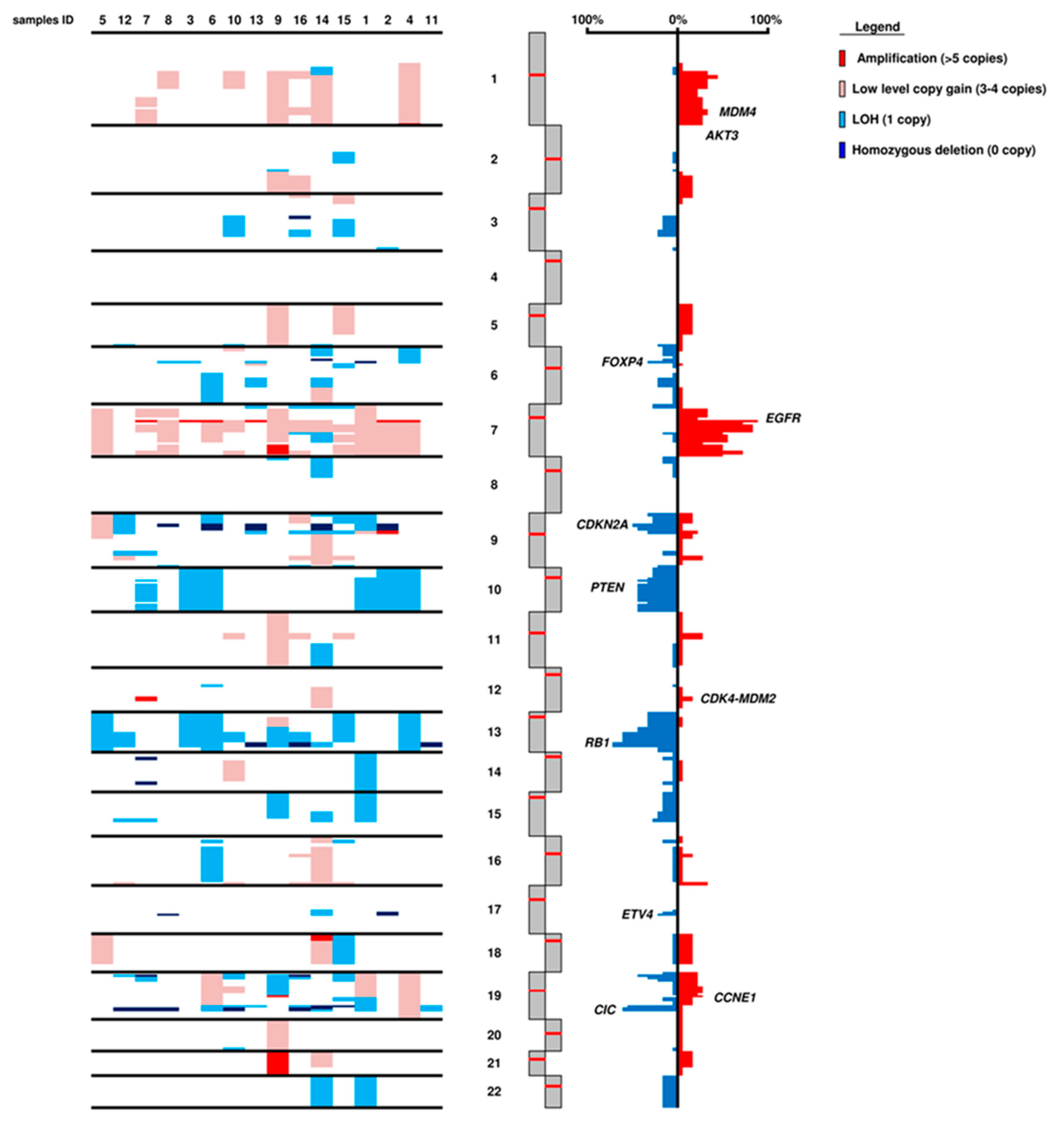
2.3. Gene and Chromosomal Copy Number Alterations
2.4. Tumour Mutational Load
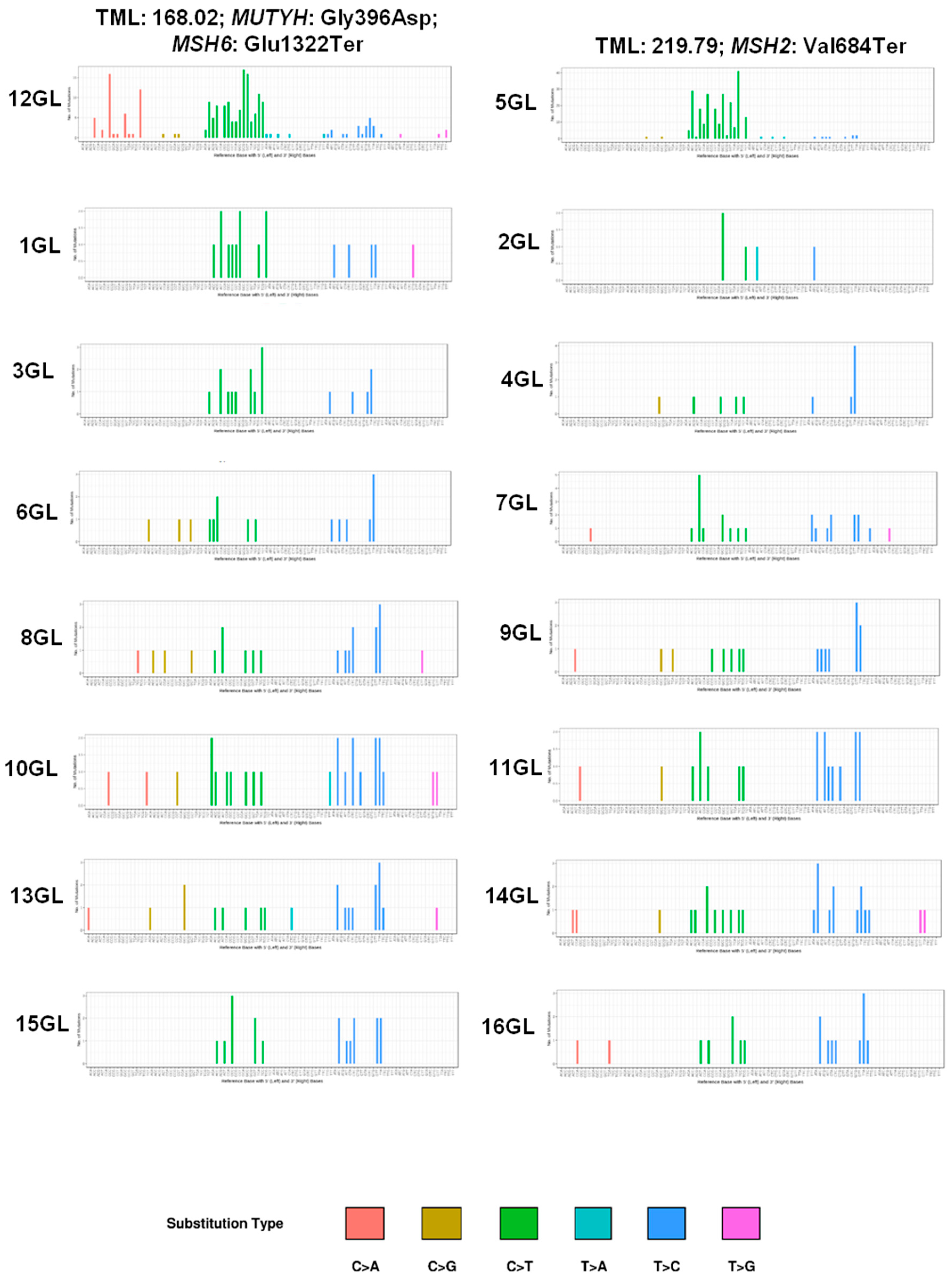
2.5. Mutational Spectrum
2.6. POLE and POLD1 Mutations
2.7. Microsatellite Instability
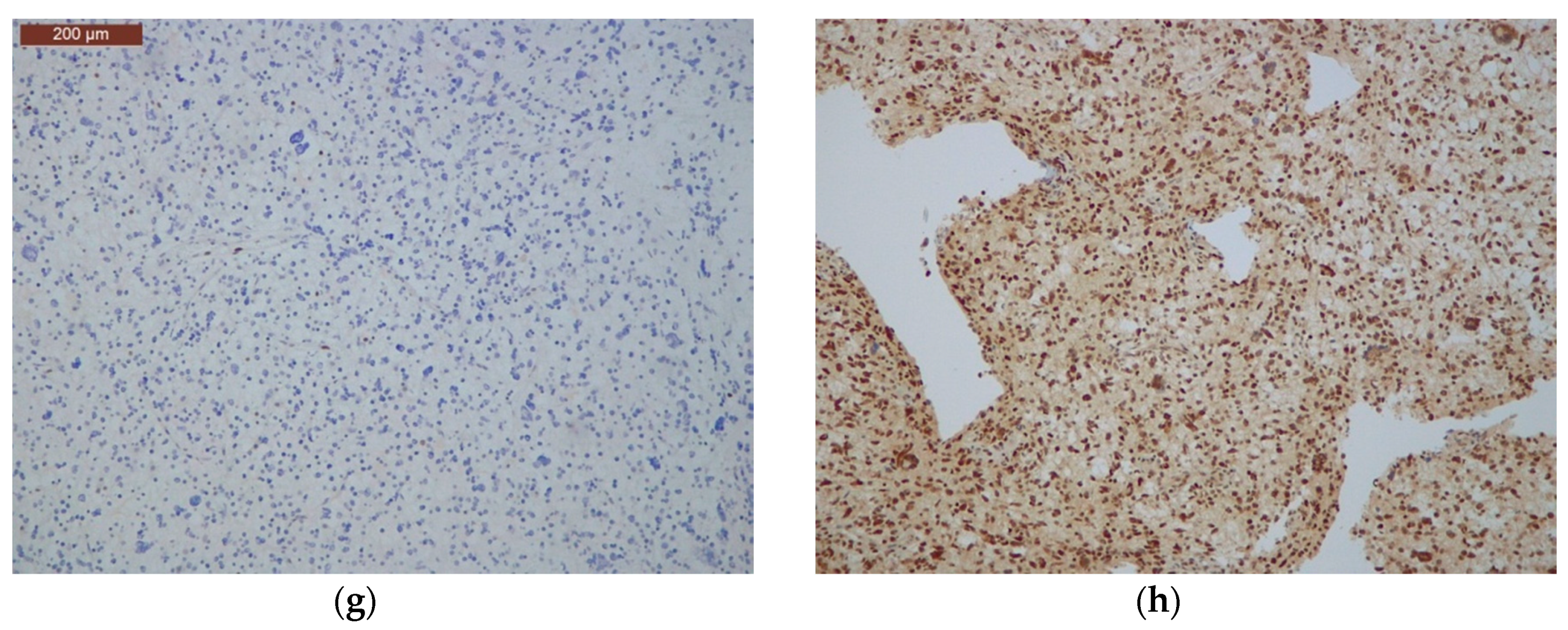
2.8. Immunohistochemical Analysis of Mismatch Repair Proteins
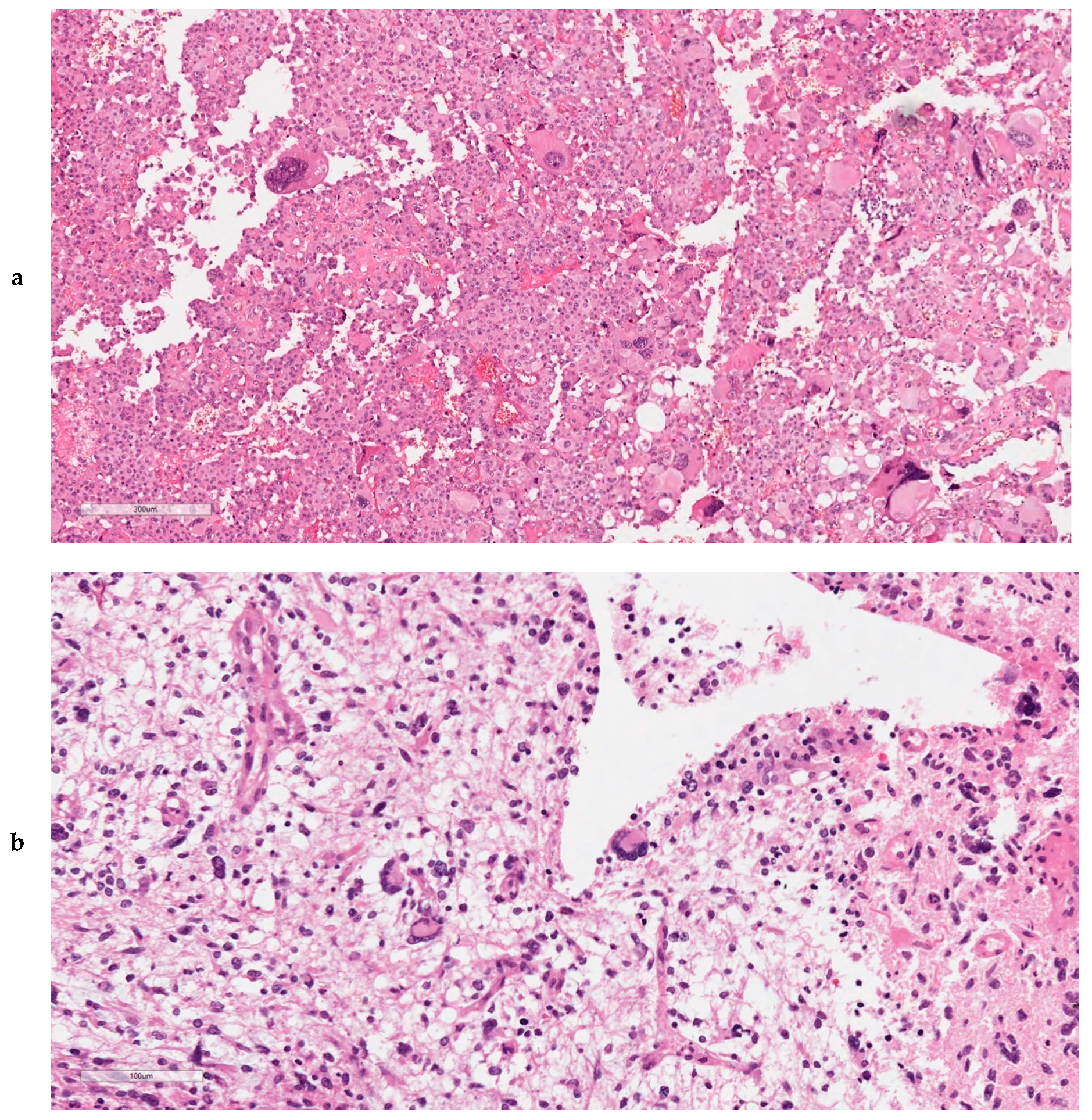
2.9. Ultra-Mutated IDH-wt GBMs Featured High Content of Giant Cells, MMR Protein Loss, and MSI
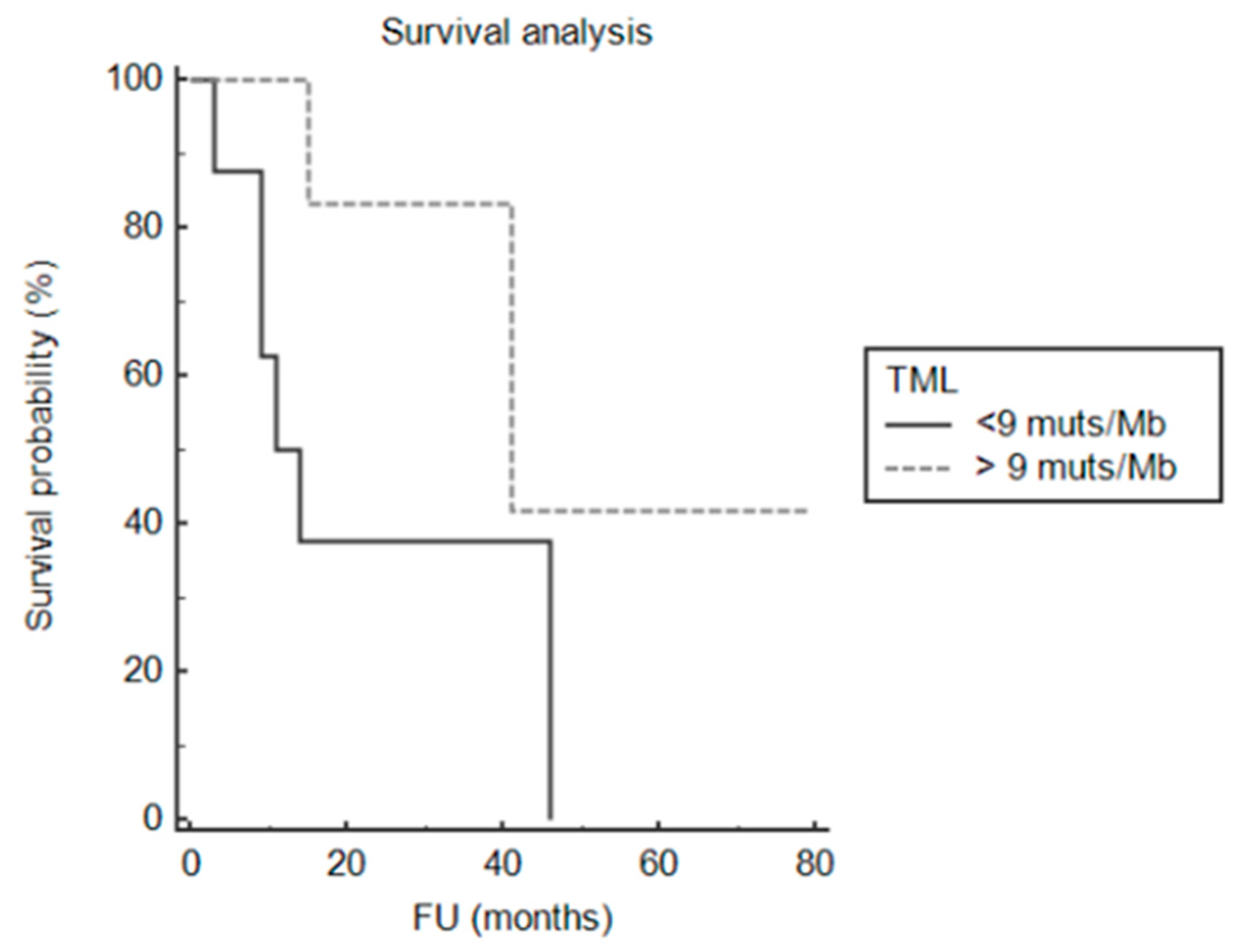
2.10. Survival Analysis
2.11. Ultra-Mutated GBMs in the TCGA PanCancer Atlas
3. Discussion
4. Materials and Methods
4.1. Cases
4.2. Mutational and Copy Number Variation Status of 409 Cancer Genes
4.3. Tumour Mutational Load and Mutational Signatures
4.4. Mutational Analysis of POLE and POLD1 Genes
4.5. Microsatellite Instability Analysis
4.6. Immunohistochemistry of DNA Mismatch Repair Proteins
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumours Diagnosed in the United States in 2011–2015. Neuro-Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide Versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5–year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 Wild Type Anaplastic Astrocytomas Exhibit Worse Prognosis than IDH1-mutated Glioblastomas, and IDH1 Mutation Status Accounts for the Unfavorable Prognostic Effect of Higher Age: Implications for Classification of Gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Yao, Y.; Chan, A.K.; Qin, Z.Y.; Chen, L.C.; Zhang, X.; Pang, J.C.; Li, H.M.; Wang, Y.; Mao, Y.; Ng, H.K.; et al. Mutation Analysis of IDH1 in Paired Gliomas Revealed IDH1 Mutation was not Associated with Malignant Progression but Predicted Longer Survival. PLoS ONE 2013, 8, e67421. [Google Scholar] [CrossRef]
- Reuss, D.E.; Sahm, F.; Schrimpf, D.; Wiestler, B.; Capper, D.; Koelsche, C.; Schweizer, L.; Korshunov, A.; Jones, D.T.; Hovestadt, V.; et al. ATRX and IDH1-R132H Immunohistochemistry with Subsequent Copy Number Analysis and IDH Sequencing As a Basis for an “Integrated” Diagnostic Approach for Adult Astrocytoma, Oligodendroglioma and Glioblastoma. Acta Neuropathol. 2015, 129, 133–146. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hanggi, D.; Wick, W.; et al. Distribution of EGFR Amplification, Combined Chromosome 7 Gain and Chromosome 10 Loss, and TERT Promoter Mutation in Brain Tumours and Their Potential for the Reclassification of IDH-wt Astrocytoma to Glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, Molecular Mechanisms and Markers. Acta Neuropathol 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Luois, D.N.; Ohgaki, H.; Wisteler, O.D.; Cavenee, W.K.; Ellison, D.W.; Figarella-Branger, D.; Perry, A.; Refeinberger, G.; von Deimling, A. WHO Classification of Tumours of the Central Nervous System; IARC: Lyon, France, 2016. [Google Scholar]
