
Neuroendocrine Assessment of Dopaminergic Function during Antidepressant Treatment in Major Depressed Patients

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Procedures
2.3. Assays
2.4. Antidepressant Treatment
2.5. Clinical Response
2.6. Data Analysis
3. Results
3.1. Comparison between Depressed Patients and Control Subjects
3.2. Apomorphine-Induced Cortisol Stimulation and Clinical Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boadie, W.D.; Nemeroff, C.B. The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 2007, 64, 327–337. [Google Scholar] [CrossRef]
- Belujon, P.; Grace, A.A. Dopamine system dysregulation in major depressive disorders. Int. J. Neuropsychopharmacol. 2017, 20, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Ebert, D.; Feistel, H.; Loew, T.; Pirner, A. Dopamine and depression—striatal dopamine D2 receptor SPECT before and after antidepressant therapy. Psychopharmacology 1996, 126, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Schneier, F.R.; Slifstein, M.; Whitton, A.E.; Pizzagalli, D.A.; Reinen, J.; McGrath, P.J.; Iosifescu, D.V.; Abi-Dargham, A. Dopamine release in antidepressant-naive major depressive disorder: A multimodal [(11)C]-(+)-PHNO positron emission tomography and functional magnetic resonance imaging study. Biol. Psychiatry 2018, 84, 563–573. [Google Scholar] [CrossRef] [PubMed]
- D’Haenen, H.A.; Bossuyt, A. Dopamine D2 receptors in depression measured with single photon emission computed tomography. Biol. Psychiatry 1994, 35, 128–132. [Google Scholar] [CrossRef]
- Shah, P.J.; Ogilvie, A.D.; Goodwin, G.M.; Ebmeier, K.P. Clinical and psychometric correlates of dopamine D2 binding in depression. Psychol. Med. 1997, 27, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.H.; McNeely, H.E.; Sagrati, S.; Boovariwala, A.; Martin, K.; Verhoeff, N.P.; Wilson, A.A.; Houle, S. Elevated putamen D(2) receptor binding potential in major depression with motor retardation: An [11C]raclopride positron emission tomography study. Am. J. Psychiatry 2006, 163, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Peciña, M.; Sikora, M.; Avery, E.T.; Heffernan, J.; Pecinña, S.; Mickey, B.J.; Zubieta, J.K. Striatal dopamine D2/3 receptor-mediated neurotransmission in major depression: Implications for anhedonia, anxiety and treatment response. Eur. Neuropsychopharmacol. 2017, 27, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Bouras, N.; Bridges, P.K. Bromocriptine in depression. Curr. Med. Res. Opin. 1982, 8, 150–153. [Google Scholar] [CrossRef]
- Barowsky, J.; Schwartz, T.L. An evidence-based approach to augmentation and combination strategies for treatment-resistant depression. Psychiatry 2006, 3, 42–61. [Google Scholar]
- Tundo, A.; de Filippis, R.; De Crescenzo, F. Pramipexole in the treatment of unipolar and bipolar depression. A systematic review and meta-analysis. Acta Psychiatr. Scand. 2019, 140, 116–125. [Google Scholar] [CrossRef]
- Kapur, S.A.; Mann, J.J. Role of the dopaminergic system in depression. Biol. Psychiatry 1992, 32, 1–17. [Google Scholar] [CrossRef]
