
Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells

 , , , , ,
, , , , ,  and
and
Abstract
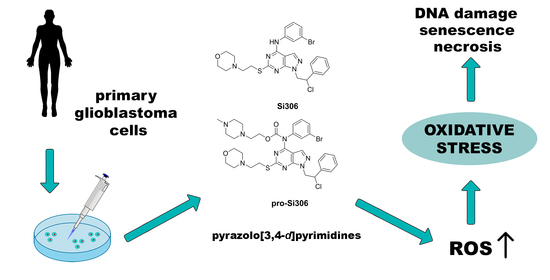
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
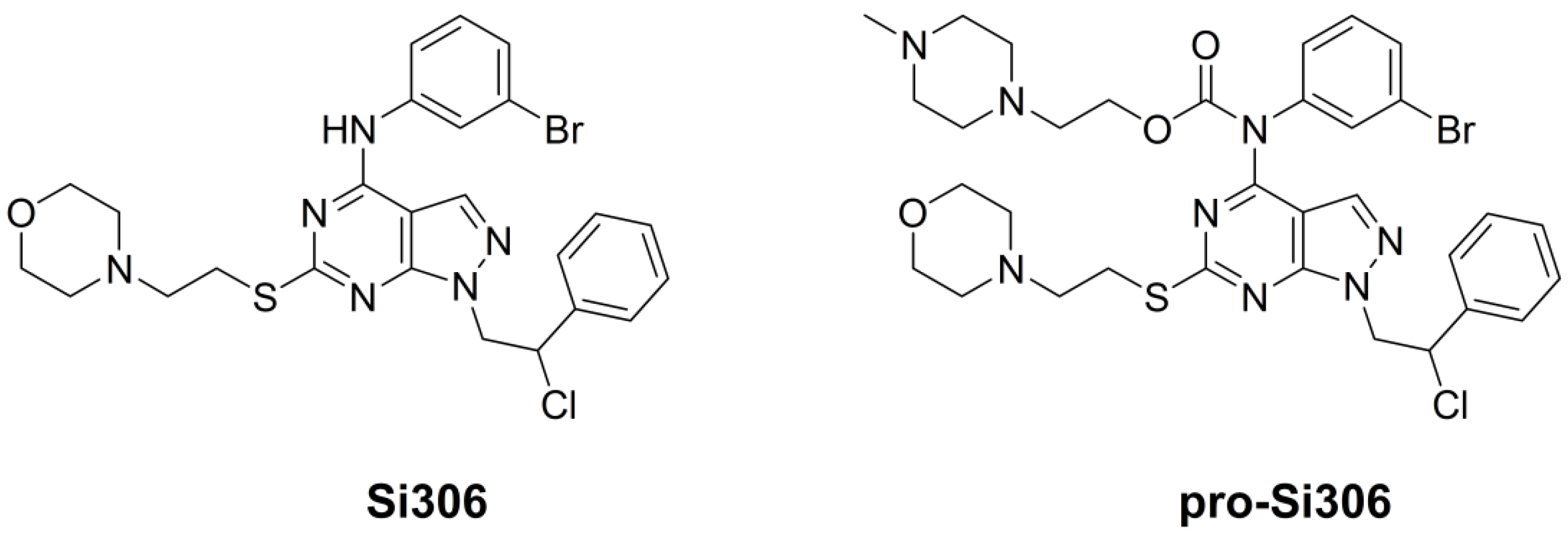
2.1. Drugs
2.2. Chemicals
2.3. Cell Lines
2.4. Primary Glioblastoma Culture
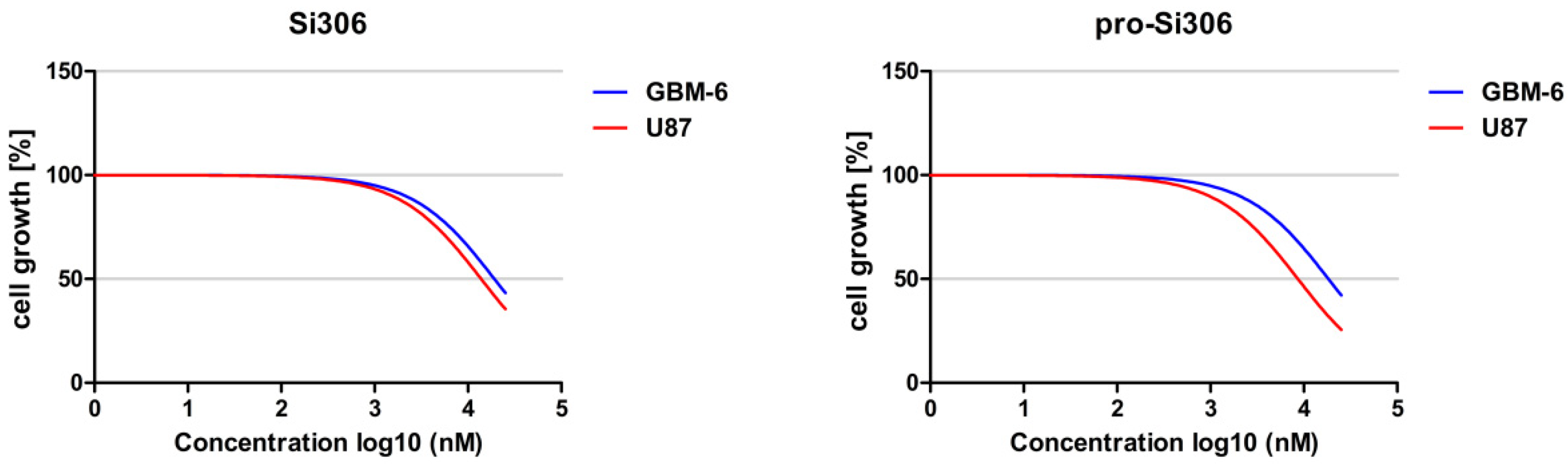
2.5. MTT Assay
2.6. Reactive Oxygen and Nitrogen Species Detection by Flow Cytometry
2.7. Mitochondrial Transmembrane Potential Detection by Flow Cytometry
2.8. Western Blot Analysis
2.9. Double Strand DNA Breaks Detection by Flow Cytometry
2.10. Double Strand DNA Breaks Detection by Immunocytochemistry
2.11. Cell Death Detection by Flow Cytometry
2.12. β-Galactosidase Activity Assay
2.13. Statistical Analysis
3. Results
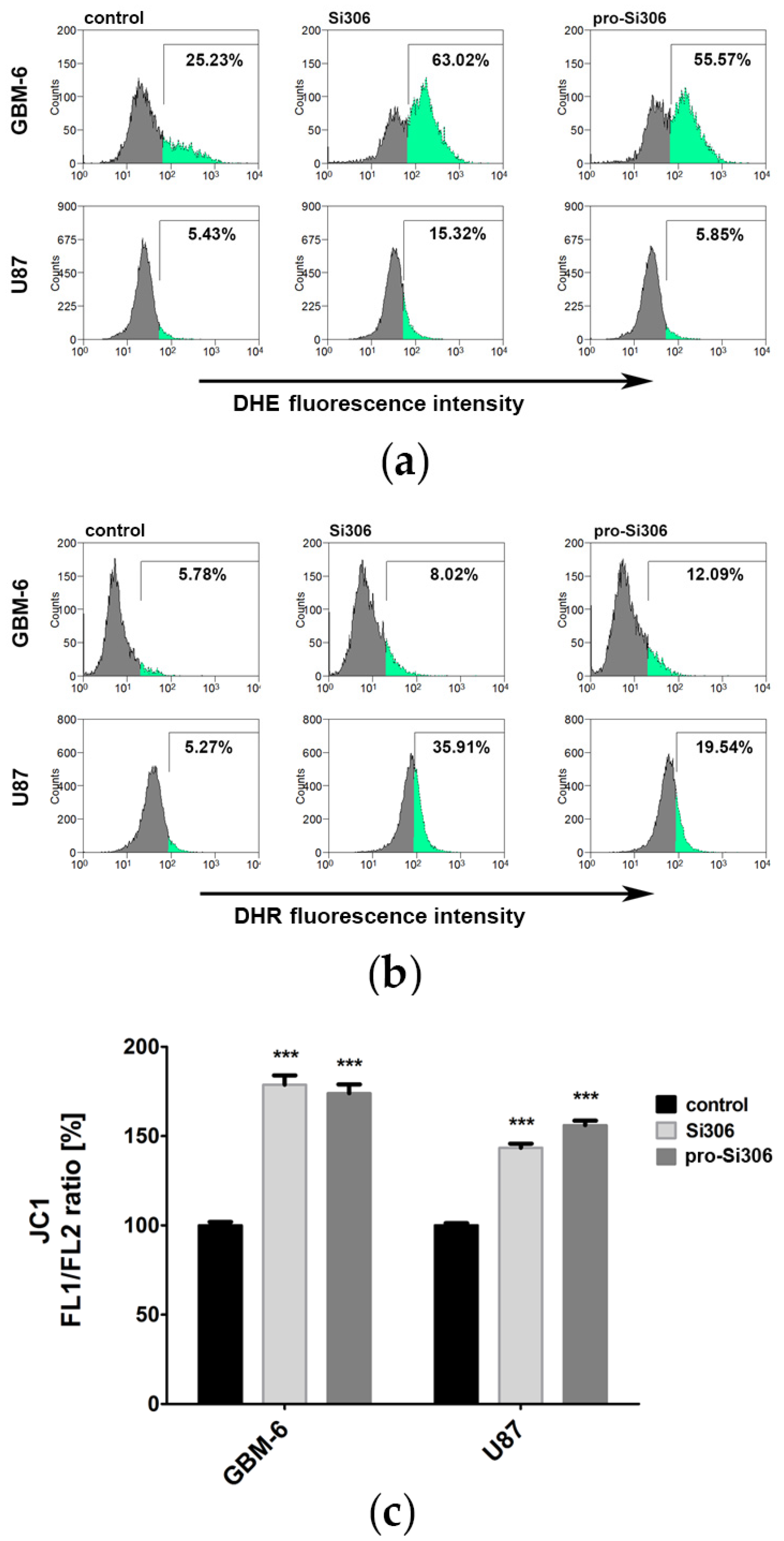
3.1. Si306 and Pro-Si306 Induce Oxidative Stress in Glioblastoma Cells
3.2. Si306 and Pro-Si306 Cause Depolarization of Mitochondria in Glioblastoma Cells
3.3. Si306 and Pro-Si306 Increase the Expression of SOD1 and SOD2 in Glioblastoma Cells
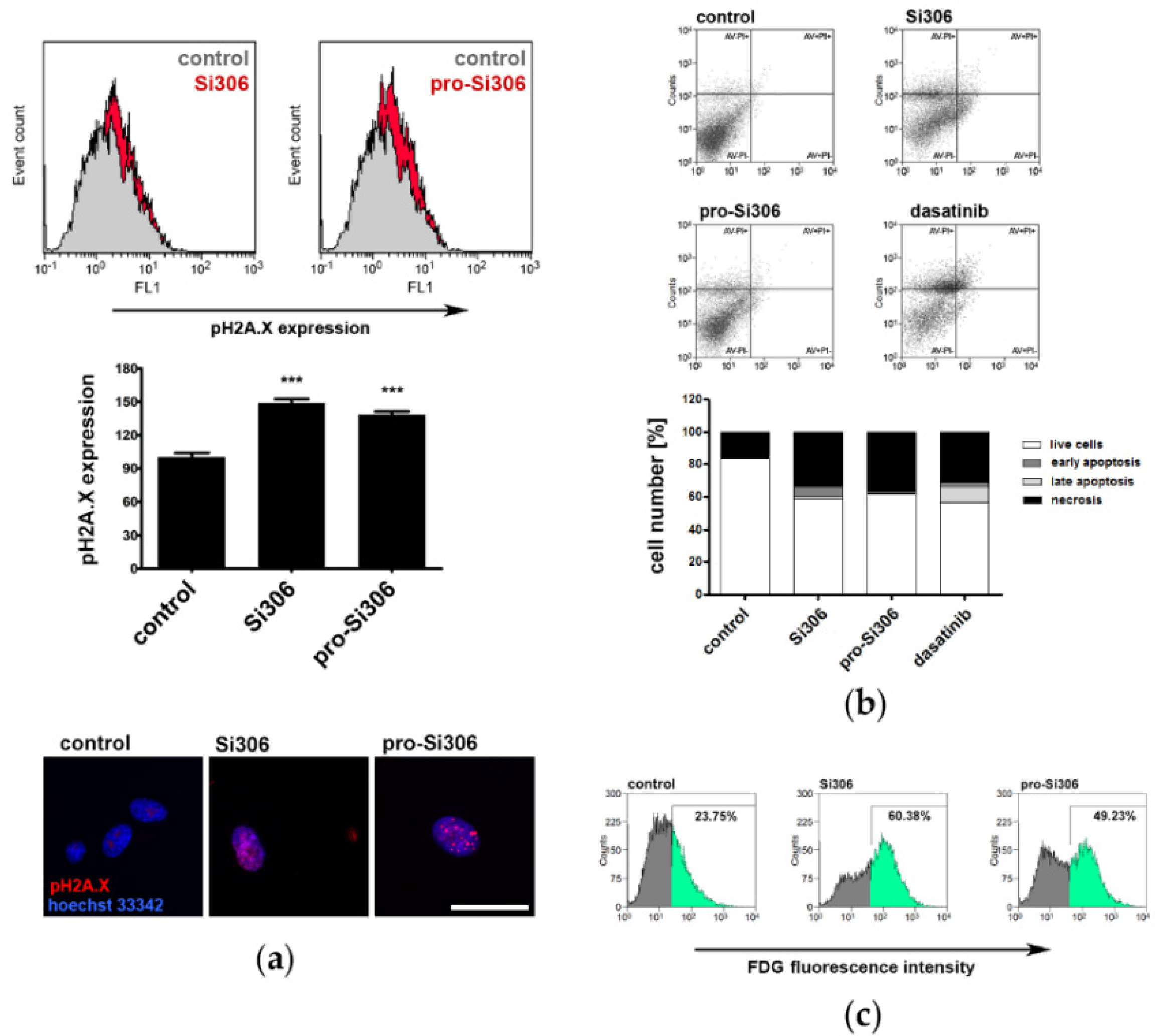
3.4. Si306 and Pro-Si306 Induce DNA Damage in Primary Glioblastoma Cells
3.5. Si306 and Pro-Si306 Induce Necrosis in Primary Glioblastoma Cells
3.6. Si306 and Pro-Si306 Induce Senescence in Primary Glioblastoma Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- de Witt Hamer, P.C. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro Oncol. 2010, 12, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, G.F.; Dunn, G.P.; Nance, E.A.; Hanes, J.; Brem, H. Emerging insights into barriers to effective brain tumor therapeutics. Front. Oncol. 2014, 4, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.M.