
Long-Term Pharmacological Inhibition of the Activity of All NOS Isoforms Rather Than Genetic Knock-Out of Endothelial NOS Leads to Impaired Spatial Learning and Memory in C57BL/6 Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Tissue Collection
2.2. Spatial Learning and Memory
2.3. Blood Pressure Measurements
2.4. Echocardiography
2.5. Non-Invasive Pulse Wave Velocity (PWV) Measurements of the Aortic Abdominal Aorta (aPWV)
2.6. Rodent Oscillatory Tension Set-Up for Arterial Compliance (ROTSAC)
2.7. Statistical Analysis
3. Results
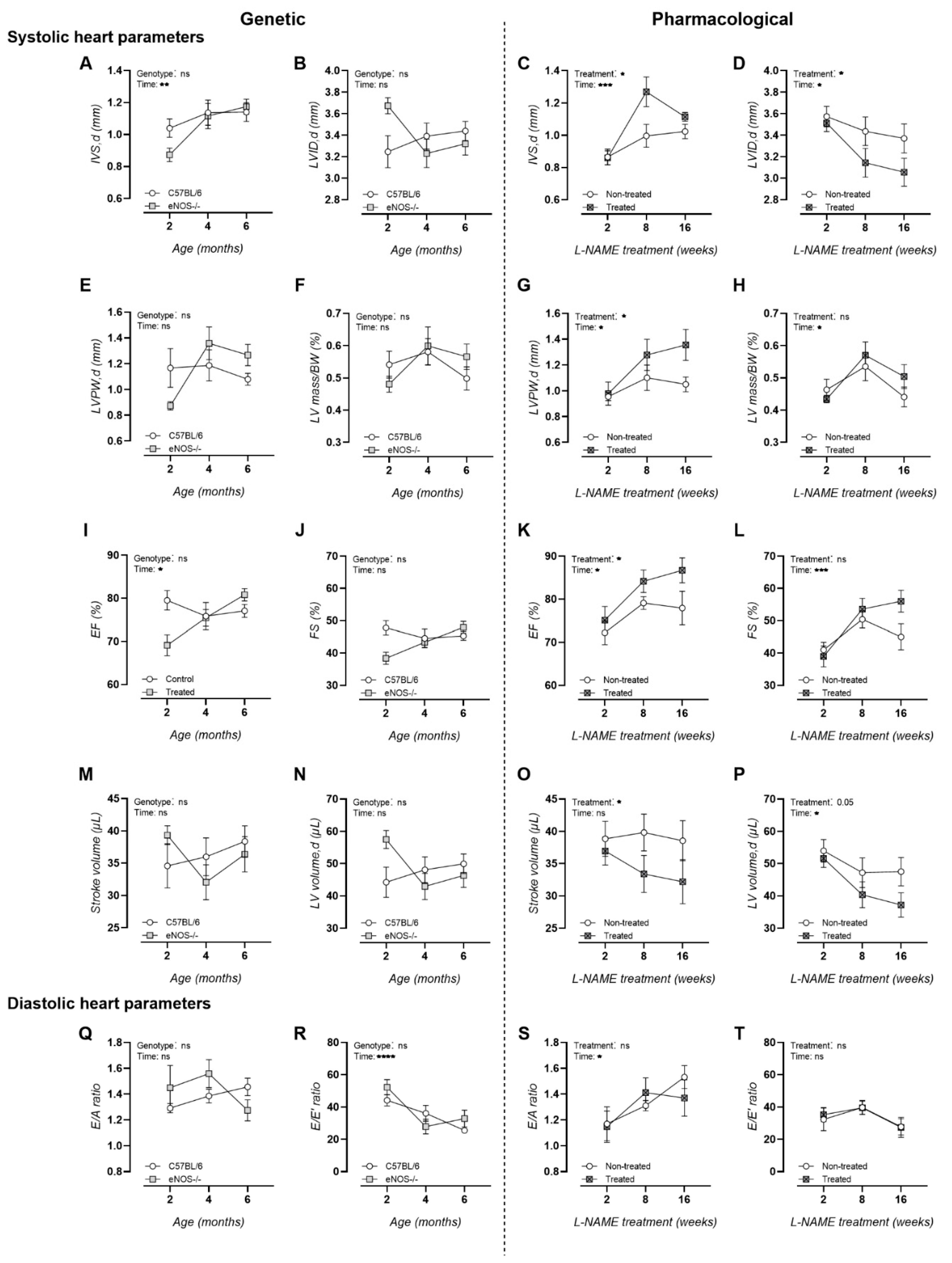
3.1. Chronic L-NAME Treatment Induces Hypertrophic Cardiomyopathy
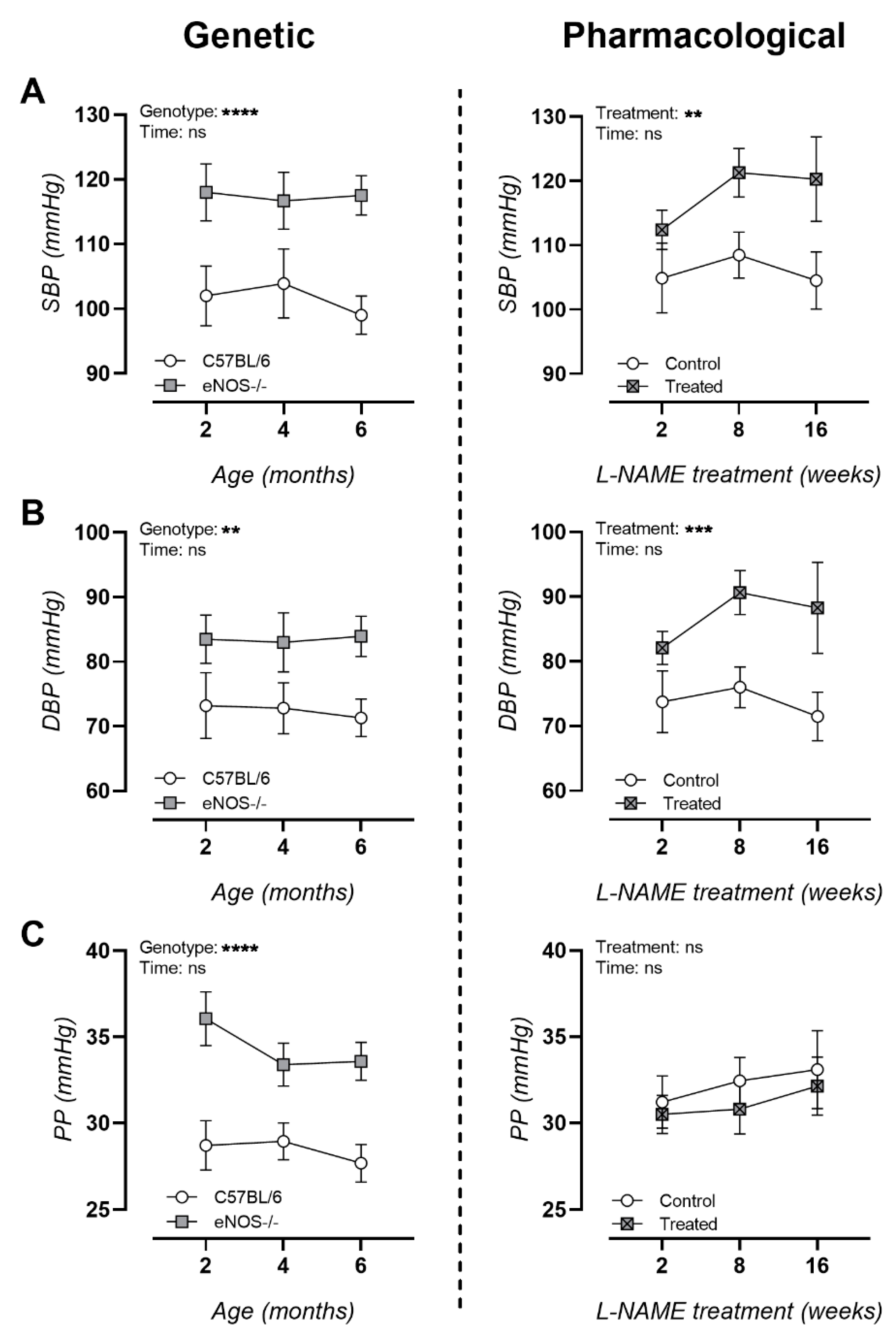
3.2. NO Dysfunction Is Associated with Hypertension
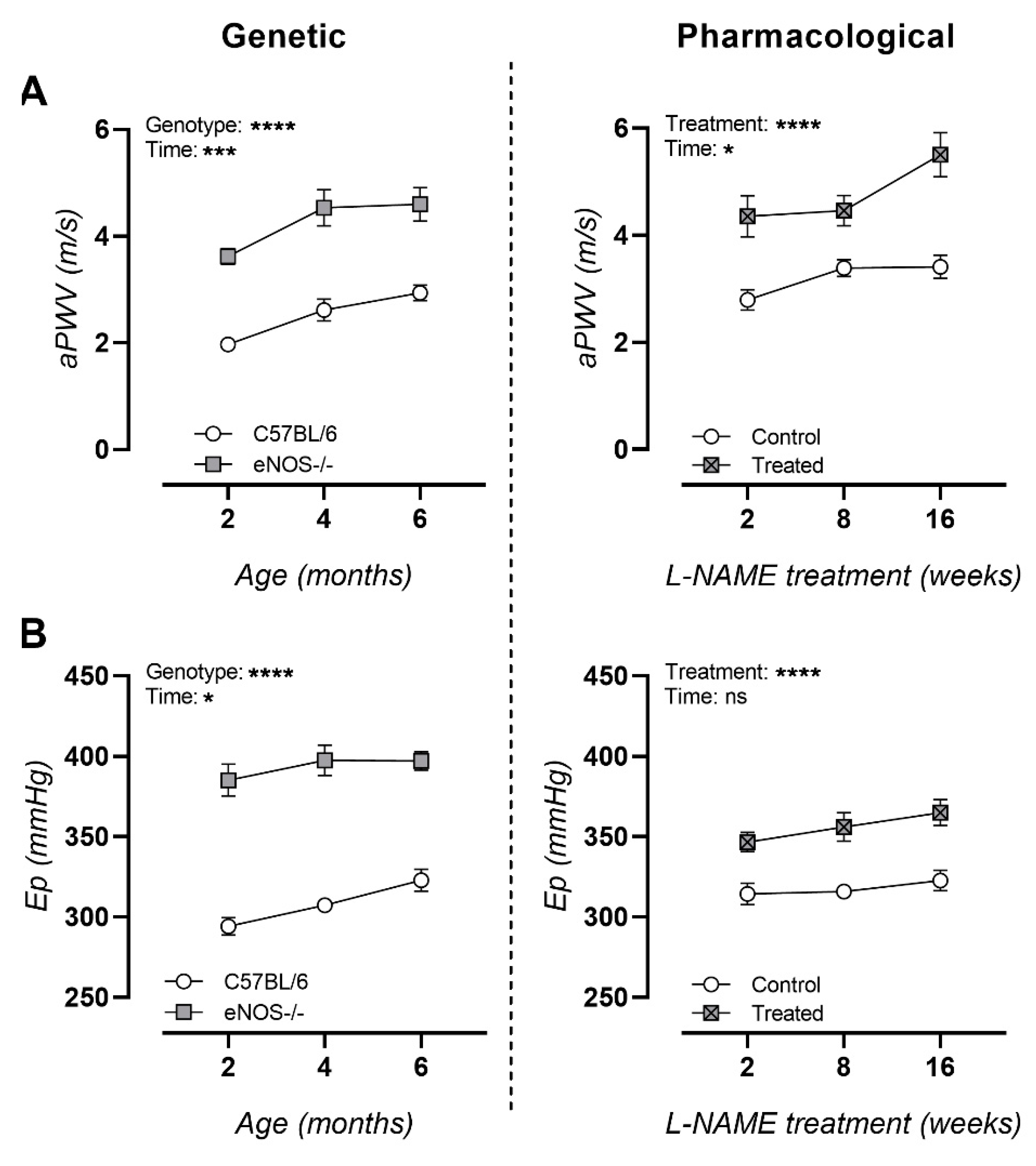
3.3. NO Dysfunction Is Characterized with Arterial Stiffness In Vivo and Ex Vivo
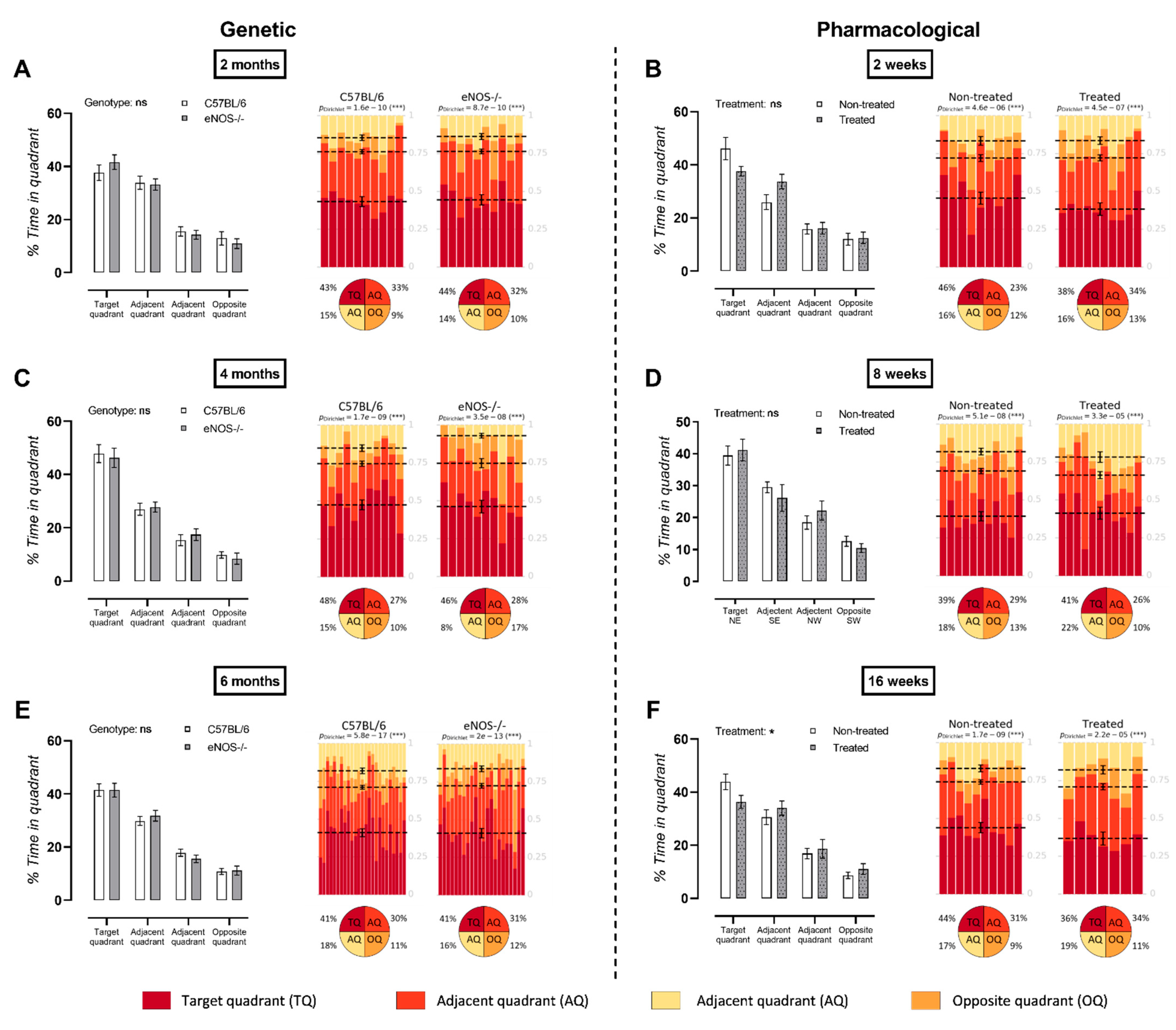
3.4. Long-Term L-NAME Treatment Leads to Deteriorated MWM Probe Trial Performance
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rabkin, S.W. Arterial stiffness: Detection and consequences in cognitive impairment and dementia of the elderly. J. Alzheimer’s Dis. 2012, 32, 541–549. [Google Scholar] [CrossRef] [PubMed]
- van Sloten, T.T.; Protogerou, A.D.; Henry, R.M.; Schram, M.T.; Launer, L.J.; Stehouwer, C.D. Association between arterial stiffness, cerebral small vessel disease and cognitive impairment: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2015, 53, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iulita, M.F.; Noriega de la Colina, A.; Girouard, H. Arterial stiffness, cognitive impairment and dementia: Confounding factor or real risk? J. Neurochem. 2018, 144, 527–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickx, J.O.; Martinet, W.; Van Dam, D.; De Meyer, G.R. Inflammation, Nitro-Oxidative Stress, Impaired Autophagy, and Insulin Resistance as a Mechanistic Convergence Between Arterial Stiffness and Alzheimer’s Disease. Front. Mol. Biosci. 2021, 8, 185. [Google Scholar] [CrossRef]
