
The Influence of Arsenic Co-Exposure in a Model of Alcohol-Induced Neurodegeneration in C57BL/6J Mice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Ethanol and Arsenic Exposure
2.3. Blood Ethanol Concentration Determination
2.4. Immunohistochemistry & Immunoreactivity Quantification
2.5. Fluoro-Jade C Staining & Quantification
2.6. CYP2E1 ELISA
2.7. Reactive Oxygen/Nitrogen Species Assay
2.8. Inductively Coupled Plasma Mass Spectrometry & Arsenic Concentrations
2.9. Statistics
3. Results
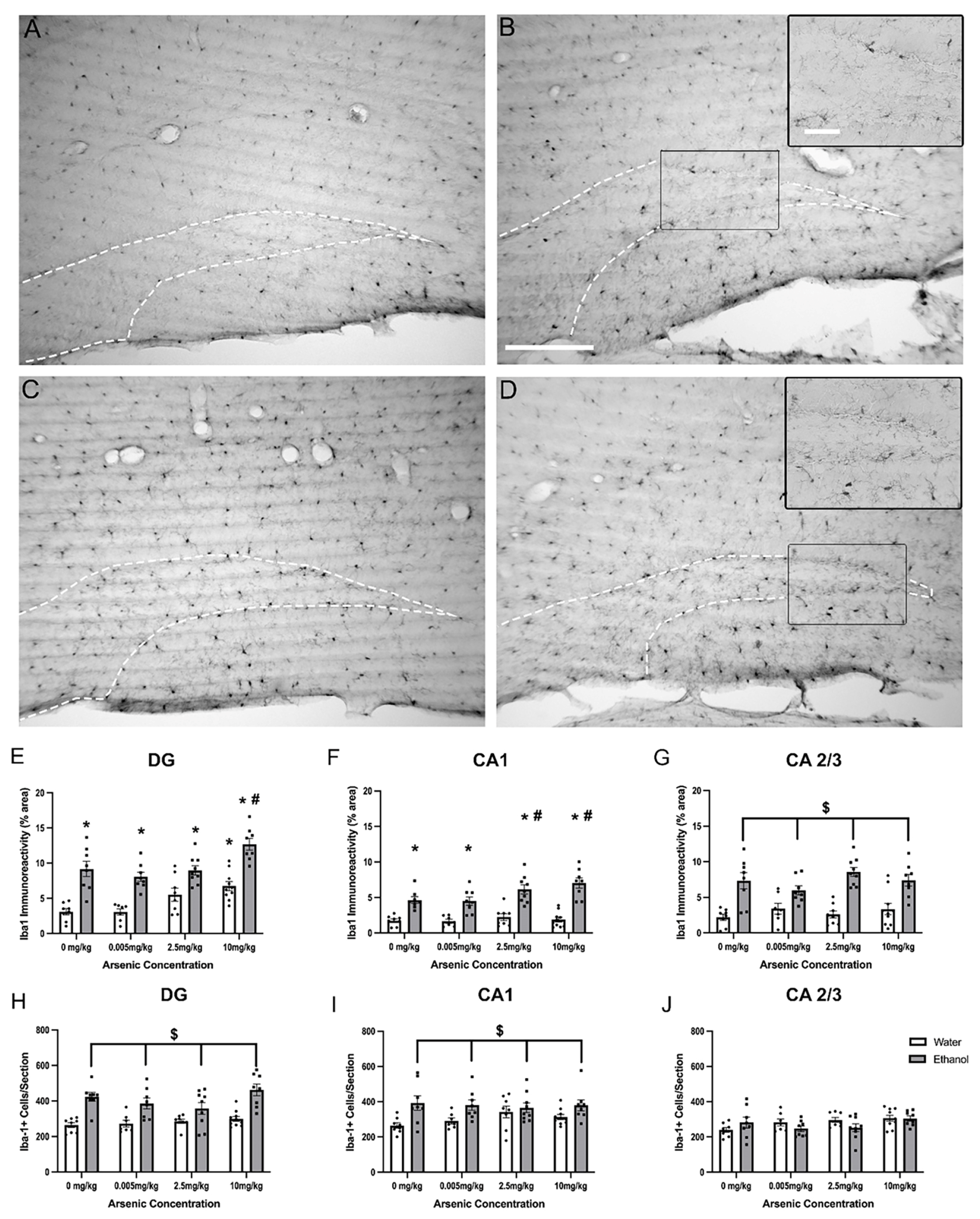
3.1. Effects of Ethanol & Arsenic on Microglial Activation
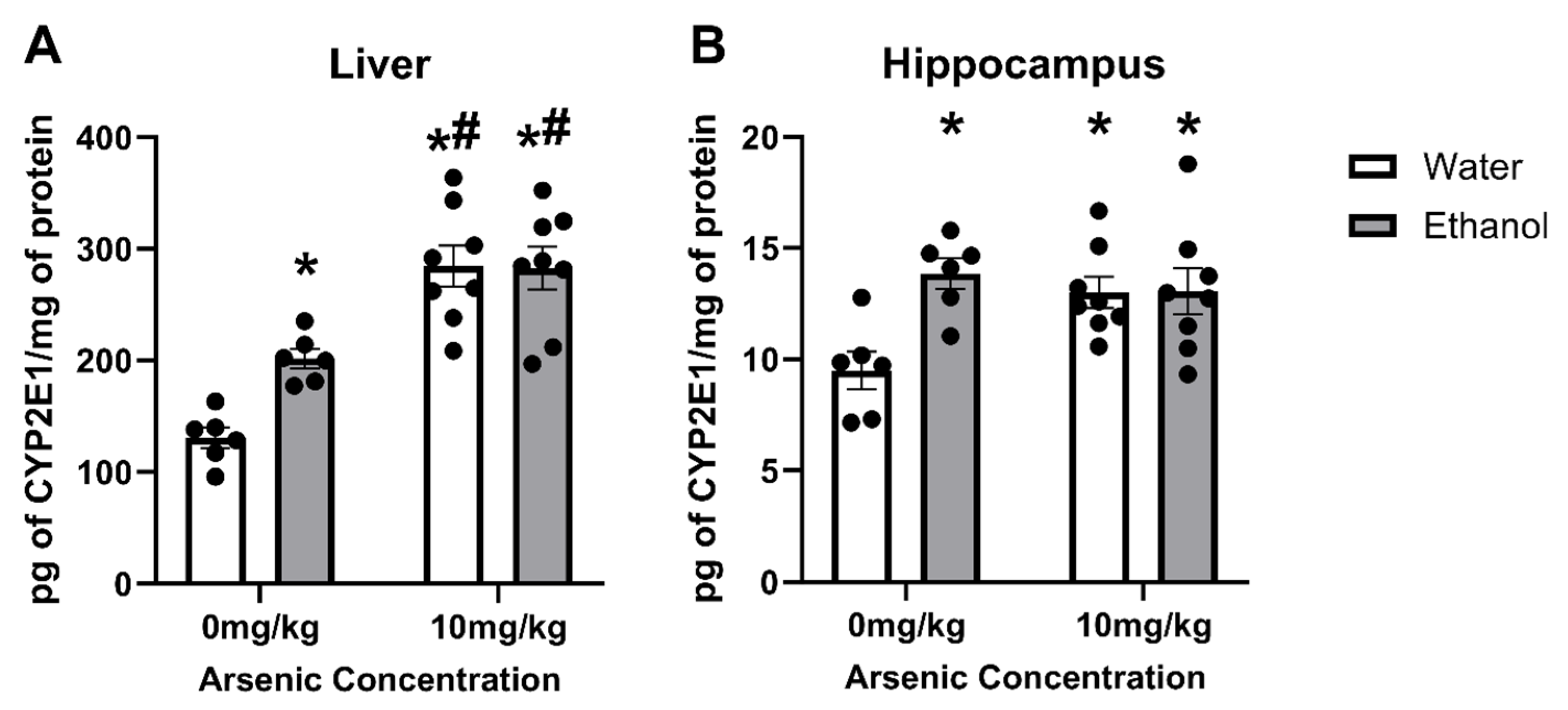
3.2. Arsenic & Ethanol Impacts on CYP2E1 Expression in Liver
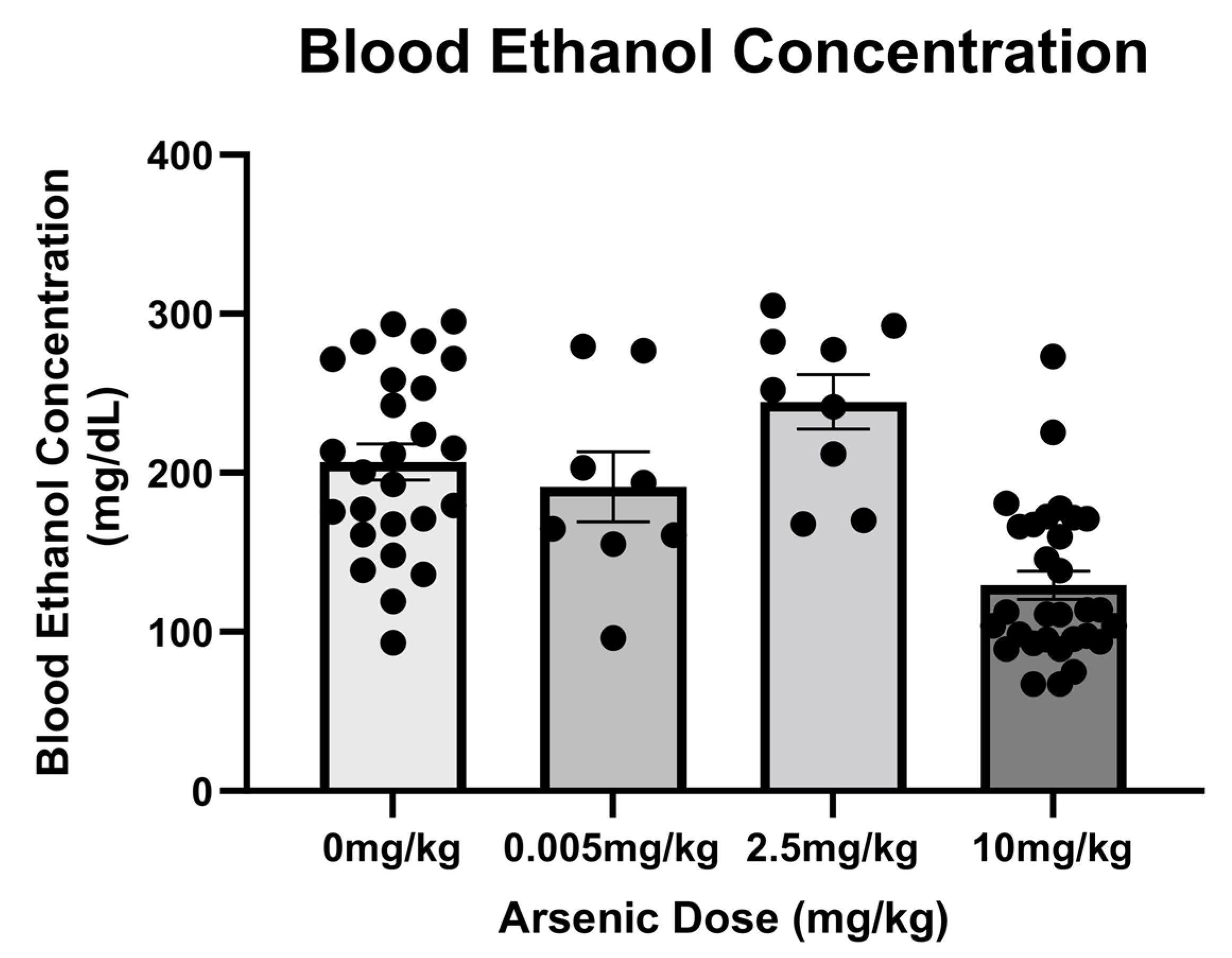
3.3. Arsenic Influences on Blood Ethanol Concentrations
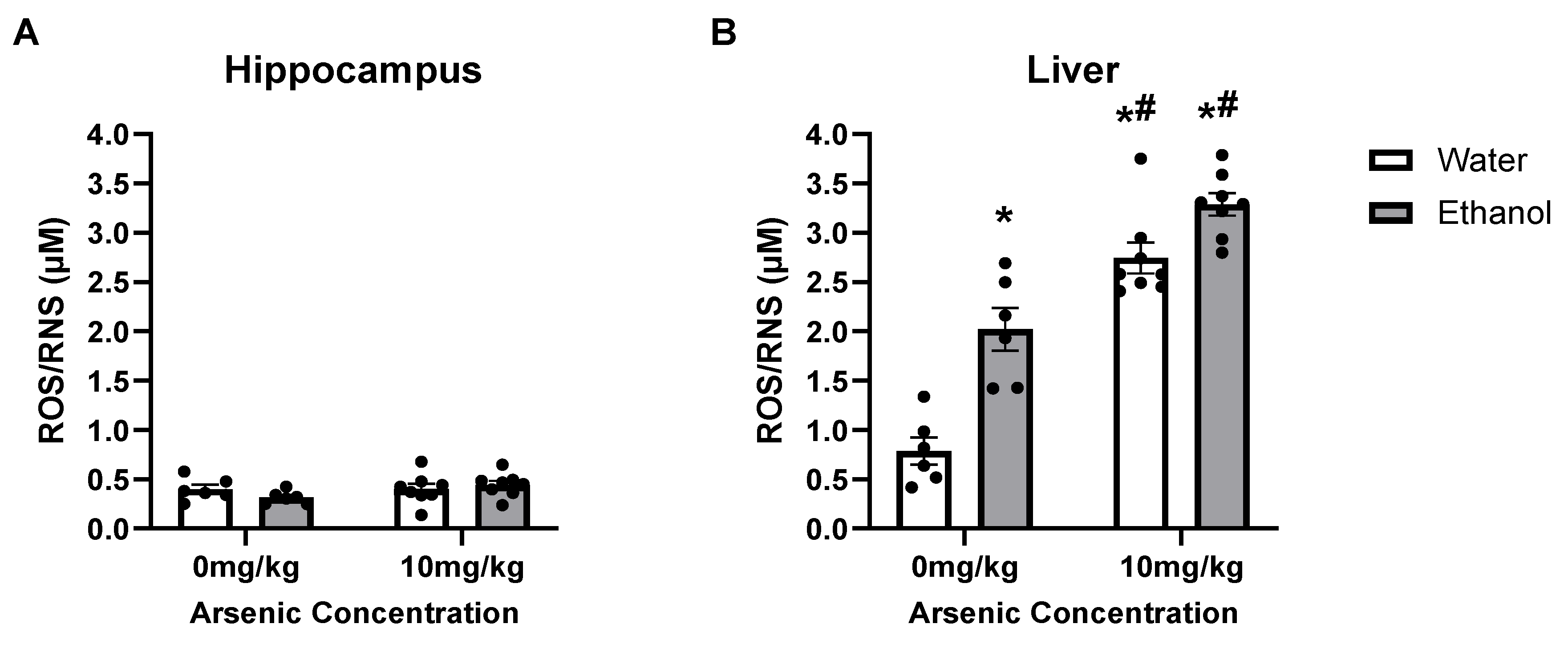
3.4. Ethanol & Arsenic-Induced ROS/RNS Production
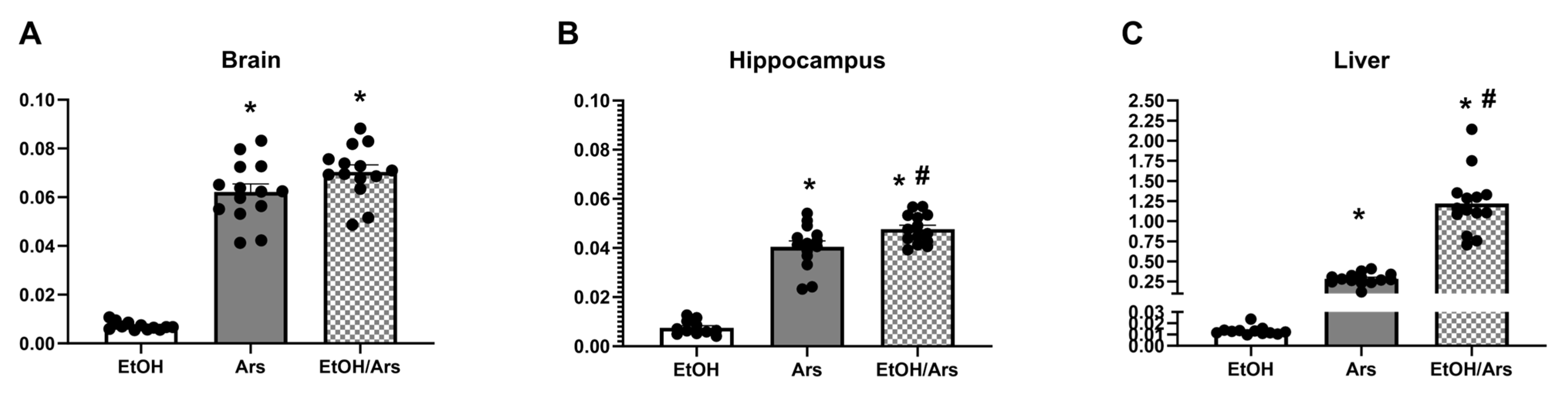
3.5. Ethanol Increases Arsenic Absorption in the Liver and Hippocampus
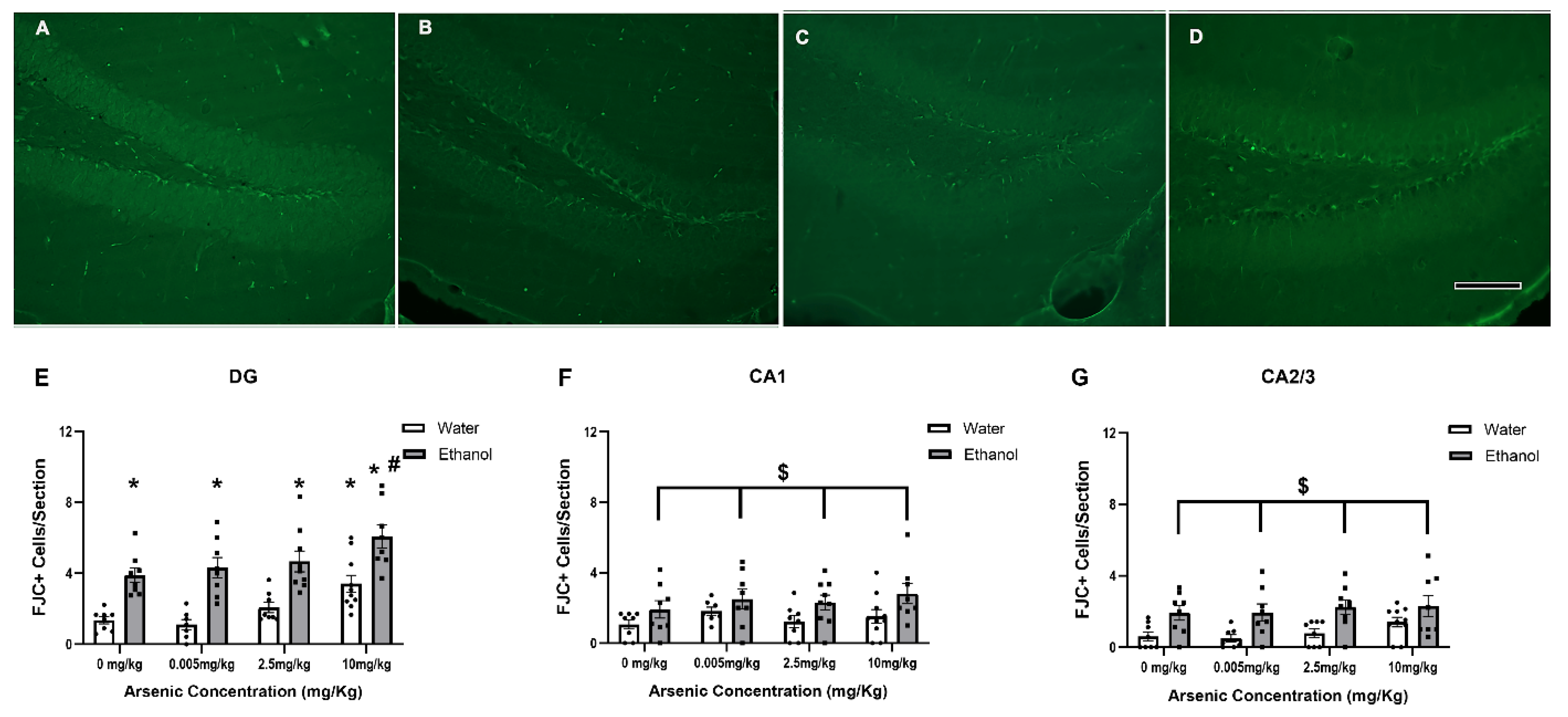
3.6. Regionally Specific Enhancement of FJC+ Cells with Ethanol & Arsenic
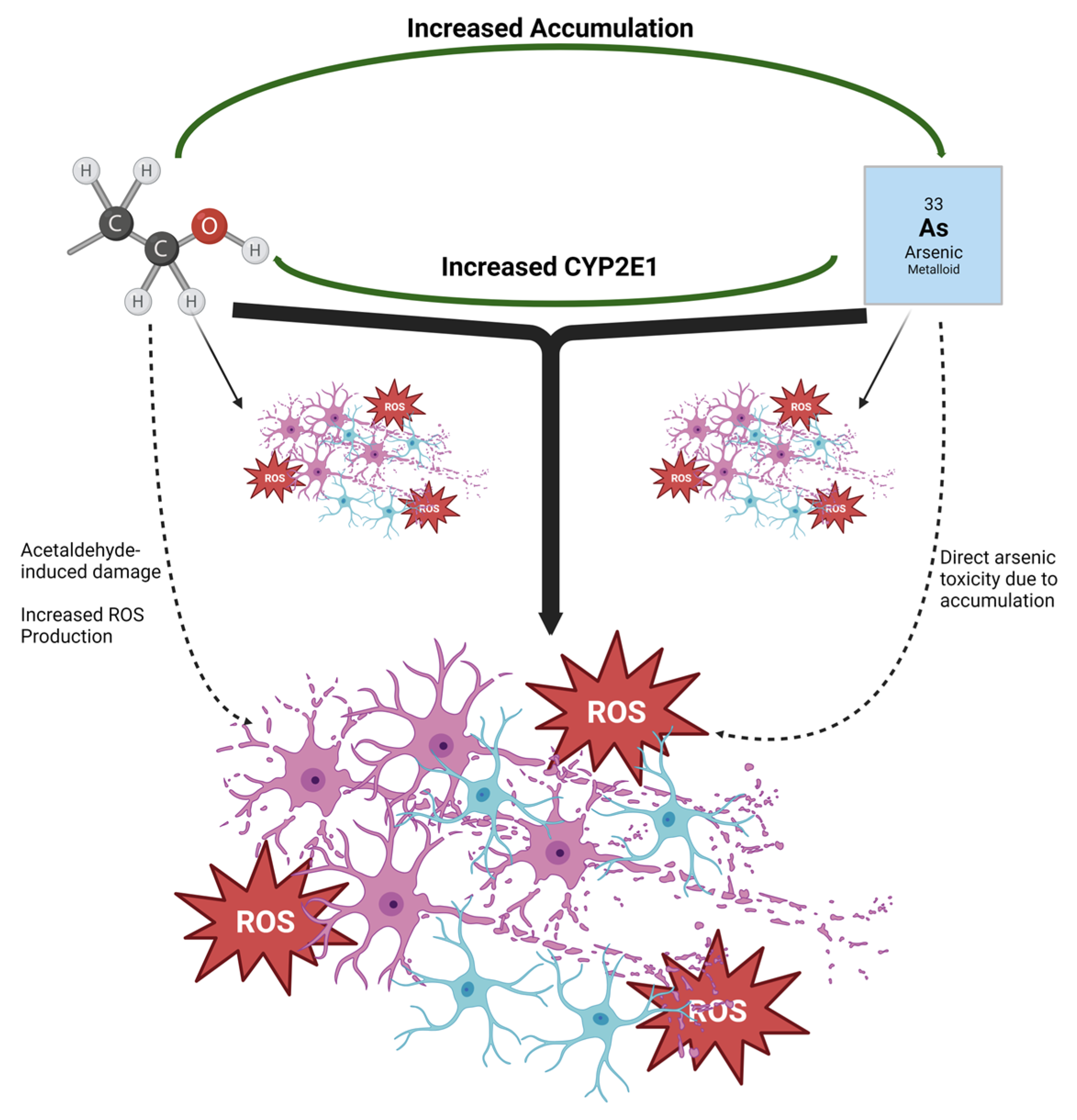
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Alcohol and Drug Use Collaborators. The global burden of disease attributable to alcohol and drug use in 195 countries and territories, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Psychiatry 2018, 5, 987–1012. [Google Scholar] [CrossRef]
- Robinson, E.; Humphreys, G.; Jones, A. Alcohol, calories, and obesity: A rapid systematic review and meta-analysis of consumer knowledge, support, and behavioral effects of energy labeling on alcoholic drinks. Obes. Rev. 2021, 22, e13198. [Google Scholar] [CrossRef]
- Nielsen, S.J.; Kit, B.K.; Fakhouri, T.; Ogden, C.L. Calories consumed from alcoholic beverages by U.S. adults, 2007–2010. NCHS Data Brief 2012, 110, 1–8. [Google Scholar]
- Huang, W.J.; Zhang, X.; Chen, W.W. Association between alcohol and Alzheimer’s disease. Exp. Ther. Med. 2016, 12, 1247–1250. [Google Scholar] [CrossRef]
- Zahr, N.M.; Kaufman, K.L.; Harper, C.G. Clinical and pathological features of alcohol-related brain damage. Nat. Rev. Neurol. 2011, 7, 284–294. [Google Scholar] [CrossRef]
- Geil, C.R.; Hayes, D.M.; McClain, J.A.; Liput, D.J.; Marshall, S.A.; Chen, K.Y.; Nixon, K. Alcohol and adult hippocampal neurogenesis: Promiscuous drug, wanton effects. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 54, 103–113. [Google Scholar] [CrossRef]
- Zahr, N.M.; Pfefferbaum, A. Alcohol’s Effects on the Brain: Neuroimaging Results in Humans and Animal Models. Alcohol. Res. 2017, 38, 183–206. [Google Scholar]
- Pandey, R.; Rai, V.; Mishra, J.; Mandrah, K.; Kumar Roy, S.; Bandyopadhyay, S. From the Cover: Arsenic Induces Hippocampal Neuronal Apoptosis and Cognitive Impairments via an Up-Regulated BMP2/Smad-Dependent Reduced BDNF/TrkB Signaling in Rats. Toxicol. Sci. 2017, 159, 137–158. [Google Scholar] [CrossRef]
- Taheri Zadeh, Z.; Esmaeilpour, K.; Aminzadeh, A.; Heidari, M.R.; Joushi, S. Resveratrol Attenuates Learning, Memory, and Social Interaction Impairments in Rats Exposed to Arsenic. Biomed. Res. Int. 2021, 2021, 9993873. [Google Scholar] [CrossRef]
- Paustenbach, D.J.; Insley, A.L.; Maskrey, J.R.; Bare, J.L.; Unice, K.M.; Conrad, V.B.; Iordanidis, L.; Reynolds, D.W.; DiNatale, K.S.; Monnot, A.D. Analysis of Total Arsenic Content in California Wines and Comparison to Various Health Risk Criteria. Am. J. Enol. Vitic. 2016, 67, 179–187. [Google Scholar] [CrossRef]
