
Tumor Marker B7-H6 Bound to the Coiled Coil Peptide-Polymer Conjugate Enables Targeted Therapy by Activating Human Natural Killer Cells

 , , , ,
, , , , 
Abstract
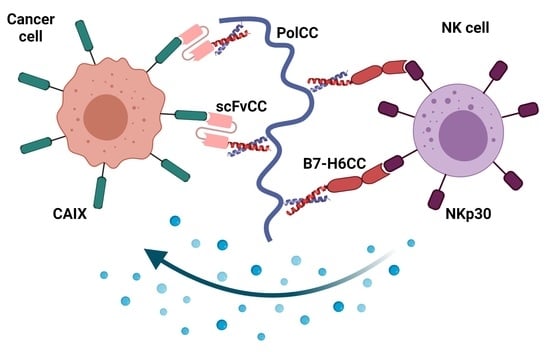
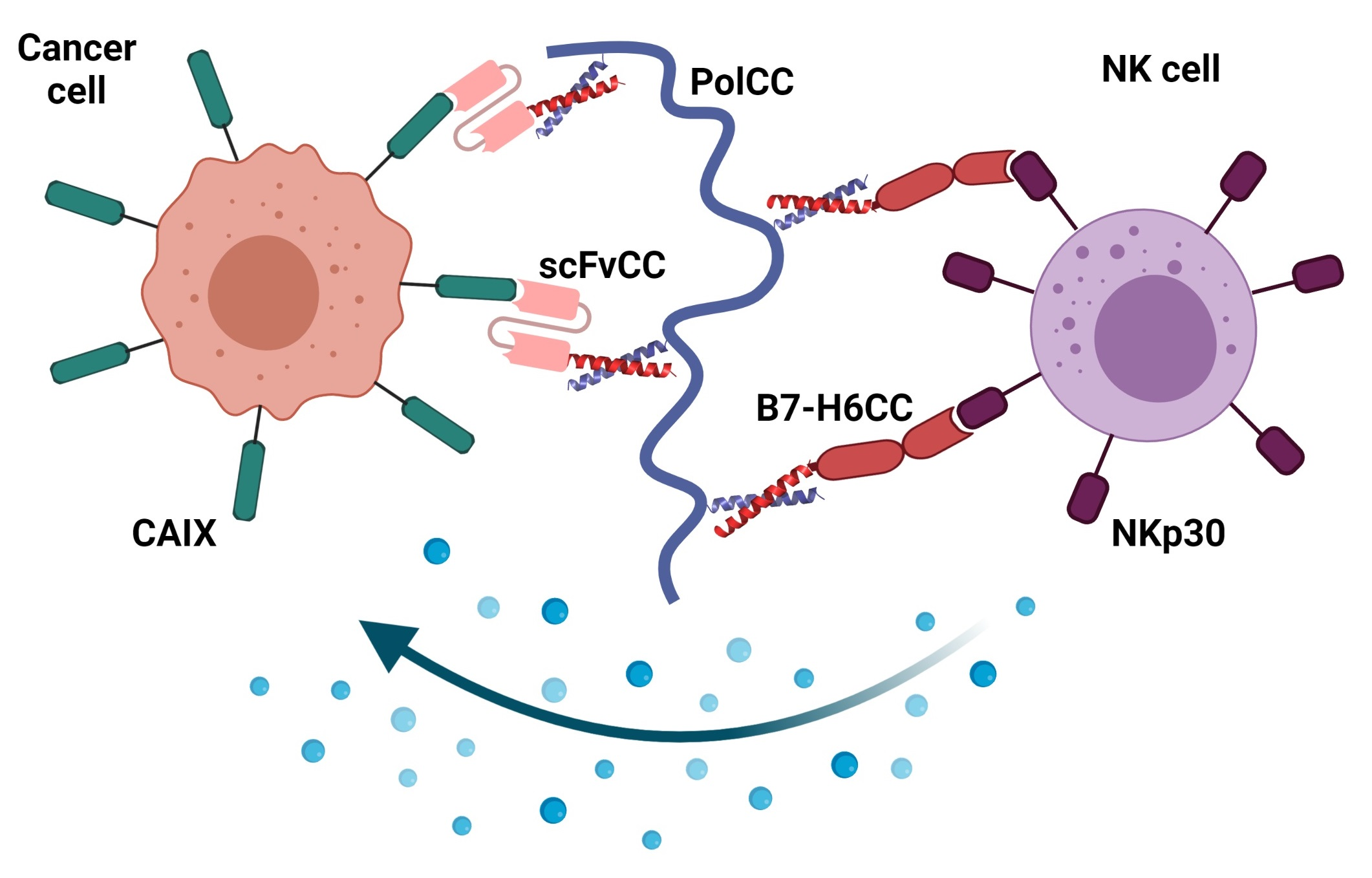
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
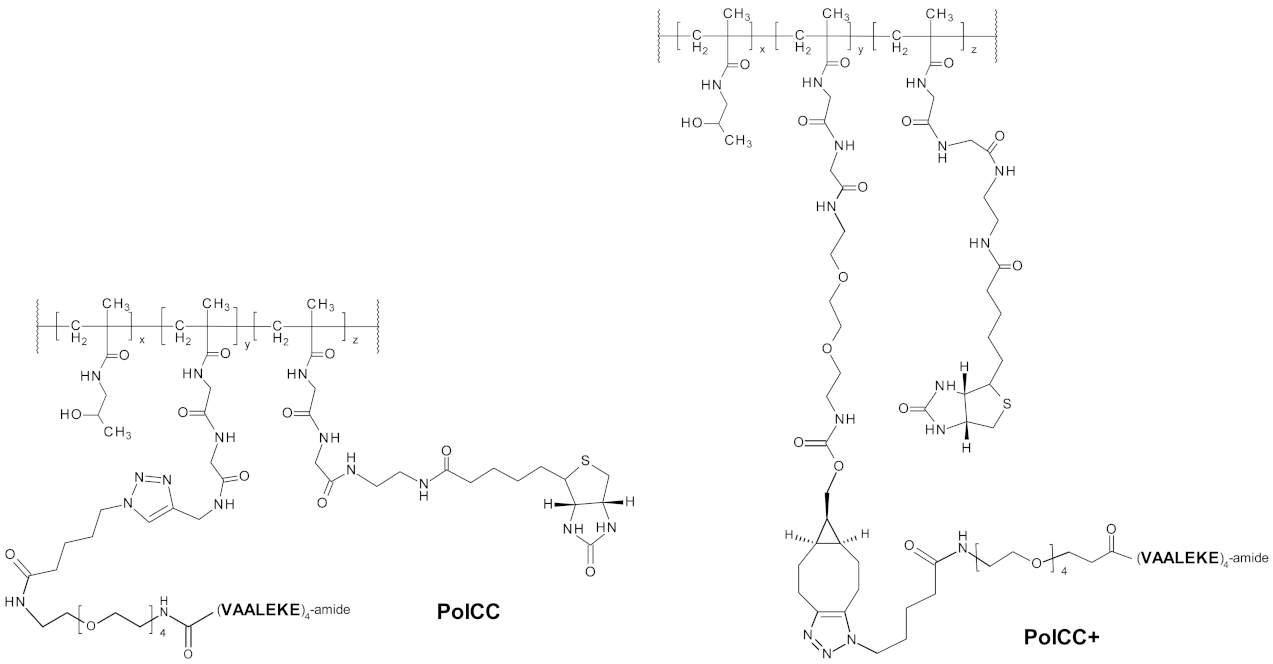
2.1. Preparation of Polymer-Peptide Conjugates
2.2. Cell Lines
2.3. Vector Design
2.4. Protein Expression and Purification
2.5. Isothermal Titration Calorimetry
2.6. Sedimentation Analysis
2.7. NK Cell Activation Assay
2.8. Cell Staining with Polymer–Protein Complexes
3. Results and Discussion
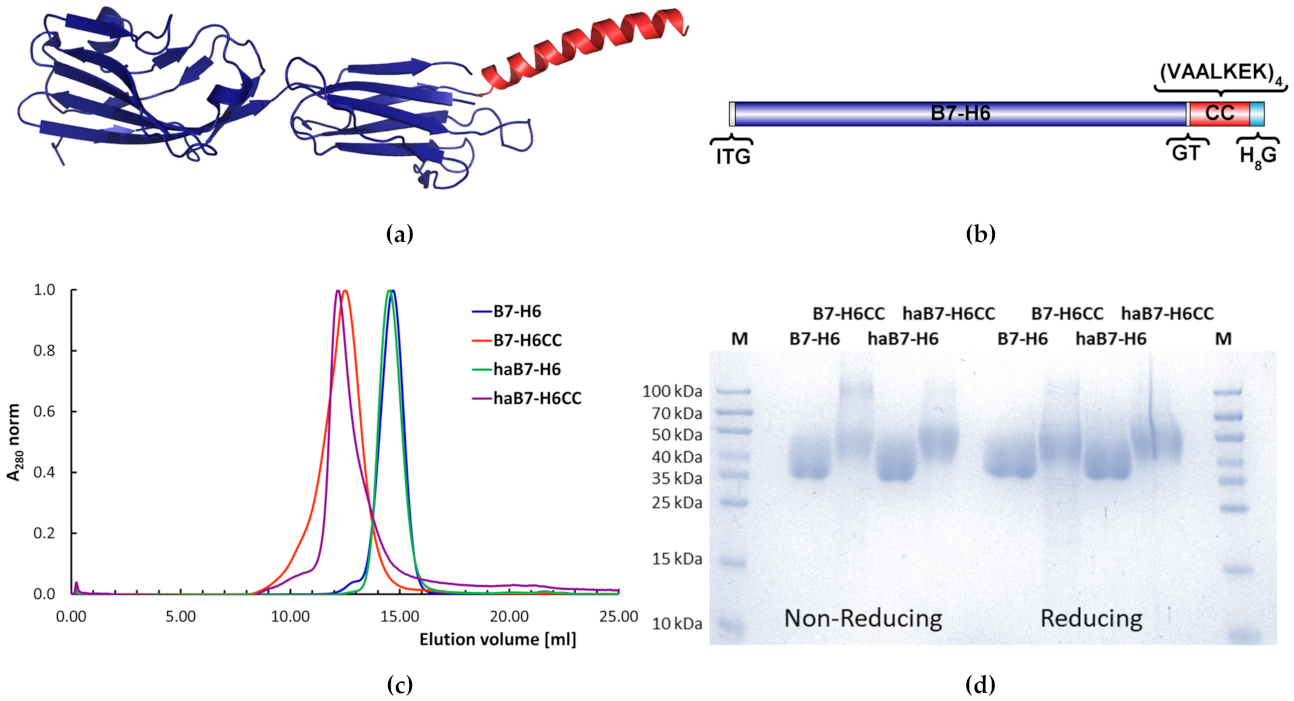
3.1. Design and Production of Coiled Coil B7-H6
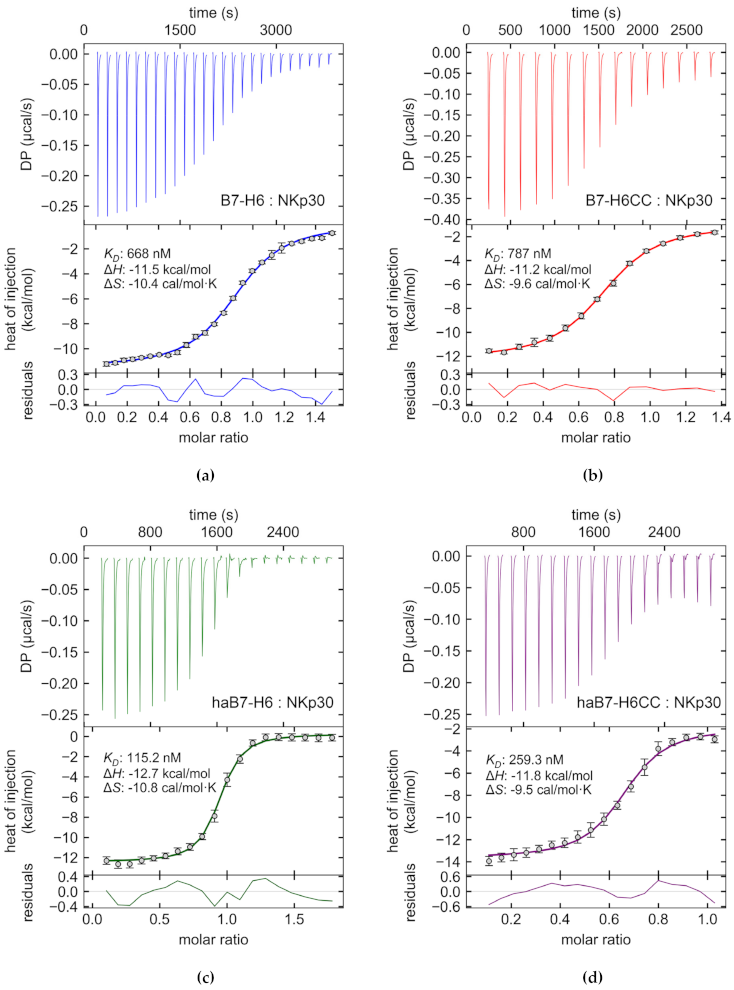
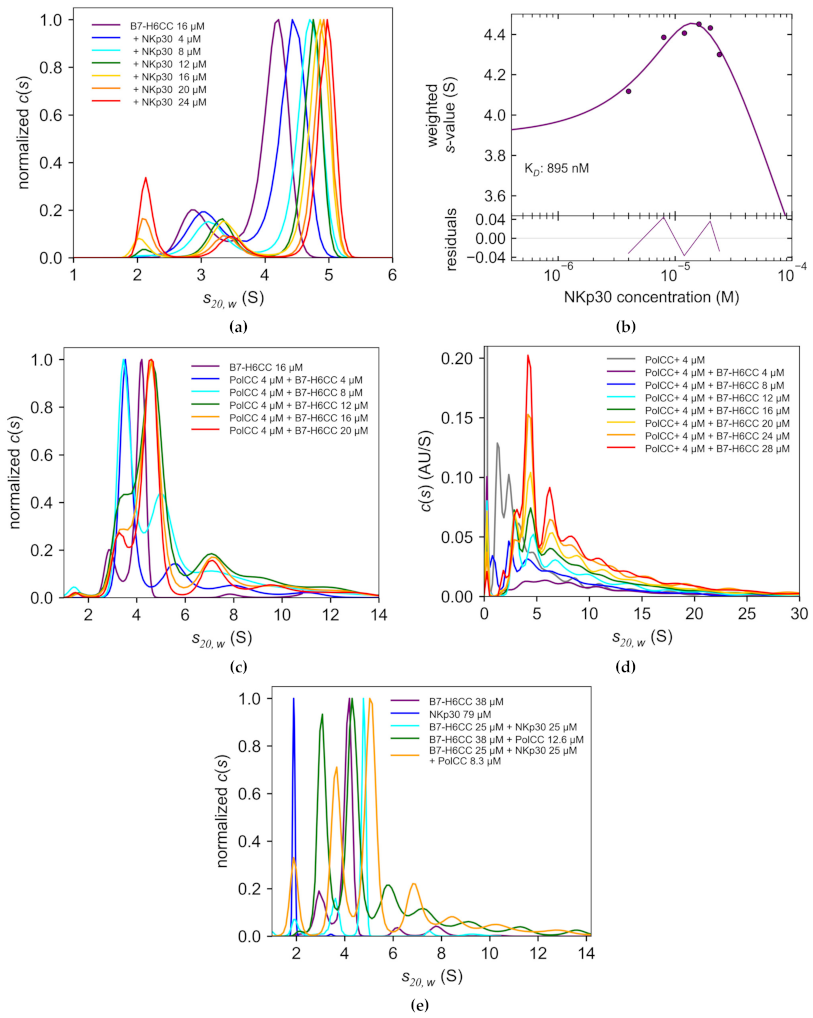
3.2. Coiled Coil B7-H6 Binds NKp30 Receptor with an Undisturbed Affinity
3.3. B7-H6CC Binds to the Polymer-Peptide Conjugate
3.4. B7-H6CC and B7-H6CC:PolCC Complexes Activate NK Cells
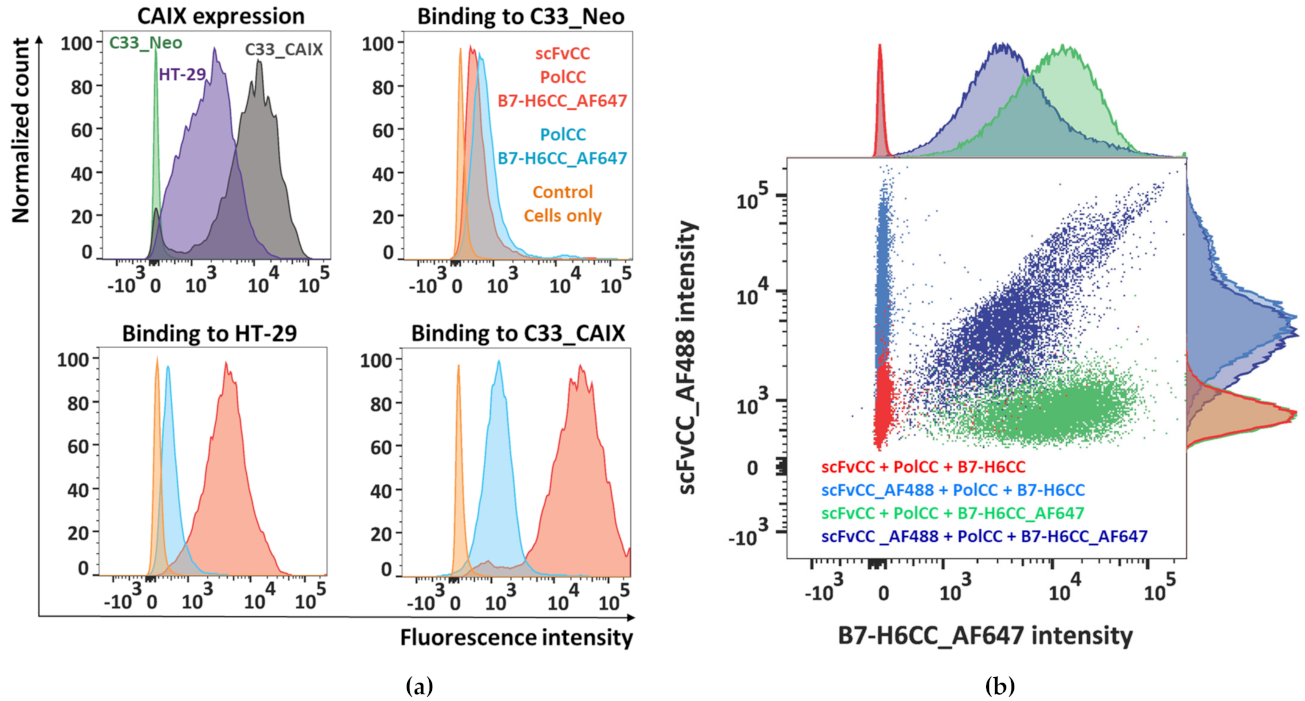
3.5. Polymer–Protein Complexes Bind to the Target Tumor Cell Line
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Sup-pressing Tumor Development and Shaping Tumor Immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, O.A.; Mesci, A.; Ma, J.; Chen, P.; Kirkham, C.L.; Hundrieser, J.; Voigt, S.; Allan, D.S.; Carlyle, J.R. Modulation of Clr Ligand Expression and NKR-P1 Receptor Function during Murine Cytomegalovirus Infection. J. Innate Immun. 2015, 7, 584–600. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2015, 94, 509–522. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Pascal, V.; Schleinitz, N.; Brunet, C.; Ravet, S.; Bonnet, E.; Lafarge, X.; Touinssi, M.; Reviron, D.; Viallard, J.F.; Moreau, J.F.; et al. Comparative analysis of NK cell subset distribution in normal and lymphoproliferative disease of granular lymphocyte conditions. Eur. J. Immunol. 2004, 34, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; Björkström, N.K.; Mjösberg, J.; Ljunggren, H. Natural Killer Cells. In The Autoimmune Diseases; Elsevier: Amsterdam, The Netherlands, 2020; pp. 229–242. [Google Scholar]