- Bauer, V.; Sotníková, R. Nitric oxide—The endothelium-derived relaxing factor and its role in endothelial functions. Gen. Physiol. Biophys. 2010, 29, 319. [Google Scholar] [CrossRef]
- Hasegawa, N.; Fujie, S.; Horii, N.; Miyamoto-Mikami, E.; Tsuji, K.; Uchida, M.; Hamaoka, T.; Tabata, I.; Iemitsu, M. Effects of Different Exercise Modes on Arterial Stiffness and Nitric Oxide Synthesis. Med. Sci. Sports Exerc. 2018, 50, 1177–1185. [Google Scholar] [CrossRef]
- Doherty, G.H. Nitric oxide in neurodegeneration: Potential benefits of non-steroidal anti-inflammatories. Neurosci. Bull. 2011, 27, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Sah, A.N.; Bawari, S.; Nabavi, S.F.; Dehpour, A.R.; Shirooie, S.; Braidy, N.; Fiebich, B.L.; Vacca, R.A.; Nabavi, S.M. Role of Nitric Oxide in Neurodegeneration: Function, Regulation, and Inhibition. Curr. Neuropharmacol. 2021, 19, 114–126. [Google Scholar] [CrossRef]
- Liu, C.; Liang, M.C.; Soong, T.W. Nitric oxide, iron and neurodegeneration. Front. Neurosci. 2019, 13, 114. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Dawson, V.L. Nitric oxide signaling in neurodegeneration and cell death. Adv. Pharmacol. 2018, 82, 57–83. [Google Scholar]
- Isabelle, M.; Simonet, S.; Ragonnet, C.; Sansilvestri-Morel, P.; Clavreul, N.; Vayssettes-Courchay, C.; Verbeuren, T.J. Chronic reduction of nitric oxide level in adult spontaneously hypertensive rats induces aortic stiffness similar to old spontaneously hypertensive rats. J. Vasc. Res. 2012, 49, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Leloup, A.J.; Fransen, P.; Van Hove, C.E.; Demolder, M.; De Keulenaer, G.W.; Schrijvers, D.M. Applanation tonometry in mice: A novel noninvasive technique to assess pulse wave velocity and arterial stiffness. Hypertension 2014, 64, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Structure of cerebral arterioles in mice deficient in expression of the gene for endothelial nitric oxide synthase. Circ. Res. 2004, 95, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraci, F.M.; Sigmund, C.D.; Shesely, E.G.; Maeda, N.; Heistad, D.D. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am. J. Physiol.-Heart Circ. Physiol. 1998, 274, H564–H570. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef]
- Nagano, K.; Ishida, J.; Unno, M.; Matsukura, T.; Fukamizu, A. Apelin elevates blood pressure in ICR mice with L-NAME-induced endothelial dysfunction. Mol. Med. Rep. 2013, 7, 1371–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, O.; Tsutsui, M.; Morishita, T.; Tanimoto, A.; Horiuchi, M.; Tasaki, H.; Huang, P.L.; Sasaguri, Y.; Yanagihara, N.; Nakashima, Y. Long-term treatment with Nω-nitro-L-arginine methyl ester causes arteriosclerotic coronary lesions in endothelial nitric oxide synthase-deficient mice. Circulation 2002, 106, 1729–1735. [Google Scholar] [CrossRef] [Green Version]
- Kilkenny, C.; Browne, W.; Cuthill, I.; Emerson, M.; Altman, D.G. The ARRIVE guidelines. ReqartoCom. PLoS Biol. 2010, 8, e100041. [Google Scholar]
- Underwood, W.; Anthony, R. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition; American Veterinary Medical Association: Schaumburg, IL, USA, 2020. [Google Scholar]
- Van Dam, D.; D’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef]
- Van Dam, D.; Lenders, G.; De Deyn, P.P. Effect of Morris water maze diameter on visual-spatial learning in different mouse strains. Neurobiol. Learn. Mem. 2006, 85, 164–172. [Google Scholar] [CrossRef]
- Feng, M.; Whitesall, S.; Zhang, Y.; Beibel, M.; Alecy, L.D.; DiPetrillo, K. Validation of volume–pressure recording tail-cuff blood pressure measurements. Am. J. Hypertens. 2008, 21, 1288–1291. [Google Scholar] [CrossRef]