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.R.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Dumont, R.A.; Hildebrandt, I.; Su, H.; Haubner, R.; Reischl, G.; Czernin, J.G.; Mischel, P.S.; Weber, W.A. Noninvasive imaging of alphaVbeta3 function as a predictor of the antimigratory and antiproliferative effects of dasatinib. Cancer Res. 2009, 69, 3173–3179. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Zhang, W.; Yang, X.; Wheeler, C.G.; Langford, C.P.; Wu, L.; Filippova, N.; Friedman, G.K.; Ding, Q.; Fathallah-Shaykh, H.M.; et al. The role of Src family kinases in growth and migration of glioma stem cells. Int. J. Oncol. 2014, 45, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis-Tuffin, L.J.; Feathers, R.; Hari, P.; Durand, N.; Li, Z.; Rodriguez, F.J.; Bakken, K.; Carlson, B.; Schroeder, M.; Sarkaria, J.N.; et al. Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9, 1783–1798. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Kemble, D.J. To C or not to C: Direct and indirect redox regulation of Src protein tyrosine kinase. Cell Cycle 2009, 8, 2353–2355. [Google Scholar] [CrossRef]
- Jin, Y.; Cai, Q.; Shenoy, A.K.; Lim, S.; Zhang, Y.; Charles, S.; Tarrash, M.; Fu, X.; Kamarajugadda, S.; Trevino, J.G.; et al. Src drives the Warburg effect and therapy resistance by inactivating pyruvate dehydrogenase through tyrosine-289 phosphorylation. Oncotarget 2016, 7, 25113–25124. [Google Scholar] [CrossRef] [Green Version]