- Morvan, M.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2015, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Habif, G.; Crinier, A.; André, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef]
- Chauhan, S.K.S.; Koehl, U.; Kloess, S. Harnessing NK Cell Checkpoint-Modulating Immunotherapies. Cancers 2020, 12, 1807. [Google Scholar] [CrossRef]
- Dahlberg, C.I.M.; Sarhan, D.; Chrobok, M.; Duru, A.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [Green Version]
- Pfefferle, A.; Huntington, N.D. You Have Got a Fast CAR: Chimeric Antigen Receptor NK Cells in Cancer Therapy. Cancers 2020, 12, 706. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.L.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, S.S.; Carol, H.; Biro, M. TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Hum. Vaccines Immunother. 2016, 12, 2790–2796. [Google Scholar] [CrossRef] [Green Version]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. In Natural Killer Cells; Humana Press: New York, NY, USA, 2016; Volume 1441, pp. 333–346. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Märklin, M.; Hagelstein, I.; Koerner, S.P.; Rothfelder, K.; Pfluegler, M.S.; Schumacher, A.; Grosse-Hovest, L.; Jung, G.; Salih, H.R. Bispecific NKG2D-CD3 and NKG2D-CD16 fusion proteins for induction of NK and T cell reactivity against acute myeloid leukemia. J. Immunother. Cancer 2019, 7, 143. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Sun, F.; Zhang, X.; Wang, T.; Jiang, J.; Cai, J.; Gao, Q.; Hezam, K.; Liu, Y.; Xie, J.; et al. CD24 targeting bi-specific antibody that simultaneously stimulates NKG2D enhances the efficacy of cancer immunotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Fritsch, K.; Kohrt, H.E. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front. Immunol. 2015, 6, 601. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.; Steinle, A.; Watzl, C.; Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013, 34, 182–191. [Google Scholar] [CrossRef]