- Di Lascio, N.; Stea, F.; Kusmic, C.; Sicari, R.; Faita, F. Non-invasive assessment of pulse wave velocity in mice by means of ultrasound images. Atherosclerosis 2014, 237, 31–37. [Google Scholar] [CrossRef]
- Leloup, A.J.; Hove, C.E.; Kurdi, A.; Moudt, S.; Martinet, W.; Meyer, G.R.; Schrijvers, D.M.; Keulenaer, G.W.; Fransen, P. A novel set-up for the ex vivo analysis of mechanical properties of mouse aortic segments stretched at physiological pressure and frequency. J. Physiol. 2016, 594, 6105–6115. [Google Scholar] [CrossRef] [Green Version]
- Maugard, M.; Doux, C.; Bonvento, G. A new statistical method to analyze Morris Water Maze data using Dirichlet distribution. F1000Research 2019, 8, 1601. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.E.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.B.; Grout, J.; Corlay, S. Jupyter Notebooks—A Publishing Format for Reproducible Computational Workflows. In Proceedings of the 20th International Conference on Electronic Publishing, Göttingen, Germany, 7–6 June 2016; Volume 2016. [Google Scholar]
- Cui, J.-M.; Li, X.-L.; Fu, F.; He, J.-P. Influence of swimming exercise in learning and memory, amino acid content and nNOS expression in prefrontal cortex of aging rats. J. Jilin Univ. (Med. Ed.) 2013, 39, 737–742. [Google Scholar]
- Izumi, Y.; Clifford, D.B.; Zorumski, C.F. Inhibition of long-term potentiation by NMDA-mediated nitric oxide release. Science 1992, 257, 1273–1276. [Google Scholar] [CrossRef]
- Roselló-Lletí, E.; Carnicer, R.; Tarazón, E.; Ortega, A.; Gil-Cayuela, C.; Lago, F.; González-Juanatey, J.R.; Portolés, M.; Rivera, M. Human ischemic cardiomyopathy shows cardiac Nos1 translocation and its increased levels are related to left ventricular performance. Sci. Rep. 2016, 6, 1–9. [Google Scholar]
- Saraiva, R.M.; Minhas, K.M.; Raju, S.V.; Barouch, L.A.; Pitz, E.; Schuleri, K.H.; Vandegaer, K.; Li, D.; Hare, J.M. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: Role of nitroso-redox equilibrium. Circulation 2005, 112, 3415–3422. [Google Scholar] [CrossRef] [Green Version]