- Spyraki, C.; Fibiger, H.C. Behavioural evidence for supersensitivity of postsynaptic dopamine receptors in the mesolimbic system after chronic administration of desipramine. Eur. J. Pharmacol. 1981, 74, 195–206. [Google Scholar] [CrossRef]
- Collu, M.; Poggiu, A.S.; Devoto, P.; Serra, G. Behavioural sensitization of mesolimbic dopamine D2 receptors in chronic fluoxetine-treated rats. Eur. J. Pharmacol. 1997, 322, 123–127. [Google Scholar] [CrossRef]
- Maj, J.; Rogóz, Z. Pharmacological effects of venlafaxine, a new antidepressant, given repeatedly, on the alpha 1-adrenergic, dopamine and serotonin systems. J. Neural. Transm. 1999, 106, 197–211. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, P.S.; Collu, M.; Gessa, G.L.; Serra, G. The role of dopamine in the mechanism of action of antidepressant drugs. Eur. J. Pharmacol. 2000, 405, 365–373. [Google Scholar] [CrossRef]
- Dziedzicka-Wasylewska, M.; Rogoz, Z.; Skuza, G.; Dlaboga, D.; Maj, J. Effect of repeated treatment with tianeptine and fluoxetine on central dopamine D(2)/D(3) receptors. Behav. Pharmacol. 2002, 13, 127–138. [Google Scholar] [CrossRef]
- Gershon, A.A.; Vishne, T.; Grunhaus, L. Dopamine D2-like receptors and the antidepressant response. Biol. Psychiatry 2007, 61, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Willner, P.; Hale, A.; Argyropoulos, S. Dopamine mechanism of antidepressant action in depressed patients. J. Aff. Disord. 2005, 86, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Tiihonen, J.; Kuoppamäki, M.; Någren, K.; Bergman, J.; Eronen, E.; Syvälahti, E.; Hietala, J. Serotonergic modulation of striatal D2 dopamine receptor binding in humans measured with positron emission tomography. Psychopharmacology 1996, 126, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Moresco, R.M.; Colombo, C.; Fazio, F.; Bonfanti, A.; Lucignani, G.; Messa, C.; Gobbo, C.; Galli, L.; Del Sole, A.; Lucca, A.; et al. Effects of fluvoxamine treatment on the in vivo binding of [F-18]FESP in drug naive depressed patients: A PET study. Neuroimage 2000, 12, 452–465. [Google Scholar] [CrossRef]
- Penttila, J.; Kajander, J.; Aalto, S.; Hirvonen, J.; Nagren, K.; Ilonen, T.; Syvalahti, E.; Hietala, J. Effects of fluoxetine on dopamine D2 receptors in the human brain: A positron emission tomography study with [11C] raclopride. Int. J. Neuropsychopharmacol. 2004, 7, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.S.; Ma, Y.; Dhawan, V.; Chaly, T.; Eidelberg, D. Selective serotonin reuptake inhibitor (SSRI) modulation of striatal dopamine measured with [11C]-raclopride and positron emission tomography. Synapse 2009, 63, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirvonen, J.; Hietala, J.; Kajander, J.; Markkula, J.; Rasi-Hakala, H.; Salminen, J.K.; Någren, K.; Aalto, S.; Karlsson, H. Effects of antidepressant drug treatment and psychotherapy on striatal and thalamic dopamine D2/3 receptors in major depressive disorder studied with [11C]raclopride PET. J. Psychopharmacol. 2011, 25, 1329–1336. [Google Scholar] [CrossRef]