- Memmer, S.; Weil, S.; Beyer, S.; Zöller, T.; Peters, E.; Hartmann, J.; Steinle, A.; Koch, J. The Stalk Domain of NKp30 Contributes to Ligand Binding and Signaling of a Preassembled NKp30-CD3ζ Complex. J. Biol. Chem. 2016, 291, 25427–25438. [Google Scholar] [CrossRef] [Green Version]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Binici, J.; Koch, J. BAG-6, a jack of all trades in health and disease. Cell. Mol. Life Sci. 2013, 71, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2013, 92, 221–229. [Google Scholar] [CrossRef]
- Wang, W.; Guo, H.; Geng, J.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Tumor-released Galectin-3, a Soluble Inhibitory Ligand of Human NKp30, Plays an Important Role in Tumor Escape from NK Cell Attack. J. Biol. Chem. 2014, 289, 33311–33319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaifu, T.; Escalière, B.; Gastinel, L.N.; Vivier, E.; Baratin, M. B7-H6/NKp30 interaction: A mechanism of alerting NK cells against tumors. Cell. Mol. Life Sci. 2011, 68, 3531–3539. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Q.; Mariuzza, R.A. Structure of the human activating natural cytotoxicity receptor NKp30 bound to its tumor cell ligand B7-H6. J. Exp. Med. 2011, 208, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Skořepa, O.; Pazicky, S.; Kalousková, B.; Bláha, J.; Abreu, C.; Ječmen, T.; Rosůlek, M.; Fish, A.; Sedivy, A.; Harlos, K.; et al. Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation. Cancers 2020, 12, 1998. [Google Scholar] [CrossRef]