- Dawson, D.; Lygate, C.A.; Zhang, M.-H.; Hulbert, K.; Neubauer, S.; Casadei, B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation 2005, 112, 3729–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danson, E.J.; Choate, J.K.; Paterson, D.J. Cardiac nitric oxide: Emerging role for nNOS in regulating physiological function. Pharmacol. Ther. 2005, 106, 57–74. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Jin, C.Z.; Jang, J.H.; Wang, Y. Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J. Physiol. 2014, 592, 3189–3200. [Google Scholar] [CrossRef]
- Tandai-Hiruma, M.; Kato, K.; Kemuriyama, T.; Ohta, H.; Tashiro, A.; Hagisawa, K.; Nishida, Y. High blood pressure enhances brain stem neuronal nitric oxide synthase activity in Dahl salt-sensitive rats. Clin. Exp. Pharmacol. Physiol. 2013, 40, 197–204. [Google Scholar] [CrossRef]
- Togashi, H.; Sakuma, I.; Yoshioka, M.; Kobayashi, T.; Yasuda, H.; Kitabatake, A.; Saito, H.; Gross, S.; Levi, R. A central nervous system action of nitric oxide in blood pressure regulation. J. Pharmacol. Exp. Ther. 1992, 262, 343–347. [Google Scholar]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal nitric oxide synthase in vascular physiology and diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Ma, H.; Chintala, R.; Miyake, K.; Fulton, D.J.; Barman, S.A.; White, R.E. Nongenomic, endothelium-independent effects of estrogen on human coronary smooth muscle are mediated by type I (neuronal) NOS and PI3-kinase-Akt signaling. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H314–H321. [Google Scholar] [CrossRef] [Green Version]
- Melikian, N.; Seddon, M.D.; Casadei, B.; Chowienczyk, P.J.; Shah, A.M. Neuronal nitric oxide synthase and human vascular regulation. Trends Cardiovasc. Med. 2009, 19, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, M.R.; Pasterkamp, G.; Yeung, A.C.; Borst, C. Arterial remodeling: Mechanisms and clinical implications. Circulation 2000, 102, 1186–1191. [Google Scholar] [CrossRef]