- Redan, B.W.; Jablonski, J.E.; Halverson, C.; Jaganathan, J.; Mabud, M.A.; Jackson, L.S. Factors Affecting Transfer of the Heavy Metals Arsenic, Lead, and Cadmium from Diatomaceous-Earth Filter Aids to Alcoholic Beverages during Laboratory-Scale Filtration. J. Agric. Food Chem. 2019, 67, 2670–2678. [Google Scholar] [CrossRef]
- Passaghe, P.; Bertoli, S.; Tubaro, F.; Buiatti, S. Monitoring of some selected heavy metals throughout the brewing process of craft beers by inductively coupled plasma mass spectrometry. Eur. Food Res. Technol. 2015, 241, 199–215. [Google Scholar] [CrossRef]
- Flanagan, S.V.; Spayd, S.E.; Procopio, N.A.; Marvinney, R.G.; Smith, A.E.; Chillrud, S.N.; Braman, S.; Zheng, Y. Arsenic in private well water part 3 of 3: Socioeconomic vulnerability to exposure in Maine and New Jersey. Sci. Total Environ. 2016, 562, 1019–1030. [Google Scholar] [CrossRef]
- Nigra, A.E.; Chen, Q.; Chillrud, S.N.; Wang, L.; Harvey, D.; Mailloux, B.; Factor-Litvak, P.; Navas-Acien, A. Inequalities in Public Water Arsenic Concentrations in Counties and Community Water Systems across the United States, 2006–2011. Environ. Health Perspect. 2020, 128, 127001. [Google Scholar] [CrossRef]
- Ayotte, J.D.; Medalie, L.; Qi, S.L.; Backer, L.C.; Nolan, B.T. Estimating the High-Arsenic Domestic-Well Population in the Conterminous United States. Environ. Sci. Technol. 2017, 51, 12443–12454. [Google Scholar] [CrossRef]
- Ringwald, P.; Chapin, C.; Iceman, C.; Tighe, M.E.; Sisk, M.; Peaslee, G.F.; Peller, J.; Wells, E.M. Characterization and within-site variation of environmental metal concentrations around a contaminated site using a community-engaged approach. Chemosphere 2021, 272, 129915. [Google Scholar] [CrossRef]
- Wuana, R.A.; Okieimen, F.E. Heavy Metals in Contaminated Soils: A Review of Sources, Chemistry, Risks and Best Available Strategies for Remediation. ISRN Ecol. 2011, 2011, 402647. [Google Scholar] [CrossRef]
- Shaji, E.; Santosh, M.; Sarath, K.V.; Prakash, P.; Deepchand, V.; Divya, B.V. Arsenic contamination of groundwater: A global synopsis with focus on the Indian Peninsula. Geosci. Front. 2021, 12, 101079. [Google Scholar] [CrossRef]
- Kumar, V.; Tripathi, V.K.; Jahan, S.; Agrawal, M.; Pandey, A.; Khanna, V.K.; Pant, A.B. Lead Intoxication Synergies of the Ethanol-Induced Toxic Responses in Neuronal Cells—PC12. Mol. Neurobiol. 2015, 52, 1504–1520. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Coronel, I.; Zhang, Y.; Hernández, M.; Boehm Ii, S.L. Low-level developmental lead exposure does not predispose to adult alcohol self-administration, but does increase the risk of relapsing to alcohol seeking in mice: Contrasting role of GLT1 and xCT brain expression. Neuropharmacology 2020, 181, 108339. [Google Scholar] [CrossRef]
- Lukács, A.; Lengyel, Z.; Institóris, L.; Szabó, A. Subchronic heavy metal and alcohol treatment in rats: Changes in the somatosensory evoked cortical activity. Acta Biol. Hung. 2007, 58, 259–267. [Google Scholar] [CrossRef]
- Virgolini, M.B.; Mattalloni, M.S.; Albrecht, P.A.; Deza-Ponzio, R.; Cancela, L.M. Modulation of Ethanol-Metabolizing Enzymes by Developmental Lead Exposure: Effects in Voluntary Ethanol Consumption. Front. Behav. Neurosci. 2017, 11, 95. [Google Scholar] [CrossRef]