- Duval, F.; Mokrani, M.C.; Crocq, M.A. What future for neuroendocrinology in psychiatry? Psychoneuroendocrinology 2013, 38, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Lal, S. Apomorphine in the evaluation of dopaminergic function in man. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1988, 12, 117–164. [Google Scholar] [CrossRef]
- Mokrani, M.C.; Duval, F.; Crocq, M.A.; Bailey, P.E.; Macher, J.P. Multihormonal responses to apomorphine in mental illness. Psychoneuroendocrinology 1995, 20, 365–375. [Google Scholar] [CrossRef]
- Duval, F.; Mokrani, M.C.; Erb, A.; Danila, V.; Gonzalez Lopera, F.; Foucher, J.; Jeanjean, L.C. Thyroid axis activity and dopamine function in major depression. Psychoneuroendocrinology 1999, 24, 695–712, under review. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Kolakowska, T.; Fang, V.S.; Fogg, L.; Robertson, A.; Lewine, R.; Strahilevitz, M.; Busch, D. Growth hormone and prolactin response to apomorphine in schizophrenia and the major affective disorders. Arch. Gen. Psychiatry 1984, 41, 512–519. [Google Scholar] [CrossRef]
- Wieck, A.; Kumar, R.; Hirst, A.D.; Marks, N.M.; Campbell, I.C.; Chekley, S.A. Increased sensitivity of dopamine receptors and recurrence of affective psychosis after childbirth. Br. Med. J. 1991, 303, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Pitchot, W.; Hansenne, M.; Moreno, A.; Ansseau, M. Effect of previous antidepressant therapy on the growth hormone response to apomorphine. Neuropsychobiology 1995, 32, 19–22. [Google Scholar] [CrossRef]
- Pitchot, W.; Hansenne, M.; Gonzalez Moreno, A.; Pinto, E.; Reggers, J.; Fuchs, S.; Pirard, S.; Ansseau, M. Reduced dopamine function in depressed patients is related to suicidal behavior but not its lethality. Psychoneuroendocrinology 2001, 26, 689–696. [Google Scholar] [CrossRef]
- McPherson, H.; Walsh, A.; Silverstone, T. Growth hormone and prolactin response to apomorphine in bipolar and unipolar depression. J. Aff. Disord. 2003, 76, 121–125. [Google Scholar] [CrossRef]
- Scantamburlo, G.; Hansenne, M.; Fuchs, S.; Pitchot, W.; Pinto, E.; Reggers, J.; Ansseau, M.; Legros, J.J. AVP- and OT-neurophysins response to apomorphine and clonidine in major depression. Psychoneuroendocrinology 2005, 30, 839–845. [Google Scholar] [CrossRef]
- Duval, F.; Mokrani, M.C.; Monreal-Ortiz, J.A.; Fattah, S.; Champeval, C.; Schulz, P.; Macher, J.P. Cortisol hypersecretion in unipolar major depression with melancholic and psychotic features: Dopaminergic, noradrenergic and thyroid correlates. Psychoneuroendocrinology 2006, 31, 876–888. [Google Scholar] [CrossRef]
- Monreal, J.A.; Duval, F.; Mokrani, M.C.; Fattah, S.; Palao, D. Differences in multihormonal responses to the dopamine agonist apomorphine between unipolar and bipolar depressed patients. J. Psychiatr. Res. 2019, 112, 18–22. [Google Scholar] [CrossRef]
- Duval, F.; Mokrani, M.C.; Erb, A.; Danila, V.; Gonzalez Lopera, F.; Jeanjean, L. Dopaminergic, noradrenergic, adrenal, and thyroid abnormalities in psychotic and affective disorders. Front. Psychiatry 2020, 11, 533872. [Google Scholar] [CrossRef] [PubMed]