- Humphrey, J.D. Mechanisms of arterial remodeling in hypertension: Coupled roles of wall shear and intramural stress. Hypertension 2008, 52, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, J.; Buttery, L.; O’Shaughnessy, M.; Afzal, F.; de Marticorena, I.F.; Hukkanen, M.; Huang, P.; MacIntyre, I.; Polak, J. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am. J. Pathol. 2001, 158, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.-L.; Xue, Y.-Q.; Ma, T.; Wang, X.; Li, J.J.; Lan, L.; Malik, K.U.; McDonald, M.P.; Dopico, A.M.; Liao, F.-F. Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol. Neurodegener. 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji, H.; Takao, K.; Hattori, S.; Miyakawa, T. Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol. Brain 2016, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, J.O.; De Moudt, S.; Calus, E.; De Deyn, P.P.; Van Dam, D.; De Meyer, G.R. Age-related cognitive decline in spatial learning and memory of C57BL/6J mice. Behav. Brain Res. 2021, 418, 113649. [Google Scholar] [CrossRef] [PubMed]
- Montaser, A.B.; Jarvinen, J.; Löffler, S.; Huttunen, J.; Auriola, S.; Lehtonen, M.; Jalkanen, A.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Enables the Efficient Brain Delivery of Small-Sized Prodrug across the Blood–Brain Barrier and into Human and Mouse Brain Parenchymal Cells. ACS Chem. Neurosci. 2020, 11, 4301–4315. [Google Scholar] [CrossRef]
- Majzúnová, M.; Pakanová, Z.; Kvasnička, P.; Bališ, P.; Čačányiová, S.; Dovinová, I. Age-dependent redox status in the brain stem of NO-deficient hypertensive rats. J. Biomed. Sci. 2017, 24, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, S.; Leopold, E.; Schmidt, K.; Brunner, F.; Mayer, B. Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): Requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br. J. Pharmacol. 1996, 118, 1433–1440. [Google Scholar] [CrossRef]