- Verma, S.K.; Dua, R.; Gill, K.D. Impaired energy metabolism after co-exposure to lead and ethanol. Basic. Clin. Pharmacol. Toxicol. 2005, 96, 475–479. [Google Scholar] [CrossRef]
- Bao, L.; Shi, H. Potential molecular mechanisms for combined toxicity of arsenic and alcohol. J. Inorg. Biochem. 2010, 104, 1229–1233. [Google Scholar] [CrossRef]
- Grifasi, I.R.; McIntosh, S.E.; Thomas, R.D.; Lysle, D.T.; Thiele, T.E.; Marshall, S.A. Characterization of the Hippocampal Neuroimmune Response to Binge-Like Ethanol Consumption in the Drinking in the Dark Model. Neuroimmunomodulation 2019, 26, 19–32. [Google Scholar] [CrossRef]
- Marshall, S.A.; McClain, J.A.; Kelso, M.L.; Hopkins, D.M.; Pauly, J.R.; Nixon, K. Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiol. Dis. 2013, 54, 239–251. [Google Scholar] [CrossRef]
- Singh, V.; Kushwaha, S.; Gera, R.; Ansari, J.A.; Mishra, J.; Dewangan, J.; Patnaik, S.; Ghosh, D. Sneaky Entry of IFNgamma Through Arsenic-Induced Leaky Blood-Brain Barrier Reduces CD200 Expression by Microglial pro-Inflammatory Cytokine. Mol. Neurobiol. 2019, 56, 1488–1499. [Google Scholar] [CrossRef]
- Singh, V.; Mitra, S.; Sharma, A.K.; Gera, R.; Ghosh, D. Isolation and Characterization of Microglia from Adult Mouse Brain: Selected Applications for ex Vivo Evaluation of Immunotoxicological Alterations Following in Vivo Xenobiotic Exposure. Chem. Res. Toxicol. 2014, 27, 895–903. [Google Scholar] [CrossRef]
- Ahmed, Z.; Shaw, G.; Sharma, V.P.; Yang, C.; McGowan, E.; Dickson, D.W. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J. Histochem. Cytochem. 2007, 55, 687–700. [Google Scholar] [CrossRef]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Li, H.; Xu, M.; Frank, J.; Luo, J. Binge ethanol exposure induces endoplasmic reticulum stress in the brain of adult mice. Toxicol. Appl. Pharmacol. 2018, 356, 172–181. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef]
- Ashok, A.; Rai, N.K.; Tripathi, S.; Bandyopadhyay, S. Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 2015, 143, 64–80. [Google Scholar] [CrossRef]
- Das, A.K.; Dewanjee, S.; Sahu, R.; Dua, T.K.; Gangopadhyay, M.; Sinha, M.K. Protective effect of Corchorus olitorius leaves against arsenic-induced oxidative stress in rat brain. Environ. Toxicol. Pharmacol. 2010, 29, 64–69. [Google Scholar] [CrossRef]
- El-Ghiaty, M.A.; El-Kadi, A.O.S. Arsenic: Various species with different effects on cytochrome P450 regulation in humans. Excli J. 2021, 20, 1184–1242. [Google Scholar] [CrossRef]
- Zhong, Y.; Dong, G.; Luo, H.; Cao, J.; Wang, C.; Wu, J.; Feng, Y.Q.; Yue, J. Induction of brain CYP2E1 by chronic ethanol treatment and related oxidative stress in hippocampus, cerebellum, and brainstem. Toxicology 2012, 302, 275–284. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.D.; Padmavathi, P.; Kavitha, G.; Saradamma, B.; Varadacharyulu, N. Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol. Cell Biochem. 2013, 375, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Bambino, K.; Zhang, C.; Austin, C.; Amarasiriwardena, C.; Arora, M.; Chu, J.; Sadler, K.C. Inorganic arsenic causes fatty liver and interacts with ethanol to cause alcoholic liver disease in zebrafish. Dis. Model. Mech. 2018, 11, dmm031575. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.; Pant, S.C.; Malhotra, P.R.; Kannan, G.M. Biochemical and histopathological changes in arsenic-intoxicated rats coexposed to ethanol. Alcohol 1997, 14, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Dutta, S.R.; Passi, D.; Bharti, J. Benefits of Alcohol on Arsenic Toxicity in Rats. J. Clin. Diagn. Res. 2017, 11, Bf01–Bf06. [Google Scholar] [CrossRef] [PubMed]