- Xu, X.; Narni-Mancinelli, E.; Cantoni, C.; Li, Y.; Guia, S.; Gauthier, L.; Chen, Q.; Moretta, A.; Vély, F.; Eisenstein, E.; et al. Structural Insights into the Inhibitory Mechanism of an Antibody against B7-H6, a Stress-Induced Cellular Ligand for the Natural Killer Cell Receptor NKp30. J. Mol. Biol. 2016, 428, 4457–4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnon, T.I.; Markel, G.; Bar-Ilan, A.; Hanna, J.; Fima, E.; Benchetrit, F.; Galili, R.; Cerwenka, A.; Benharroch, D.; Sion-Vardy, N.; et al. Harnessing Soluble NK Cell Killer Receptors for the Generation of Novel Cancer Immune Therapy. PLoS ONE 2008, 3, e2150. [Google Scholar] [CrossRef]
- Kellner, C.; Maurer, T.; Hallack, D.; Repp, R.; Van De Winkel, J.G.J.; Parren, P.W.H.I.; Valerius, T.; Humpe, A.; Gramatzki, M.; Peipp, M.; et al. Mimicking an Induced Self Phenotype by Coating Lymphomas with the NKp30 Ligand B7-H6 Promotes NK Cell Cytotoxicity. J. Immunol. 2012, 189, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, C.; Günther, A.; Humpe, A.; Repp, R.; Klausz, K.; Derer, S.; Valerius, T.; Ritgen, M.; Brüggemann, M.; Van De Winkel, J.G.; et al. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. OncoImmunology 2015, 5, e1058459. [Google Scholar] [CrossRef] [Green Version]
- Peipp, M.; Derer, S.; Lohse, S.; Staudinger, M.; Klausz, K.; Valerius, T.; Gramatzki, M.; Kellner, C. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget 2015, 6, 32075–32088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, M.-R.; Sentman, C.L. An NKp30-Based Chimeric Antigen Receptor Promotes T Cell Effector Functions and Antitumor Efficacy In Vivo. J. Immunol. 2012, 189, 2290–2299. [Google Scholar] [CrossRef]
- Wu, M.-R.; Zhang, T.; Demars, L.R.; Sentman, C.L. B7H6-specific chimeric antigen receptors lead to tumor elimination and host antitumor immunity. Gene Ther. 2015, 22, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, C.K.; Gacerez, A.T.; Sentman, C.L.; Ackerman, M.E. Development of unique cytotoxic chimeric antigen receptors based on human scFv targeting B7H6. Protein Eng. Des. Sel. 2017, 30, 713–721. [Google Scholar] [CrossRef]