- Reif, D.W.; McCreedy, S.A. N-nitro-L-arginine and N-monomethyl-L-arginine exhibit a different pattern of inactivation toward the three nitric oxide synthases. Arch. Biochem. Biophys. 1995, 320, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Bland-Ward, P.; Pitcher, A.; Wallace, P.; Gaffen, Z.; Babbedge, R.; Moore, P. Isoform selectivity of indazole-based nitric oxide synthase inhibitors. Br. J. Pharmacol.-Proc. Suppl. 1994, 112, 351P. [Google Scholar]
- Bland-Ward, P.A.; Moore, P.K. 7-Nitro indazole derivatives are potent inhibitors of brain, endothelium and inducible isoforms of nitric oxide synthase. Life Sci. 1995, 57, PL131–PL135. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrickx, J.O.; De Moudt, S.; Calus, E.; De Deyn, P.P.; Van Dam, D.; De Meyer, G.R.Y. Long-Term Pharmacological Inhibition of the Activity of All NOS Isoforms Rather Than Genetic Knock-Out of Endothelial NOS Leads to Impaired Spatial Learning and Memory in C57BL/6 Mice. Biomedicines 2021, 9, 1905. https://doi.org/10.3390/biomedicines9121905
Hendrickx JO, De Moudt S, Calus E, De Deyn PP, Van Dam D, De Meyer GRY. Long-Term Pharmacological Inhibition of the Activity of All NOS Isoforms Rather Than Genetic Knock-Out of Endothelial NOS Leads to Impaired Spatial Learning and Memory in C57BL/6 Mice. Biomedicines. 2021; 9(12):1905. https://doi.org/10.3390/biomedicines9121905
Chicago/Turabian StyleHendrickx, Jhana O., Sofie De Moudt, Elke Calus, Peter Paul De Deyn, Debby Van Dam, and Guido R. Y. De Meyer. 2021. "Long-Term Pharmacological Inhibition of the Activity of All NOS Isoforms Rather Than Genetic Knock-Out of Endothelial NOS Leads to Impaired Spatial Learning and Memory in C57BL/6 Mice" Biomedicines 9, no. 12: 1905. https://doi.org/10.3390/biomedicines9121905