- Heit, C.; Dong, H.; Chen, Y.; Thompson, D.C.; Deitrich, R.A.; Vasiliou, V.K. The role of CYP2E1 in alcohol metabolism and sensitivity in the central nervous system. Subcell. Biochem. 2013, 67, 235–247. [Google Scholar] [CrossRef]
- D’Souza El-Guindy, N.B.; Kovacs, E.J.; De Witte, P.; Spies, C.; Littleton, J.M.; de Villiers, W.J.; Lott, A.J.; Plackett, T.P.; Lanzke, N.; Meadows, G.G. Laboratory models available to study alcohol-induced organ damage and immune variations: Choosing the appropriate model. Alcohol. Clin. Exp. Res. 2010, 34, 1489–1511. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J. Neuroinflamm. 2012, 9, 130. [Google Scholar] [CrossRef]
- Qin, L.; He, J.; Hanes, R.N.; Pluzarev, O.; Hong, J.S.; Crews, F.T. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflamm. 2008, 5, 10. [Google Scholar] [CrossRef]
- Rodriguez, V.M.; Del Razo, L.M.; Limon-Pacheco, J.H.; Giordano, M.; Sanchez-Pena, L.C.; Uribe-Querol, E.; Gutierrez-Ospina, G.; Gonsebatt, M.E. Glutathione reductase inhibition and methylated arsenic distribution in Cd1 mice brain and liver. Toxicol. Sci. 2005, 84, 157–166. [Google Scholar] [CrossRef]
- Tyler, C.R.; Allan, A.M. The effects of arsenic exposure on neurological and cognitive dysfunction in human and rodent studies: A review. Curr. Environ. Health Rep. 2014, 1, 132–147. [Google Scholar] [CrossRef]
- Nelson, J.C.; Greengrove, E.; Nwachukwu, K.N.; Grifasi, I.R.; Marshall, S.A. Repetitive binge-like consumption based on the Drinking-in-the-Dark model alters the microglial population in the mouse hippocampus. J. Integr. Neurosci. 2021, 20, 933–943. [Google Scholar] [CrossRef]
- Grifasi, I.R.; Evans, W.A.; Rexha, A.D.; Sako, L.W.; Marshall, S.A. A comparison of hippocampal microglial responses in aged and young rodents following dependent and non-dependent binge drinking. Int. Rev. Neurobiol. 2019, 148, 305–343. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.A.; Rinker, J.A.; Harrison, L.K.; Fletcher, C.A.; Herfel, T.M.; Thiele, T.E. Assessment of the Effects of 6 Standard Rodent Diets on Binge-Like and Voluntary Ethanol Consumption in Male C57BL/6J Mice. Alcohol. Clin. Exp. Res. 2015, 39, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Palmer, J.L.; Ippolito, J.A.; Curtis, B.J.; Choudhry, M.A.; Kovacs, E.J. Intoxication by intraperitoneal injection or oral gavage equally potentiates postburn organ damage and inflammation. Mediat. Inflamm. 2013, 2013, 971481. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.A.; Casachahua, J.D.; Rinker, J.A.; Blose, A.K.; Lysle, D.T.; Thiele, T.E. IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain Behav. Immun. 2016, 51, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Nwachukwu, K.N.; King, D.M.; Healey, K.L.; Swartzwelder, H.S.; Marshall, S.A. Sex-specific effects of adolescent intermittent ethanol exposure-induced dysregulation of hippocampal glial cells in adulthood. Alcohol 2022, 100, 31–39. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: New York, NY, USA, 2009. [Google Scholar]
- Schmued, L.C.; Stowers, C.C.; Scallet, A.C.; Xu, L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005, 1035, 24–31. [Google Scholar] [CrossRef]
