Plasma Biomarkers and Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: New Tools for Better Patient Selection?

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Circulating PD-L1
2.1. Prognostic Role of sPD-L1 in NSCLC
2.2. sPD-L1 in NSCLC Patients Treated with Radiotherapy
2.3. sPD-L1 in NSCLC Patients Treated with ICIs
2.4. Source and Biological Activity of sPD-L1
3. Other Soluble Proteins
3.1. Granzyme B
3.2. PD-L2, IL-2, IFN-Gamma
4. Circulating Tumor DNA (ctDNA)
4.1. ctDNA Quantitative Approach and ICIs in NSCLC
4.2. Tumor Mutational Burden (TMB)
4.3. Current Limitations for ctDNA and bTMB Use as Biomarkers for ICIs in NSCLC
5. New Plasma Biomarkers: Blood Microbiome and Plasma Marker of the Intestinal Barrier
6. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spige, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip., E.; Holgado., E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.Y.; Koh, J.; Go, H.; Lee, D.S.; Jeon, Y.K.; Chung, D.H. Programmed death-1 ligand 1 and 2 are highly expressed in pleomorphic carcinomas of the lung: Comparison of sarcomatous and carcinomatous areas. Eur. J. Cancer 2015, 51, 2698–2707. [Google Scholar] [CrossRef]
- Ilie, M.; Long-Mira, E.; Bence, C.; Butori, C.; Lassalle, S.; Bouhlel, L.; Fazzalari, L.; Zahaf, K.; Lalvée, S.; Washetine, K.; et al. Comparative study of the PDL1 status between surgically resected specimens and matched biopsies of NSCLC patients reveal major discordances: A potential issue for anti-PD-L1 therapeutic strategies. Ann. Oncol. 2016, 27, 147–153. [Google Scholar] [CrossRef]
- Li, C.; Huang, C.; Mok, T.S.; Zhuang, W.; Xu, H.; Miao, Q.; Fan, X.; Zhu, W.; Huang, Y.; Lin, X.; et al. Comparison of 22C3 PD-L1 Expression between Surgically Resected Specimens and Paired Tissue Microarrays in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, D.; Clavé, S.; Taus, Á.; Hardy-Werbin, M.; Rocha, P.; Lorenzo, M.; Menéndez, S.; Salido, M.; Albanell, J.; Pijuan, L.; et al. Heterogeneity of Tumor and Immune Cell PD-L1 Expression and Lymphocyte Counts in Surgical NSCLC Samples. Clin. Lung Cancer 2017, 18, 682–691.e5. [Google Scholar] [CrossRef] [PubMed]
- Uruga, H.; Bozkurtlar, E.; Huynh, T.G.; Muzikansky, A.; Goto, Y.; Gomez-Caraballo, M.; Hata, A.N.; Gainor, J.F.; Mark, E.J.; Engelman, J.A.; et al. Programmed Cell Death Ligand (PD-L1) Expression in Stage II and III Lung Adenocarcinomas and Nodal Metastases. J. Thorac. Oncol. 2017, 12, 458–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinato, D.J.; Shiner, R.J.; White, S.D.; Black, J.R.; Trivedi, P.; Stebbing, J.; Sharma, R.; Mauri, F.A. Intra-tumoral heterogeneity in the expression of programmed-death (PD) ligands in isogeneic primary and metastatic lung cancer: Implications for immunotherapy. Oncoimmunology 2016, 5, e1213934, eCollection 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, A.S.; Aubry, M.C.; Moser, J.C.; Harrington, S.M.; Dronca, R.S.; Park, S.S.; Dong, H. Temporal and spatial discordance of programmed cell death-ligand 1 expression and lymphocyte tumor infiltration between paired primary lesions and brain metastases in lung cancer. Ann. Oncol. 