- Korber, V.; Yang, J.; Barah, P.; Wu, Y.; Stichel, D.; Gu, Z.; Fletcher, M.N.C.; Jones, D.; Hentschel, B.; Lamszus, K.; et al. Evolutionary Trajectories of IDH(WT) Glioblastomas Reveal a Common Path of Early Tumourigenesis Instigated Years ahead of Initial Diagnosis. Cancer Cell 2019, 35, 692–704.e12. [Google Scholar] [CrossRef]
- Ulgen, E.; Can, O.; Bilguvar, K.; Oktay, Y.; Akyerli, C.B.; Danyeli, A.E.; Yakicier, M.C.; Sezerman, O.U.; Pamir, M.N.; Ozduman, K. Whole Exome Sequencing-based Analysis to Identify DNA Damage Repair Deficiency as a Major Contributor to Gliomagenesis in Adult Diffuse Gliomas. J. Neurosurg. 2019, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO Recommendations on Microsatellite Instability Testing for Immunotherapy in Cancer, and Its Relationship with PD-1/PD-L1 Expression and Tumour Mutational Burden: A Systematic Review-based Approach. Ann. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Langella, T.; Corbetta, C.; Pellegatta, S. Hypermutations in Gliomas: A Potential Immunotherapy Target. Discov. Med. 2017, 23, 113–120. [Google Scholar] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational Burden, Immune Checkpoint Expression, and Mismatch Repair in Glioma: Implications for Immune Checkpoint Immunotherapy. Neuro. Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Severson, E.; Gay, L.; Vergilio, J.A.; Elvin, J.; Suh, J.; Daniel, S.; Covert, M.; Frampton, G.M.; Hsu, S.; et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumour Mutational Burden, and Hypermutation Signatures. Oncologist 2017, 22, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.; Perry, A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef]