- McClain, J.A.; Morris, S.A.; Deeny, M.A.; Marshall, S.A.; Hayes, D.M.; Kiser, Z.M.; Nixon, K. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav. Immun. 2011, 25 (Suppl. 1), S120–S128. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Choi, Y.; Ha, S.K.; Song, B.J. Cytochrome P450-2E1 promotes aging-related hepatic steatosis, apoptosis and fibrosis through increased nitroxidative stress. Free Radic. Biol. Med. 2016, 91, 188–202. [Google Scholar] [CrossRef]
- Shao, A.; Fei, J.; Feng, S.; Weng, J. Chikusetsu saponin IVa alleviated sevoflurane-induced neuroinflammation and cognitive impairment by blocking NLRP3/caspase-1 pathway. Pharmacol. Rep. 2020, 72, 833–845. [Google Scholar] [CrossRef]
- Giri, T.; Jiang, J.; Xu, Z.; McCarthy, R.; Halabi, C.M.; Tycksen, E.; Cahill, A.G.; England, S.K.; Palanisamy, A. Labor induction with oxytocin in pregnant rats is not associated with oxidative stress in the fetal brain. Sci. Rep. 2022, 12, 3143. [Google Scholar] [CrossRef] [PubMed]
- Siregar, A.S.; Nyiramana, M.M.; Kim, E.J.; Shin, E.J.; Woo, M.S.; Kim, J.M.; Park, S.H.; Hahm, J.R.; Choi, Y.J.; Kang, D. Oyster broth concentrate and its major component taurine alleviate acute alcohol-induced liver damage. Food Sci. Nutr. 2022, 10, 2390–2399. [Google Scholar] [CrossRef]
- Kang, S.-W.; Kim, S.J.; Kim, M.-S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.E.; Takinami, Y.; Rainczuk, A.; Baker, E.S.; Roberts, B.R. Utilizing Ion Mobility-Mass Spectrometry to Investigate the Unfolding Pathway of Cu/Zn Superoxide Dismutase. Front. Chem. 2021, 9, 614595. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.W.; Ryherd, G.L.; Scheibly, C.M.; Sasser, A.L.; Guillette, T.C.; Belcher, S.M. Gestational Cd Exposure in the CD-1 Mouse Induces Sex-Specific Hepatic Insulin Insensitivity, Obesity, and Metabolic Syndrome in Adult Female Offspring. Toxicol. Sci. 2020, 178, 264–280. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Carroll, R.J.; Harden, K.K.; Wu, G. Comparisons of treatment means when factors do not interact in two-factorial studies. Amino Acids 2012, 42, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Hough, R.L.; Fletcher, T.; Leonardi, G.S.; Goessler, W.; Gnagnarella, P.; Clemens, F.; Gurzau, E.; Koppova, K.; Rudnai, P.; Kumar, R.; et al. Lifetime exposure to arsenic in residential drinking water in Central Europe. Int. Arch. Occup. Environ. Health 2010, 83, 471–481. [Google Scholar] [CrossRef] [PubMed]
- O’Bryant, S.E.; Edwards, M.; Menon, C.V.; Gong, G.; Barber, R. Long-term low-level arsenic exposure is associated with poorer neuropsychological functioning: A Project FRONTIER study. Int. J. Environ. Res. Public. Health 2011, 8, 861–874. [Google Scholar] [CrossRef]
- Karelina, K.; Gaier, K.R.; Prabhu, M.; Wenger, V.; Corrigan, T.E.D.; Weil, Z.M. Binge ethanol in adulthood exacerbates negative outcomes following juvenile traumatic brain injury. Brain Behav. Immun. 2017, 60, 304–311. [Google Scholar] [CrossRef]
- Nixon, K.; Kim, D.H.; Potts, E.N.; He, J.; Crews, F.T. Distinct cell proliferation events during abstinence after alcohol dependence: Microglia proliferation precedes neurogenesis. Neurobiol. Dis. 2008, 31, 218–229. [Google Scholar] [CrossRef]