- Vaněk, O.; Nálezková, M.; Kavan, D.; Borovičková, I.; Pompach, P.; Novák, P.; Kumar, V.; Vannucci, L.; Hudeček, J.; Hofbauerová, K.; et al. Soluble recombinant CD69 receptors optimized to have an exceptional physical and chemical stability display prolonged circulation and remain intact in the blood of mice. FEBS J. 2008, 275, 5589–5606. [Google Scholar] [CrossRef]
- Kolenko, P.; Skálová, T.; Vaněk, O.; Štěpánková, A.; Dušková, J.; Hašek, J.; Bezouška, K.; Dohnálek, J. The high-resolution structure of the extracellular domain of human CD69 using a novel polymer. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 1258–1260. [Google Scholar] [CrossRef]
- Deming, T.J.; Klok, H.-A.; Armes, S.P.; Becker, M.L.; Champion, J.A.; Chen, E.Y.-X.; Heilshorn, S.C.; van Hest, J.C.M.; Irvine, D.J.; Johnson, J.A.; et al. Polymers at the Interface with Biology. Biomacromolecules 2018, 19, 3151–3162. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.D.; Nam, G.-H.; Kwak, G.; Yang, Y.; Kwon, I.C. Harnessing designed nanoparticles: Current strategies and future perspectives in cancer immunotherapy. Nano Today 2017, 17, 23–37. [Google Scholar] [CrossRef]
- Aikins, M.E.; Xu, C.; Moon, J.J. Engineered Nanoparticles for Cancer Vaccination and Immunotherapy. Accounts Chem. Res. 2020, 53, 2094–2105. [Google Scholar] [CrossRef]
- Thangam, R.; Patel, K.D.; Kang, H.; Paulmurugan, R. Advances in Engineered Polymer Nanoparticle Tracking Platforms towards Cancer Immunotherapy—Current Status and Future Perspectives. Vaccines 2021, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Janisova, L.; Gruzinov, A.; Zaborova, O.V.; Velychkivska, N.; Vaněk, O.; Chytil, P.; Etrych, T.; Janoušková, O.; Zhang, X.; Blanchet, C.; et al. Molecular Mechanisms of the Interactions of N-(2-Hydroxypropyl)methacrylamide Copolymers Designed for Cancer Therapy with Blood Plasma Proteins. Pharmaceutics 2020, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolovic, B.; Danial, M.; Klok, H.-A. Coiled coils: Attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem. Soc. Rev. 2010, 39, 3541–3575. [Google Scholar] [CrossRef]