- Riehmer, V.; Gietzelt, J.; Beyer, U.; Hentschel, B.; Westphal, M.; Schackert, G.; Sabel, M.C.; Radlwimmer, B.; Pietsch, T.; Reifenberger, G.; et al. Genomic Profiling Reveals Distinctive Molecular Relapse Patterns in IDH1/2 Wild-type Glioblastoma. Genes Chromosomes Cancer 2014, 53, 589–605. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e10. [Google Scholar] [CrossRef]
- Catalogue of Somatic Mutations in Cancer (COSMIC). Version 3. Available online: https://cancer.sanger.ac.uk/cosmic/signatures/SBS/ (accessed on 20 August 2019).
- Haradhvala, N.J.; Kim, J.; Maruvka, Y.E.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.M.; Leshchiner, I.; Kamburov, A.; Mouw, K.W.; et al. Distinct Mutational Signatures Characterize Concurrent Loss of Polymerase Proofreading and Mismatch Repair. Nat. Commun. 2018, 9, 1746. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumours from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutational Signatures (V3—May 2019). Available online: https://cancer.sanger.ac.uk/cosmic/signatures/SBS (accessed on 20 August 2019).
- Oh, J.E.; Ohta, T.; Nonoguchi, N.; Satomi, K.; Capper, D.; Pierscianek, D.; Sure, U.; Vital, A.; Paulus, W.; Mittelbronn, M.; et al. Genetic Alterations in Gliosarcoma and Giant Cell Glioblastoma. Brain Pathol. 2016, 26, 517–522. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.N.; Cottrell, C.E.; Vigh-Conrad, K.A.; Carter, J.H.; Heusel, J.W.; Ansstas, G.; Dahiya, S. Beyond Sequence Variation: Assessment of Copy Number Variation in Adult Glioblastoma Through Targeted Tumour Somatic Profiling. Hum. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Erson-Omay, E.Z.; Caglayan, A.O.; Schultz, N.; Weinhold, N.; Omay, S.B.; Ozduman, K.; Koksal, Y.; Li, J.; Serin Harmanci, A.; Clark, V.; et al. Somatic POLE Mutations Cause an Ultramutated Giant Cell High-grade Glioma Subtype with Better Prognosis. Neuro. Oncol. 2015, 17, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Vande Perre, P.; Siegfried, A.; Corsini, C.; Bonnet, D.; Toulas, C.; Hamzaoui, N.; Selves, J.; Chipoulet, E.; Hoffmann, J.S.; Uro-Coste, E.; et al. Germline Mutation p.N363K in POLE is Associated with an Increased Risk of Colorectal Cancer and Giant Cell Glioblastoma. Fam. Cancer 2019, 18, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.F.; Li, K.K.; Kwan, J.S.H.; Yang, R.R.; Aibaidula, A.; Tang, Q.; Bao, Y.; Mao, Y.; Chen, H.; Ng, H.K. Whole-exome Sequencing Revealed Mutational Profiles of Giant Cell Glioblastomas. Brain Pathol. 2019. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. ImmuneCheckpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2019, 34, 2206–2211. [Google Scholar] [CrossRef]
- Simbolo, M.; Gottardi, M.; Corbo, V.; Fassan, M.; Mafficini, A.; Malpeli, G.; Lawlor, R.T.; Scarpa, A. DNA Qualification Workflow for Next Generation Sequencing of Histopathological Samples. PLoS ONE 2013, 8, e62692. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the Consequences of Genomic Variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Lienard, M.; Toffoli, S.; Kamal, M.; Le Tourneau, C.; Gentien, D.; Servant, N.; Gestraud, P.; Rio Frio, T.; et al. Multi-factor Data Normalization Enables the Detection of Copy Number Aberrations in Amplicon Sequencing Data. Bioinformatics 2014, 30, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gay, M.; Vila-Casadesus, M.; Franch-Exposito, S.; Hernandez-Illan, E.; Lozano, J.J.; Castellvi-Bel, S. Mutational Signatures in Cancer (MuSiCa): A Web Application to Implement Mutational Signatures Analysis In Cancer Samples. BMC Bioinformatics 2018, 19, 224. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Gender | Age | Site | OS (months) | IDH1/2 | EGFR | ATRX | TP53 | PTEN | TML | MMR Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 GL | M | 49 | fronto-temporal right | DOD (11) | wt | ampl | 5.28 | ||||
| 6 GL | M | 41 | fronto-temporal right | DOD (3) | wt | ampl | 5.66 | ||||
| 2 GL | F | 49 | temporo-parietal left | DOD (46) | wt | ampl/mut | 6.34 | ||||
| 15 GL | M | 43 | fronto-basal | DOD (14) | wt | mut | 6.47 | ||||
| 16 GL | M | 40 | temporal left | DOD (9) | wt | mut | 7.12 | ||||
| 7 GL | M | 39 | temporo-parieto-occipital left | DOD (14) | wt | ampl | 7.33 | ||||
| 13 GL | F | 41 | temporal left | Alive (24) | wt | ampl | 7.75 | ||||
| 9 GL | M | 18 | temporo-parietal left | Alive (38) | wt | mut | mut | 8.22 | |||