- Dennis, C.V.; Sheahan, P.J.; Graeber, M.B.; Sheedy, D.L.; Kril, J.J.; Sutherland, G.T. Microglial proliferation in the brain of chronic alcoholics with hepatic encephalopathy. Metab. Brain Dis. 2014, 29, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; Murase, S.I.; Horwitz, A.F.; Mandell, J.W. Infiltrative microgliosis: Activation and long-distance migration of subependymal microglia following periventricular insults. J. Neuroinflamm. 2005, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Gall, C.; Rose, G.; Lynch, G. Proliferative and migratory activity of glial cells in the partially deafferented hippocampus. J. Comp. Neurol. 1979, 183, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Ibanez, F.; Picard, K.; Bordeleau, M.; Sharma, K.; Bisht, K.; Tremblay, M.E. Immunofluorescence Staining Using IBA1 and TMEM119 for Microglial Density, Morphology and Peripheral Myeloid Cell Infiltration Analysis in Mouse Brain. J. Vis. Exp. 2019, 152. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Tu, J.-B.; Ran, R.-T.; Zhang, W.-X.; Tan, Q.; Tang, P.; Kuang, T.; Cheng, S.-Q.; Chen, C.-Z.; Jiang, X.-J.; et al. Using the Metabolome to Understand the Mechanisms Linking Chronic Arsenic Exposure to Microglia Activation, and Learning and Memory Impairment. Neurotox. Res. 2021, 39, 720–739. [Google Scholar] [CrossRef]
- Singh, V.; Gera, R.; Kushwaha, R.; Sharma, A.K.; Patnaik, S.; Ghosh, D. Hijacking microglial glutathione by inorganic arsenic impels bystander death of immature neurons through extracellular cystine/glutamate imbalance. Sci. Rep. 2016, 6, 30601. [Google Scholar] [CrossRef]
- Howard, L.A.; Miksys, S.; Hoffmann, E.; Mash, D.; Tyndale, R.F. Brain CYP2E1 is induced by nicotine and ethanol in rat and is higher in smokers and alcoholics. Br. J. Pharmacol. 2003, 138, 1376–1386. [Google Scholar] [CrossRef]
- Upadhya, S.C.; Tirumalai, P.S.; Boyd, M.R.; Mori, T.; Ravindranath, V. Cytochrome P4502E (CYP2E) in brain: Constitutive expression, induction by ethanol and localization by fluorescence in situ hybridization. Arch. Biochem. Biophys. 2000, 373, 23–34. [Google Scholar] [CrossRef]
- Leung, T.M.; Lu, Y. Alcoholic Liver Disease: From CYP2E1 to CYP2A5. Curr. Mol. Pharmacol. 2017, 10, 172–178. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, J.; Chen, X.; Dong, Z.; Yin, X.M. Diverse Consequences in Liver Injury in Mice with Different Autophagy Functional Status Treated with Alcohol. Am. J. Pathol. 2019, 189, 1744–1762. [Google Scholar] [CrossRef]
- Anwar-Mohamed, A.; El-Sherbeni, A.; Kim, S.H.; Elshenawy, O.H.; Althurwi, H.N.; Zordoky, B.N.; El-Kadi, A.O. Acute arsenic treatment alters cytochrome P450 expression and arachidonic acid metabolism in lung, liver and kidney of C57Bl/6 mice. Xenobiotica 2013, 43, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W.; Liangpunsakul, S. Acetaldehyde generating enzyme systems: Roles of alcohol dehydrogenase, CYP2E1 and catalase, and speculations on the role of other enzymes and processes. Novartis Found. Symp. 2007, 285, 4–16; discussion 16–22, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A. 6-Genetic polymorphisms of alcohol metabolizing enzymes associated with protection from or increased risk of alcohol abuse. In Alcohol, Drugs, Genes and the Clinical Laboratory; Dasgupta, A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 107–116. [Google Scholar]
- Druesne-Pecollo, N.; Tehard, B.; Mallet, Y.; Gerber, M.; Norat, T.; Hercberg, S.; Latino-Martel, P. Alcohol and genetic polymorphisms: Effect on risk of alcohol-related cancer. Lancet Oncol. 2009, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflamm. 2012, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, M.C.; Borelli, A.C.; Beggiato, S.; Tanganelli, S.; Loche, A.; Cacciaglia, R.; Ferraro, L.; Antonelli, T. GET73 Prevents Ethanol-Induced Neurotoxicity in Primary Cultures of Rat Hippocampal Neurons. Alcohol Alcohol. 2015, 51, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef]