- Utterström, J.; Naeimipour, S.; Selegård, R.; Aili, D. Coiled coil-based therapeutics and drug delivery systems. Adv. Drug Deliv. Rev. 2020, 170, 26–43. [Google Scholar] [CrossRef]
- Pola, R.; Laga, R.; Ulbrich, K.; Sieglová, I.; Král, V.; Fábry, M.; Kabešová, M.; Kovář, M.; Pechar, M. Polymer Therapeutics with a Coiled Coil Motif Targeted against Murine BCL1 Leukemia. Biomacromolecules 2013, 14, 881–889. [Google Scholar] [CrossRef]
- Wu, K.; Liu, J.; Johnson, R.N.; Yang, J.; Kopeček, J. Drug-Free Macromolecular Therapeutics: Induction of Apoptosis by Coiled-Coil-Mediated Cross-Linking of Antigens on the Cell Surface. Angew. Chem. Int. Ed. 2010, 49, 1451–1455. [Google Scholar] [CrossRef]
- Wu, K.; Yang, J.; Liu, J.; Kopeček, J. Coiled-coil based drug-free macromolecular therapeutics: In vivo efficacy. J. Control. Release 2012, 157, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.-W.; Yang, J.; Zhang, R.; Sima, M.; Kopeček, J. Cell Surface Self-Assembly of Hybrid Nanoconjugates via Oligonucleotide Hybridization Induces Apoptosis. ACS Nano 2014, 8, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.-W.; Zhang, R.; Yang, J.; Chao, M.P.; Shami, P.J.; Kopeček, J. A Two-Step Pretargeted Nanotherapy for CD20 Cross-linking May Achieve Superior Anti-Lymphoma Efficacy to Rituximab. Theranostics 2015, 5, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Fang, Y.; Yang, J.; Kopeček, J. Drug-free macromolecular therapeutics: Impact of structure on induction of apoptosis in Raji B cells. J. Control. Release 2017, 263, 139–150. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y.; Li, L.; Yang, J.; Radford, D.C.; Kopeček, J. Human Serum Albumin-Based Drug-Free Macromolecular Therapeutics: Apoptosis Induction by Coiled-Coil-Mediated Cross-Linking of CD20 Antigens on Lymphoma B Cell Surface. Macromol. Biosci. 2018, 18, e1800224. [Google Scholar] [CrossRef] [PubMed]