- Paech, F.; Bouitbir, J.; Krahenbuhl, S. Hepatocellular Toxicity Associated with Tyrosine Kinase Inhibitors: Mitochondrial Damage and Inhibition of Glycolysis. Front. Pharmacol. 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, T.; Luo, P.; Zhu, H.; Zhao, Y.; Wu, H.; Gai, R.; Wu, Y.; Yang, B.; Yang, X.; He, Q. Oxidative stress is involved in Dasatinib-induced apoptosis in rat primary hepatocytes. Toxicol. Appl. Pharmacol. 2012, 261, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Radi, M.; Musumeci, F.; Brullo, C.; Botta, M. Biologically driven synthesis of pyrazolo[3,4-d]pyrimidines as protein kinase inhibitors: An old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev. 2014, 114, 7189–7238. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Musumeci, F.; Botta, M. Novel dual Src/Abl inhibitors for hematologic and solid malignancies. Expert Opin. Investig. Drugs 2010, 19, 931–945. [Google Scholar] [CrossRef]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C.; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef]
- Calgani, A.; Vignaroli, G.; Zamperini, C.; Coniglio, F.; Festuccia, C.; Di Cesare, E.; Gravina, G.L.; Mattei, C.; Vitale, F.; Schenone, S.; et al. Suppression of SRC Signaling Is Effective in Reducing Synergy between Glioblastoma and Stromal Cells. Mol. Cancer Ther. 2016, 15, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Fallacara, A.L.; Zamperini, C.; Podolski-Renic, A.; Dinic, J.; Stankovic, T.; Stepanovic, M.; Mancini, A.; Rango, E.; Iovenitti, G.; Molinari, A.; et al. A New Strategy for Glioblastoma Treatment: In Vitro and In Vivo Preclinical Characterization of Si306, a Pyrazolo[3,4-d]Pyrimidine Dual Src/P-Glycoprotein Inhibitor. Cancers 2019, 11, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarra, M.; Celano, M.; Maiuolo, J.; Schenone, S.; Botta, M.; Angelucci, A.; Bramanti, P.; Russo, D. Antiproliferative and pro-apoptotic