- Tapia, J.A.; Salido, G.M.; González, A. Ethanol consumption as inductor of pancreatitis. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 3–8. [Google Scholar] [CrossRef]
- Bailey, S.M.; Cunningham, C.C. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology 1998, 28, 1318–1326. [Google Scholar] [CrossRef]
- Prystupa, A.; Błażewicz, A.; Kiciński, P.; Sak, J.J.; Niedziałek, J.; Załuska, W. Serum Concentrations of Selected Heavy Metals in Patients with Alcoholic Liver Cirrhosis from the Lublin Region in Eastern Poland. Int. J. Environ. Res. Public. Health 2016, 13, 582. [Google Scholar] [CrossRef]
- Baj, J.; Teresiński, G.; Forma, A.; Flieger, M.; Proch, J.; Niedzielski, P.; Grochowski, C.; Blicharska, E.; Buszewicz, G.; Bogucki, J.; et al. Chronic Alcohol Abuse Alters Hepatic Trace Element Concentrations-Metallomic Study of Hepatic Elemental Composition by Means of ICP-OES. Nutrients 2022, 14, 546. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Gorantla, S.; Zheng, J.; Persidsky, Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J. Neurochem. 2007, 100, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Jiang, Y.; Gupta, S.; Benlhabib, E. Effects of chronic ethanol drinking on the blood brain barrier and ensuing neuronal toxicity in alcohol-preferring rats subjected to intraperitoneal LPS injection. Alcohol Alcohol. 2007, 42, 385–399. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Choudhary, S.; Holmes, E.W.; Yong, S.; Banan, A.; Jakate, S.; Fields, J.Z. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J. Pharmacol. Exp. Ther. 2001, 299, 442–448. [Google Scholar] [PubMed]
- Tang, Y.; Forsyth, C.B.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Oats supplementation prevents alcohol-induced gut leakiness in rats by preventing alcohol-induced oxidative tissue damage. J. Pharmacol. Exp. Ther. 2009, 329, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Brzóska, M.M.; Moniuszko-Jakoniuk, J.; Piłat-Marcinkiewicz, B.; Sawicki, B. Liver and kidney function and histology in rats exposed to cadmium and ethanol. Alcohol Alcohol. 2003, 38, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Juhasz, A.L.; Zhang, Y.; Zhang, L.; Ma, L.Q.; Zhou, D.; Li, H. Alcohol consumption promotes arsenic absorption but reduces tissue arsenic accumulation in mice. Eco-Environ. Health 2023, 2, 107–116. [Google Scholar] [CrossRef]
- Grochowski, C.; Blicharska, E.; Bogucki, J.; Proch, J.; Mierzwińska, A.; Baj, J.; Litak, J.; Podkowiński, A.; Flieger, J.; Teresiński, G.; et al. Increased Aluminum Content in Certain Brain Structures is Correlated with Higher Silicon Concentration in Alcoholic Use Disorder. Molecules 2019, 24, 1721. [Google Scholar] [CrossRef]
- Zahr, N.M.; Mayer, D.; Rohlfing, T.; Hasak, M.P.; Hsu, O.; Vinco, S.; Orduna, J.; Luong, R.; Sullivan, E.V.; Pfefferbaum, A. Brain injury and recovery following binge ethanol: Evidence from in vivo magnetic resonance spectroscopy. Biol. Psychiatry 2010, 67, 846–854. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Firdaus, F.; Zafeer, M.F.; Anis, E.; Ahmad, M.; Afzal, M. Ellagic acid attenuates arsenic induced neuro-inflammation and mitochondrial dysfunction associated apoptosis. Toxicol. Rep. 2018, 5, 411–417. [Google Scholar] [CrossRef]