2016, 27, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Fang, W.; Yu, J.; Chen, N.; Zhan, J.; Ma, Y.; Yang, Y.; Huang, Y.; Zhao, H.; Zhang, L. Expression of programmed death ligand-1 on tumor cells varies pre and post chemotherapy in non-small cell lung cancer. Sci. Rep. 2016, 6, 20090. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Hong, M.; Ahn, S.; Choi, Y.L.; Kim, K.M.; Oh, D.; Ahn, Y.C.; Jung, S.H.; Ahn, M.J.; Park, K.; et al. Changes in tumour expression of programmed death-ligand 1 after neoadjuvant concurrent chemoradiotherapy in patients with squamous oesophageal cancer. Eur. J. Cancer 2016, 52, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Frigola, X.; Inman, B.A.; Lohse, C.M.; Krco, C.J.; Cheville, J.C.; Thompson, R.H.; Leibovich, B.; Blute, M.L.; Dong, H.; Kwon, E.D. Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin. Cancer Res. 2011, 17, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, J.; Li, Y.; Nie, J.; Dai, L.; Hu, W.; Chen, X.; Han, J.; Ma, X.; Tian, G.; et al. Circulating PD-L1 in NSCLC patients and the correlation between the level of PD-L1 expression and the clinical characteristics. Thorac. Cancer 2015, 6, 534–538. [Google Scholar] [CrossRef]
- Okuma, Y.; Hosomi, Y.; Nakahara, Y.; Watanabe, K.; Sagawa, Y.; Homma, S. High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer 2017, 104, 1–6. [Google Scholar] [CrossRef]
- Ding, Y.; Sun, C.; Li, J.; Hu, L.; Li, M.; Liu, J.; Pu, L.; Xiong, S. The prognostic significance of soluble programmed death ligand 1 expression in cancers: A systematic review and meta-anlysis. Scand. J. Immunol. 2017, 86, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Xu, B.; Wang, Y.; Wu, C.; Jiang, J.; Wu, C. Prognostic significance of circulating soluble programmed death ligand 1 in patients with solid tumors: A meta-analysis. Medicine (Baltimore) 2018, 97, e9617. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, P.; Wang, J.; Xi, Q.; Zhao, X.; Ji, M.; Hu, G. Plasma levels of soluble programmed death ligand-1 may be associated with overall survival in nonsmall cell lung cancer patients receiving thoracic radiotherapy. Medicine (Baltimore) 2017, 96, e6102. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Wakui, H.; Utsumi, H.; Sagawa, Y.; Hosomi, Y.; Kuwano, K.; Homma, S. Soluble Programmed Cell Death Ligand 1 as a Novel Biomarker for Nivolumab Therapy for Non-Small-cell Lung Cancer. Clin. Lung Cancer 2018, 19, 410–417.e1. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Julie, C.; Dumenil, C.; Hélias-Rodzewicz, Z.; Tisserand, J.; Dumoulin, J.; Giraud, V.; Labrune, S.; Chinet, T.; Emile, J.F.; et al. Predictive role of plasmatic biomarkers in advanced non-small cell lung cancer treated by nivolumab. Oncoimmunology 2018, 7, e1452581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigola, X.; Inman, B.A.; Krco, C.J.; Liu, X.; Harrington, S.M.; Bulur, P.A.; Dietz, A.B.; Dong, H.; Kwon, E.D. Soluble B7-H1: Differences in production between dendritic and T cells. Immunol. Lett. 2012, 142, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Kiyotani, K.; Sakata, S.; Nagano, S.; Kumehara, S.; Baba, S.; Besse, B.; Yanagitani, N.; Friboulet, L.; Nishio, M.; et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J. Exp. Med. 2019, 216, 982–1000. [Google Scholar] [CrossRef]