- Gambles, M.T.; Li, J.; Wang, J.; Sborov, D.; Yang, J.; Kopeček, J. Crosslinking of CD38 Receptors Triggers Apoptosis of Malignant B Cells. Molecules 2021, 26, 4658. [Google Scholar] [CrossRef]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Pechar, M.; Pola, R.; Laga, R.; Ulbrich, K.; Bednárová, L.; Maloň, P.; Sieglová, I.; Král, V.; Fábry, M.; Vaněk, O. Coiled Coil Peptides as Universal Linkers for the Attachment of Recombinant Proteins to Polymer Therapeutics. Biomacromolecules 2011, 12, 3645–3655. [Google Scholar] [CrossRef]
- Kissel, M.; Peschke, P.; Šubr, V.; Ulbrich, K.; Strunz, A.M.; Kühnlein, R.; Debus, J.; Friedrich, E. Detection and cellular localisation of the synthetic soluble macromolecular drug carrier pHPMA. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1055–1062. [Google Scholar] [CrossRef]
- Aricescu, A.R.; Lu, W.; Jones, E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 1243–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaněk, O.; Celadova, P.; Skořepa, O.; Bláha, J.; Kalousková, B.; Dvorská, A.; Poláchová, E.; Pucholtová, H.; Kavan, D.; Pompach, P.; et al. Production of recombinant soluble dimeric C-type lectin-like receptors of rat natural killer cells. Sci. Rep. 2019, 9, 17836. [Google Scholar] [CrossRef] [Green Version]
- Csaderova, L.; Debreova, M.; Radvak, P.; Stano, M.; Vrestiakova, M.; Kopacek, J.; Pastorekova, S.; Svastova, E. The effect of carbonic anhydrase IX on focal contacts during cell spreading and migration. Front. Physiol. 2013, 4, 271. [Google Scholar] [CrossRef] [Green Version]
- Bláha, J.; Pachl, P.; Novák, P.; Vaněk, O. Expression and purification of soluble and stable ectodomain of natural killer cell receptor LLT1 through high-density transfection of suspension adapted HEK293S GnTI— cells. Protein Expr. Purif. 2015, 109, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Durocher, Y. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, E9. [Google Scholar] [CrossRef] [PubMed]