- Manthari, R.K.; Tikka, C.; Ommati, M.M.; Niu, R.; Sun, Z.; Wang, J.; Zhang, J.; Wang, J. Arsenic induces autophagy in developmental mouse cerebral cortex and hippocampus by inhibiting PI3K/Akt/mTOR signaling pathway: Involvement of blood-brain barrier’s tight junction proteins. Arch. Toxicol. 2018, 92, 3255–3275. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Balino, P.; Alfonso-Loeches, S.; Aragon, C.M.; Guerri, C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav. Immun. 2011, 25 (Suppl. 1), S80–S91. [Google Scholar] [CrossRef] [PubMed]
- Silvestre de Ferron, B.; Bennouar, K.E.; Kervern, M.; Alaux-Cantin, S.; Robert, A.; Rabiant, K.; Antol, J.; Naassila, M.; Pierrefiche, O. Two Binges of Ethanol a Day Keep the Memory Away in Adolescent Rats: Key Role for GLUN2B Subunit. Int. J. Neuropsychopharmacol. 2015, 19. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.L.; Amodeo, L.R.; Chandler, L.J.; Crews, F.T.; Ehlers, C.L.; Gomez, A.A.; Healey, K.L.; Kuhn, C.M.; Macht, V.A.; Marshall, S.A.; et al. The role of sex in the persistent effects of adolescent alcohol exposure on behavior and neurobiology in rodents. Int. Rev. Neurobiol. 2021, 160, 305–340. [Google Scholar] [CrossRef]
- Maynard, M.E.; Barton, E.A.; Robinson, C.R.; Wooden, J.I.; Leasure, J.L. Sex differences in hippocampal damage, cognitive impairment, and trophic factor expression in an animal model of an alcohol use disorder. Brain Struct. Funct. 2018, 223, 195–210. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Treatment | No Arsenic | 0.005 mg/kg | 2.5 mg/kg | 10 mg/kg |
|---|---|---|---|---|---|
| Immunohistochemistry | Ethanol | n = 8 ♀ = 4; ♂ = 4 | n = 8 ♀ = 4; ♂ = 4 | n = 9 ♀ = 4; ♂ = 5 | n = 8 ♀ = 5; ♂ = 3 |
| Water | n = 8 ♀ = 5; ♂ = 3 | n = 7 ♀ = 4; ♂ = 4 | n = 8 ♀ = 3; ♂ = 5 | n = 10 ♀ = 5; ♂ = 5 | |
| ELISA and ROS Assay | Ethanol | n = 6 ♀ = 3; ♂ = 3 | - | - | n = 8 ♀ = 4; ♂ = 4 |
| Water | n = 6 ♀ = 3; ♂ = 3 | - | - | n = 8 ♀ = 4; ♂ = 4 | |
| ICP-MS | Ethanol | n = 12 ♀ = 6; ♂ = 6 | - | - | n = 14 ♀ = 7; ♂ = 7 |
| Water | - | - | - | n = 14 ♀ = 7; ♂ = 7 |
| Experiment | Treatment | No Arsenic | 0.005 mg/kg | 2.5 mg/kg | 10 mg/kg |
|---|---|---|---|---|---|
| Immunohistochemistry | Ethanol | µ = 0.4 ± 0.3 | µ = −0.8 ± 1.7 | µ = 0.4 ± 1.5 | µ = 0.9 ± 2.5 |
| Water | µ = 1.8 ± 1.5 | µ = 1.1 ± 1.1 | µ = −1.4 ± 1.1 | µ = 0.9 ± 1.3 | |
| ELISA and ROS Assay | Ethanol | µ = 1.9 ± 2.6 | - | - | µ = −1.6 ± 1.7 |
| Water | µ = 1.6 ± 1.2 | - | - | µ = −0.2 ± 0.8 | |
| ICP-MS | Ethanol | µ = −1.6 ± 1.2 | - | - | µ = 1.0 ± 0.9 |
| Water | - | - | - | µ = −0.9 ± 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sides, T.R.; Nelson, J.C.; Nwachukwu, K.N.; Boston, J.; Marshall, S.A. The Influence of Arsenic Co-Exposure in a Model of Alcohol-Induced Neurodegeneration in C57BL/6J Mice. Brain Sci. 2023, 13, 1633. https://doi.org/10.3390/brainsci13121633
Sides TR, Nelson JC, Nwachukwu KN, Boston J, Marshall SA. The Influence of Arsenic Co-Exposure in a Model of Alcohol-Induced Neurodegeneration in C57BL/6J Mice. Brain Sciences. 2023; 13(12):1633. https://doi.org/10.3390/brainsci13121633
Chicago/Turabian StyleSides, Tori R., James C. Nelson, Kala N. Nwachukwu, Jhana Boston, and S. Alex Marshall. 2023. "The Influence of Arsenic Co-Exposure in a Model of Alcohol-Induced Neurodegeneration in C57BL/6J Mice" Brain Sciences 13, no. 12: 1633. https://doi.org/10.3390/brainsci13121633