- Trapani, J.A.; Sutton, V.R. Granzyme B: Pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 2003, 15, 533–543. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sutton, V.R.; Sedelies, K.A.; Ciccone, A.; Jenkins, M.; Turner, S.J.; Bird, P.I.; Trapani, J.A. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J. Cell Biol. 2006, 173, 133–144. [Google Scholar] [CrossRef]
- Larimer Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Larimer, B.M.; Bloch, E.; Nesti, S.; Austin, E.E.; Wehrenberg-Klee, E.; Boland, G.; Mahmood, U. The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging. Clin. Cancer Res. 2019, 25, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Oyanagi, J.; Koh, Y.; Sato, K.; Mori, K.; Teraoka, S.; Akamatsu, H.; Kanai, K.; Hayata, A.; Tokudome, N.; Akamatsu, K.; et al. Predictive value of serum protein levels in patients with advanced non-small cell lung cancer treated with nivolumab. Lung Cancer 2019, 132, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; Rodriguez-Ruiz, M.E.; Oñate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Proudhon, C.; Romano, E.; Girard, N.; Lantz, O.; Stern, M.H.; Pierga, J.Y.; Bidard, F.C. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2018, 15, 639–650. [Google Scholar] [CrossRef]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: A proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef]
- Goldberg, S.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef]
- Iijima, Y.; Hirotsu, Y.; Amemiya, K.; Ooka, Y.; Mochizuki, H.; Oyama, T.; Nakagomi, T.; Uchida, Y.; Kobayashi, Y.; Tsutsui, T.; et al. Very early response of circulating tumour-derived DNA in plasma predicts efficacy of nivolumab treatment in patients with non-small cell lung cancer. Eur. J. Cancer 2017, 86, 349–357. [Google Scholar] [CrossRef]
- Giroux Leprieur, E.; Herbretau, G.; Dumenil, C.; Julie, C.; Giraud, V.; Labrune, S.; Dumoulin, J.; Tisserand, J.; Emile, J.F.; Blons, H.; et al. Circulating tumor DNA evaluated by next-generation sequencing is predictive of tumor response and prolonged clinical benefit with nivolumab in advanced non-small cell lung cancer. Oncoimmunology 2018, 7, e1424675. [Google Scholar] [CrossRef]
- Guibert, N.; Mazieres, J.; Delaunay, M.; Casanova, A.; Farella, M.; Keller, L.; Favre, G.; Pradines, A. Monitoring of KRAS-mutated ctDNA to discriminate pseudo-progression from true progressionduring anti-PD-1 treatment of lung adenocarcinoma. Oncotarget 2017, 8, 38056–38060. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology: Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 2018, 33, 853–861.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients with Non-Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef]
- Ouaknine Krief, J.; Helly de Tauriers, P.; Dumenil, C.; Neveux, N.; Dumoulin, J.; Giraud, V.; Labrune, S.; Tisserand, J.; Julie, C.; Emile, J.F.; et al. Role of antibiotic use, plasma citrulline and blood microbiome in advanced non-small cell lung cancer patients treated with nivolumab. J. Immunother. Cancer 2019, 7, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crenn, P.; Messing, B.; Cynober, L. Citrulline as a biomarker of intestinal failure due to enterocyte mass reduction. Clin. Nutr. 2008, 27, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]