effects afforded by novel Src-kinase inhibitors in human neuroblastoma cells. BMC Cancer 2010, 10, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccherini, E.; Indovina, P.; Zamperini, C.; Dreassi, E.; Casini, N.; Cutaia, O.; Forte, I.M.; Pentimalli, F.; Esposito, L.; Polito, M.S.; et al. SRC family kinase inhibition through a new pyrazolo[3,4-d]pyrimidine derivative as a feasible approach for glioblastoma treatment. J. Cell. Biochem. 2015, 116, 856–863. [Google Scholar] [CrossRef]
- Nesovic, M.; Divac Rankov, A.; Podolski-Renic, A.; Nikolic, I.; Tasic, G.; Mancini, A.; Schenone, S.; Pesic, M.; Dinic, J. Src Inhibitors Pyrazolo[3,4-d]pyrimidines, Si306 and Pro-Si306, Inhibit Focal Adhesion Kinase and Suppress Human Glioblastoma Invasion In Vitro and In Vivo. Cancers 2020, 12, 1570. [Google Scholar] [CrossRef]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of Pyrazolo[3,4-d]pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef] [PubMed]
- Zawilska, J.B.; Wojcieszak, J.; Olejniczak, A.B. Prodrugs: A challenge for the drug development. Pharmacol. Rep. 2013, 65, 1–14. [Google Scholar] [CrossRef]
- Vogel, T.W.; Zhuang, Z.; Li, J.; Okamoto, H.; Furuta, M.; Lee, Y.S.; Zeng, W.; Oldfield, E.H.; Vortmeyer, A.O.; Weil, R.J. Proteins and protein pattern differences between glioma cell lines and glioblastoma multiforme. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 3624–3632. [Google Scholar] [CrossRef] [Green Version]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture conditions defining glioblastoma cells behavior: What is the impact for novel discoveries? Oncotarget 2017, 8, 69185–69197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reers, M.; Smiley, S.T.; Mottola-Hartshorn, C.; Chen, A.; Lin, M.; Chen, L.B. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995, 260, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, S.; Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Simic, T.; Nicoletti, F.; Maksimovic-Ivanic, D. The Double-Faced Role of Nitric Oxide and Reactive Oxygen Species in Solid Tumors. Antioxidants 2020, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Ung, P.M.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, F.; Shao, Z.; Jiang, S.; Cheng, Z. Erlotinib induces the human non-small-cell lung cancer cells apoptosis via activating ROS-dependent JNK pathways. Cancer Med. 2016, 5, 3166–3175. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.P.; Shen, S.C.; Lee, W.R.; Yang, L.L.; Chen, Y.C. Imatinib mesylate induction of ROS-dependent apoptosis in melanoma B16F0 cells. J. Dermatol. Sci. 2011, 62, 183–191. [Google Scholar] [CrossRef]
- Mallis, R.J.; Buss, J.E.; Thomas, J.A. Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem. J. 2001, 355, 145–153. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A. Mechanism of cadmium induced apoptosis in the immunocyte. Toxicol. Lett. 2008, 177, 83–89. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Pawlowska, E.; Szczepanska, J.; Szatkowska, M.; Blasiak, J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme-Role in Pathogenesis and Therapeutic Perspective. Int. J. Mol. Sci. 2018, 19, 889. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Krawczynski, K.; Godlewski, J.; Bronisz, A. Oxidative Stress-Part of the Solution or Part of the Problem in the Hypoxic Environment of a Brain Tumor. Antioxidants 2020, 9, 747. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014, 127, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of Glutathione Metabolism in IDH1-Mutated Glioma. Mol. Cancer Ther. 2020, 19, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Molecular mechanisms of liver injury: Apoptosis or necrosis. Exp. Toxicol. Pathol. 2014, 66, 351–356. [Google Scholar] [CrossRef]
- Wang, J.Y.J. Cell Death Response to DNA Damage. Yale J. Biol. Med. 2019, 92, 771–779. [Google Scholar]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Cardinali, C.; Santoni, M.; Bernardini, G.; Santoni, A.; Santoni, G. Axitinib induces senescence-associated cell death and necrosis in glioma cell lines: The proteasome inhibitor, bortezomib, potentiates axitinib-induced cytotoxicity in a p21(Waf/Cip1) dependent manner. Oncotarget 2017, 8, 3380–3395. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostić, A.; Jovanović Stojanov, S.; Podolski-Renić, A.; Nešović, M.; Dragoj, M.; Nikolić, I.; Tasić, G.; Schenone, S.; Pešić, M.; Dinić, J. Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sci. 2021, 11, 884. https://doi.org/10.3390/brainsci11070884
Kostić A, Jovanović Stojanov S, Podolski-Renić A, Nešović M, Dragoj M, Nikolić I, Tasić G, Schenone S, Pešić M, Dinić J. Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sciences. 2021; 11(7):884. https://doi.org/10.3390/brainsci11070884
Chicago/Turabian StyleKostić, Ana, Sofija Jovanović Stojanov, Ana Podolski-Renić, Marija Nešović, Miodrag Dragoj, Igor Nikolić, Goran Tasić, Silvia Schenone, Milica Pešić, and Jelena Dinić. 2021. "Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells" Brain Sciences 11, no. 7: 884. https://doi.org/10.3390/brainsci11070884