- Healy, E.; McKeon, P. Dopaminergic sensitivity and prediction of antidepressant response. J. Psychopharmacol. 2000, 14, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Cowen, P.J.; Braddock, L.E.; Gosden, B. The effect of amitriptyline treatment on the growth hormone response to apomorphine. Psychopharmacology 1984, 83, 378–379. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), 4th ed.; American Psychiatric Press: Washington, DC, USA, 1994; pp. 273–315. [Google Scholar]
- Hamilton, M.A. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960, 23, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Pfennig, A.; Severus, E.; Whybrow, P.C.; Angst, J.; Möller, H.J. World Federation of Societies of Biological Psychiatry. Task force on unipolar depressive disorders. World J. Biol. Psychiatry 2013, 14, 334–385. [Google Scholar] [CrossRef] [PubMed]
- Gassaway, M.; Rives, M.; Kruegel, A.; Javittch, J.A.; Sames, D. The atypical antidepressant and neurorestorative agent tianeptine is a μ-opioid receptor agonist. Transl. Psychiatry 2014, 4, e411. [Google Scholar] [CrossRef]
- Frank, E.; Prien, R.F.; Jarrett, R.B.; Keller, M.B.; Kupfer, D.J.; Lavori, P.W.; Rush, A.J.; Weissman, M.M. Conceptualization and rationale for consensus definitions of terms in major depressive disorder: Remission, recovery, relapse, and recurrence. Arch. Gen. Psychiatry 1991, 48, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.J.; Feinberg, M.; Greden, J.F.; Tarika, J.; Albala, A.A.; Haskett, R.F.; James, N.M.; Kronfol, Z.; Lohr, N.; Steiner, M.; et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch. Gen. Psychiatry 1981, 38, 15–22. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2005; ISBN 3-900051-07-0. [Google Scholar]
- Metz, C.E. Basic principles of ROC analysis. Semin. Nucl. Med. 1978, 8, 283–298. [Google Scholar] [CrossRef]
- Millan, M.J.; Maiofiss, L.; Cussac, D.; Audinot, V.; Boutin, J.A.; Newman-Tancredi, A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J. Pharmacol. Exp. Therapeut. 2002, 303, 791–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvernmo, T.; Hartter, S.; Burger, E. A review of the receptor-binding and pharmacokinetic properties of dopamine agonists. Clin. Ther. 2006, 28, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Duval, F.; Mokrani, M.C.; Crocq, M.A.; Bailey, P.; Diep, T.S.; Correa, H.; Macher, J.P. Dopaminergic function and the cortisol response to dexamethasone in psychotic depression. Prog. Neuro Psychoharmacol. Biol. Psychiat. 2000, 24, 204–225. [Google Scholar] [CrossRef]
- Duval, F.; Mokrani, M.C.; Monreal, J.; Weiss, T.; Fattah, S.; Hamel, B.; Macher, J.P. Interaction between the serotonergic system and HPA and HPT axes in patients with major depression: Implications for pathogenesis of suicidal behavior. Dialogues Clin. Neurosci. 2002, 4, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Borowski, B.; Kuhn, C. D1 and D2 dopamine receptors stimulate hypothalamo-pituitary-adrenal activity in rats. Neuropharmacology 1992, 31, 671–678. [Google Scholar] [CrossRef]
- Eaton, M.J.; Cheung, S.; Moore, K.E.; Lookingland, K.J. Dopamine receptor-mediated regulation of corticotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Brain Res. 1996, 738, 60–66. [Google Scholar] [CrossRef]