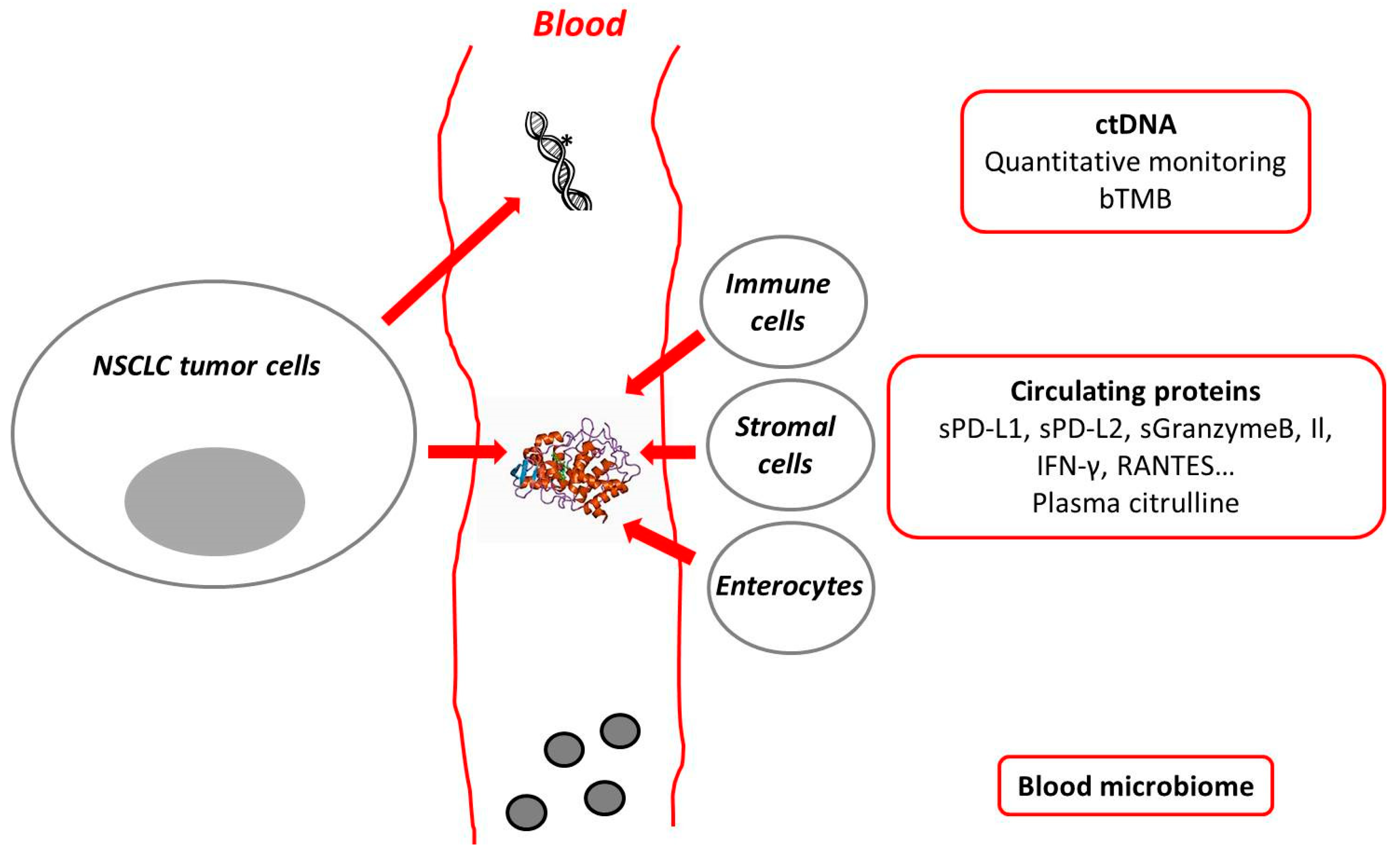{kind=link}
| Study | Patients (n) | Time of sPD-L1 Measure | sPD-L1 Cut-Off | Treatment | Main Results |
|---|---|---|---|---|---|
| Zhang et al. [19] | Advanced NSCLC (109), healthy controls (65) | Diagnosis | 0.636 ng/mL | NS | Higher sPD-L1 levels in patients than controls Shorter OS in high sPD-L1 patients |
| Okuma et al. [20] | Advanced or postsurgical recurrent lung cancer (96) | Before initiation of CT or at least 3–4 weeks after last CT | 7.32 ng/mL | CT | Shorter OS in high sPD-L1 patients |
| Zhao et al. [23] | Locally advanced or inoperable NSCLC (126) | Diagnosis, Week 2 and 4 of treatment | 0.0965 ng/mL | TRT ± CT | Decrease of sPD-L1 during TRT Shorter OS in high sPD-L1 patients |
| Okuma et al. [24] | Advanced or recurrent NSCLC (39) | Baseline | 3.357 ng/mL | nivolumab | Shorter OS TTF in high sPD-L1 patients Higher rates of CR, PR, and SD in low sPD-L1 patients |
| Costantini et al. [25] | Advanced NSCLC (43) | At initial diagnosis, at nivolumab initiation, at first tumor evaluation | 0.0337 ng/mL | nivolumab | sPD-L1 at first tumor evaluation higher in non-responders Patients with clinical benefit had lower sPD-L1 levels at first tumor evaluation Patients with increasing sPD-L1 levels had worse ORR, lower rates of clinical benefit, shorter PFS and OS |
| Study | Patients (n) | Variable Measured | Cut-Off Used for bTMB | Treatment | Positive Findings |
|---|---|---|---|---|---|
| Cabel et al. [34] | NSCLC, uveal melanoma, MSI colorectal cancer (10) | ctDNA concentrations | NA | nivolumab, pembrolizuamb | PFS, OS |
| Goldberg et al. [35] | Metastatic NSCLC (28) | ctDNA concentrations | NA | Anti PD-1, anti-PD-L1 alone or in combination | Time on treatment, PFS, OS |
| Iijima et al. [36] | Advanced NSCLC (14) | ctDNA concentrations | NA | nivolumab | Durable response |
| Giroux-Leprieur et al. [37] | Advanced NSCLC (15) | ctDNA concentrations by NGS | NA | nivolumab | PFS, clinical benefit |
| Wang et al. [43] | Advanced NSCLC (50) | NCC-GP150 (150 genes) | bTMB ≥ 6 | Anti-PD-1 Anti-PD-L1 | PFS ORR |
| Gandara et al. [44] | Advanced NSCLC (273) | NGS | bTMB ≥ 16 | Atezolizumab (anti-PD-L1) | PFS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costantini, A.; Takam Kamga, P.; Dumenil, C.; Chinet, T.; Emile, J.-F.; Giroux Leprieur, E. Plasma Biomarkers and Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: New Tools for Better Patient Selection? Cancers 2019, 11, 1269. https://doi.org/10.3390/cancers11091269
Costantini A, Takam Kamga P, Dumenil C, Chinet T, Emile J-F, Giroux Leprieur E. Plasma Biomarkers and Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: New Tools for Better Patient Selection? Cancers. 2019; 11(9):1269. https://doi.org/10.3390/cancers11091269
Chicago/Turabian StyleCostantini, Adrien, Paul Takam Kamga, Coraline Dumenil, Thierry Chinet, Jean-François Emile, and Etienne Giroux Leprieur. 2019. "Plasma Biomarkers and Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: New Tools for Better Patient Selection?" Cancers 11, no. 9: 1269. https://doi.org/10.3390/cancers11091269