| 11 GL | F | 29 | cerebellar right | DOD (15) | wt | 9.24 | |||||
| 10 GL | M | 43 | frontal | DOD (15) | wt | mut | mut | 10.1 | |||
| 8 GL | M | 46 | temporo-parietal left | Alive (41) | wt | mut | 12.35 | ||||
| 1 GL | F | 47 | temporo-parieto-occipital right | Alive (23) | wt | mut | mut | 13.07 | MLH1 mut * | ||
| 14 GL | M | 43 | frontal lobe | Alive (14) | wt | mut | 14.34 | ||||
| 3 GL | M | 24 | temporal left | Alive (24) | wt | ampl/mut | mut | 43.19 | |||
| 12 GL | M | 39 | frontal | Alive (32) | wt | mut | mut | 168.02 | MSH6 mut | ||
| 5 GL | F | 37 | frontal | Alive (79) | wt | mut | mut | 219.79 | MSH2 mut |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barresi, V.; Simbolo, M.; Mafficini, A.; Piredda, M.L.; Caffo, M.; Cardali, S.M.; Germanò, A.; Cingarlini, S.; Ghimenton, C.; Scarpa, A. Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment. Cancers 2019, 11, 1279. https://doi.org/10.3390/cancers11091279
Barresi V, Simbolo M, Mafficini A, Piredda ML, Caffo M, Cardali SM, Germanò A, Cingarlini S, Ghimenton C, Scarpa A. Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment. Cancers. 2019; 11(9):1279. https://doi.org/10.3390/cancers11091279
Chicago/Turabian StyleBarresi, Valeria, Michele Simbolo, Andrea Mafficini, Maria Liliana Piredda, Maria Caffo, Salvatore Massimiliano Cardali, Antonino Germanò, Sara Cingarlini, Claudio Ghimenton, and Aldo Scarpa. 2019. "Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment" Cancers 11, no. 9: 1279. https://doi.org/10.3390/cancers11091279