- Ran, X.; Yang, Y.; Meng, Y.; Li, Y.; Zhou, L.; Wang, Z.; Zhu, J. Distribution of D1 and D2 receptor-immunoreactive neurons in the paraventricular nucleus of the hypothalamus in the rat. J. Chem. Neuroanat. 2019, 98, 97–103. [Google Scholar] [CrossRef]
- Sekine, Y.; Suzuki, K.; Ramachandran, P.V.; Blackburn, T.P.; Ashby, C.R., Jr. Acute and repeated administration of fluoxetine, citalopram, and paroxetine significantly alters the activity of midbrain dopamine neurons in rats: An in vivo electrophysiological study. Synapse 2007, 61, 72–77. [Google Scholar] [CrossRef]
- León, L.A.; Cardenas, F.P. Contribution of the dopaminergic system to the effect of chronic fluoxetine in the rat forced swim test. Psychol. Neurosci. 2008, 1, 81–86. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Chattarji, S.; Diamond, D.M.; Jay, T.M.; Reagan, L.P.; Svenningsson, P.; Fuchs, E. The neurobiological properties of tianeptine (Stablon): From monoamine hypothesis to glutamatergic modulation. Mol. Psychiatry 2010, 15, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.F.; Cavalcante, J.L.; Castelo, M.S.; Castelo, M.S.; Lima, M.C. Augmentation strategies for treatment resistant depression: A literature review. J. Clin. Pharm. Ther. 2007, 32, 415–428. [Google Scholar] [CrossRef]
- Quitkin, F.M.; McGrath, P.J.; Stewart, J.W.; Deliyannides, D.; Taylor, B.P.; Davies, C.A.; Klein, D.F. Remission rates with 3 consecutive antidepressant trials: Effectiveness for depressed outpatients. J. Clin. Psychiatry 2005, 66, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Hig- gins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Duval, F.; Lebowitz, B.D.; Macher, J.P. Treatments in depression. Dialogues Clin. Neurosci. 2006, 8, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, F.; Jay, T.M. A new strategy for antidepressant prescription. Front. Neurosci. 2010, 4, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimitvilai, S.; Brodie, M.S. Reversal of prolonged dopamine inhibition of dopaminergic neurons of the ventral tegmental area. J. Pharmacol. Exp. Ther. 2010, 333, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimitvilai, S.; Herman, M.; You, C.; Arora, D.S.; McElvain, M.A.; Roberto, M.; Brodie, M.S. Dopamine D2 receptor desensitization by dopamine or corticotropin releasing factor in ventral tegmental area neurons is associated with increased glutamate release. Neuropharmacology 2014, 82, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Serra, G.; Argiolas, A.; Fadda, F.; Gessa, G.L. Hyposensitivity of dopamine autoreceptors induced by chronic administration of tricyclic antidepressants. Pharmacol. Res. Commun. 1980, 12, 619–624. [Google Scholar] [CrossRef]
- Chiodo, L.A.; Antelman, S.M. Repeated tricyclics induce a progressive dopamine autoreceptor subsensitivity independent of daily drug treatment. Nature 1980, 287, 451–454. [Google Scholar] [CrossRef]
- Sun, P.; Wang, J.; Gu, W.; Cheng, W.; Jin, G.Z.; Friedman, E.; Zheng, J.; Zhen, X. PSD-95 regulates D1 dopamine receptor resensitization, but not receptor-mediated Gs-protein activation. Cell Res. 2009, 19, 612–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.K.; Mohan, M.L.; Naga Prasad, S.V. G protein-coupled receptor resensitization paradigms. Int. Rev. Cell. Mol. Biol. 2018, 339, 63–91. [Google Scholar] [CrossRef]
- Duval, F. Endocrinologie et psychiatrie. EMC Psychiatr. 2004, 1, 1–28. [Google Scholar] [CrossRef]




{kind=link}
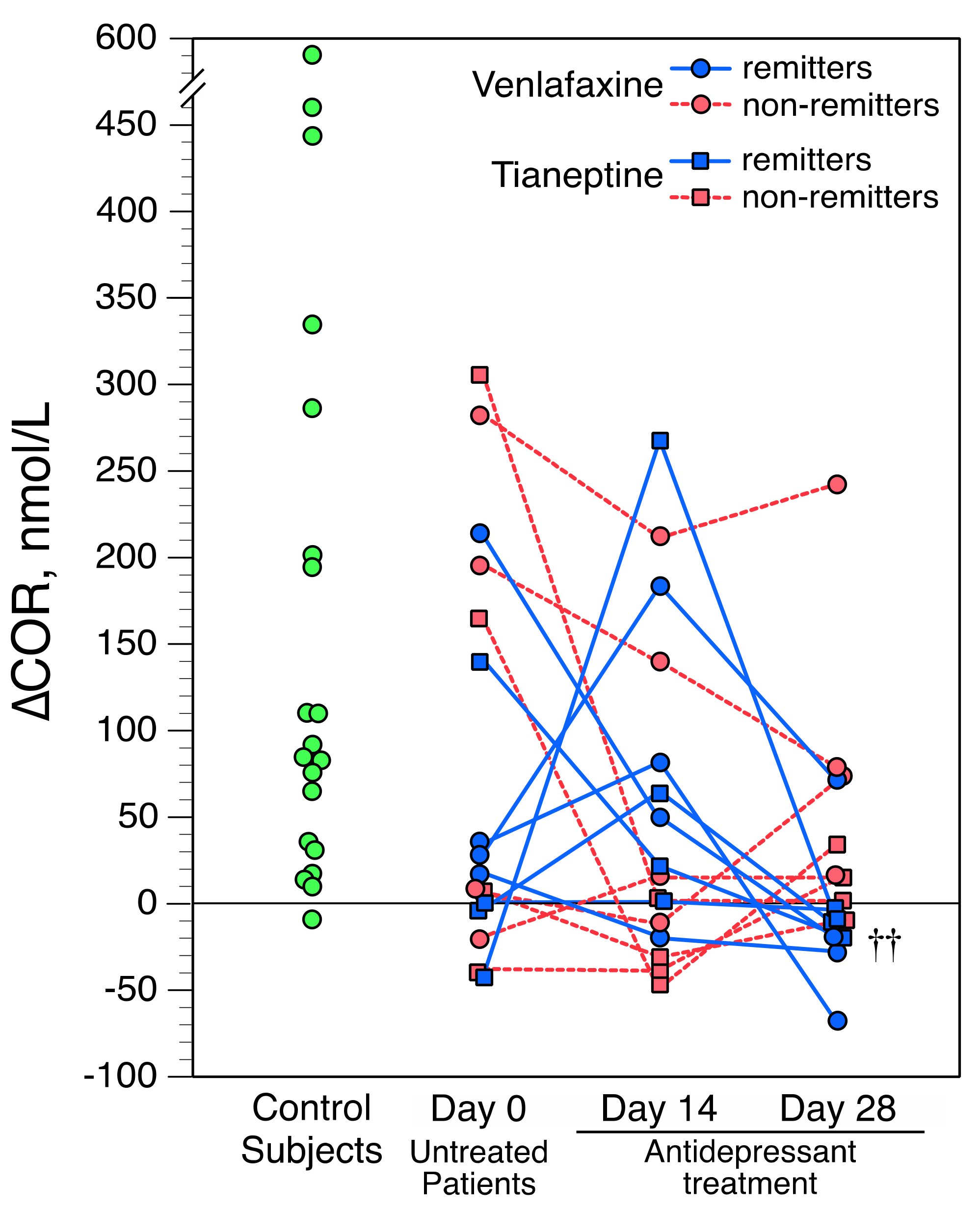{kind=link}
{kind=link}
| Tianeptine | Venlafaxine | p | |
|---|---|---|---|
| (n = 8) | (n = 8) | ||
| Age, years a | 35.6 ± 12.6 | 41.7 ± 7.6 | 0.34 |
| Sex M/F, n | 05-Mar | 05-Mar | 1 |
| HAM-D Scores | |||
| Day 0 | 25.9 ± 4.2 | 27.7 ± 5.0 | 0.59 |
| Day 14 | 16.1 ± 4.6 | 14.5 ± 11.9 | 0.63 |
| Day 28 | 11.5 ± 6.8 | 8.2 ± 6.9 | 0.39 |
| Day 42 | 9.9 ± 9.1 | 8.2 ± 7.9 | 0.79 |
| Remitters, n | 4 | 4 | 1 |
| Apomorphine test | |||
| CORBL, nmol/L | |||
| Day 0 | 200.1 ± 65.2 | 231.2 ± 78.2 | 1 |
| Day 14 | 293.2 ± 108.1 | 227.2 ± 76.9 | 0.2 |
| Day 28 | 296.4 ± 118.4 | 267.2 ± 98.2 | 0.72 |
| ∆COR, nmol/L | |||
| Day 0 | 69.4 ± 127.1 | 95.2 ± 115.6 | 0.27 |
| Day 14 | 30.5 ± 102.4 | 82.0 ±87.9 | 0.18 |
| Day 28 | −0.2 ± 17.3 | 47.2 ± 96.1 | 0.44 |
| Post-DST CORmax | |||
| Day 0 | 49.1 ± 58.5 | 23.1 ± 10.0 | 0.95 |
| Day 14 | 23.5 ± 9.2 | 28.6 ± 18.4 | 0.79 |
| Day 28 | 23.0 ± 10.7 | 25.7 ± 15.3 | 0.91 |
| Remitter Patients | Non-Remitter Patients | p | |
|---|---|---|---|
| (n = 8) | (n = 8) | ||
| Age, years a | 39.4 ± 7.8 | 38.0 ± 13.3 | 0.95 |
| Sex M/F, n | 05-Mar | 05-Mar | 1 |
| HAM-D Scores | |||
| Day 0 | 27.0 ± 5.0 | 26.6 ± 4.5 | 0.91 |
| Day 14 | 11.7 ± 5.7 | 21.9 ± 5.8 | 0.003 |
| Day 28 | 4.7 ± 3.3 | 15.0 ± 5.4 | 0.001 |
| Day 42 | 2.1 ± 2.2 | 16.0 ± 5.6 | 0.0008 |
| Venlafaxine/Tianeptine | 04-Apr | 04-Apr | |
| Apomorphine test | |||
| CORBL, nmol/L | |||
| Day 0 | 233.1 ± 87.4 | 198.5 ± 78.2 | 0.6 |
| Day 14 | 244.2 ± 75.0 | 276.2 ± 117.8 | 0.67 |
| Day 28 | 266.0 ± 107.1 | 297.6 ± 110.0 | 0.57 |
| ∆COR, nmol/L | |||
| Day 0 | 49.4 ± 84.4 | 115.2 ± 142.5 | 0.57 |
| Day 14 | 81.6 ± 97.6 | 30.9 ± 87.9 | 0.14 |
| Day 28 | −10.5 ± 39.3 | 57.5 ± 81.5 | 0.005 |
| ∆∆COR, nmol/L | |||
| Day 14–Day 0 | 32.2 ± 151.5 | −84.4 ± 119.3 | 0.16 |
| Day 28–Day 14 | −92.1 ± 90.9 | 26.6 ± 48.4 | 0.002 |
| Post-DST CORmax | |||
| Day 0 | 41.7 ± 52.6 | 30.5 ± 32.6 | 0.46 |
| Day 14 | 20.7 ± 9.1 | 33.4 ± 17.1 | 0.31 |
| Day 28 | 18.6 ± 6.4 | 30.1 ± 15.4 | 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duval, F.; Mokrani, M.-C.; Erb, A.; Gonzalez Lopera, F.; Danila, V.; Tomsa, M. Neuroendocrine Assessment of Dopaminergic Function during Antidepressant Treatment in Major Depressed Patients. Brain Sci. 2021, 11, 425. https://doi.org/10.3390/brainsci11040425
Duval F, Mokrani M-C, Erb A, Gonzalez Lopera F, Danila V, Tomsa M. Neuroendocrine Assessment of Dopaminergic Function during Antidepressant Treatment in Major Depressed Patients. Brain Sciences. 2021; 11(4):425. https://doi.org/10.3390/brainsci11040425
Chicago/Turabian StyleDuval, Fabrice, Marie-Claude Mokrani, Alexis Erb, Felix Gonzalez Lopera, Vlad Danila, and Mihaela Tomsa. 2021. "Neuroendocrine Assessment of Dopaminergic Function during Antidepressant Treatment in Major Depressed Patients" Brain Sciences 11, no. 4: 425. https://doi.org/10.3390/brainsci11040425