- Pekar, L.; Klausz, K.; Busch, M.; Valldorf, B.; Kolmar, H.; Wesch, D.; Oberg, H.-H.; Krohn, S.; Boje, A.S.; Gehlert, C.L.; et al. Affinity Maturation of B7-H6 Translates into Enhanced NK Cell–Mediated Tumor Cell Lysis and Improved Proinflammatory Cytokine Release of Bispecific Immunoligands via NKp30 Engagement. J. Immunol. 2020, 206, 225–236. [Google Scholar] [CrossRef]
- Scheuermann, T.H.; Brautigam, C.A. High-precision, automated integration of multiple isothermal titration calorimetric thermograms: New features of NITPIC. Methods 2014, 76, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Brautigam, C.A. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzym. 2015, 562, 109–133. [Google Scholar] [CrossRef]
- Rozbeský, D.; Kavan, D.; Chmelík, J.; Novák, P.; Vaněk, O.; Bezouška, K. High-level expression of soluble form of mouse natural killer cell receptor NKR-P1C(B6) in Escherichia coli. Protein Expr. Purif. 2011, 77, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P. Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Skálová, T.; Kotýnková, K.; Dušková, J.; Hašek, J.; Koval’, T.; Kolenko, P.; Novák, P.; Man, P.; Hanč, P.; Vaněk, O.; et al. Mouse Clr-g, a Ligand for NK Cell Activation Receptor NKR-P1F: Crystal Structure and Biophysical Properties. J. Immunol. 2012, 189, 4881–4889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skálová, T.; Bláha, J.; Harlos, K.; Dušková, J.; Koval’, T.; Stránský, J.; Hašek, J.; Vaněk, O.; Dohnálek, J. Four crystal structures of human LLT1, a ligand of human NKR-P1, in varied glycosylation and oligomerization states. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 578–591. [Google Scholar] [CrossRef]
- Joyce, M.G.; Tran, P.; Zhuravleva, M.A.; Jaw, J.; Colonna, M.; Sun, P.D. Crystal structure of human natural cytotoxicity receptor NKp30 and identification of its ligand binding site. Proc. Natl. Acad. Sci. USA 2011, 108, 6223–6228. [Google Scholar] [CrossRef] [Green Version]
- Bláha, J.; Kalousková, B.; Skořepa, O.; Pažický, S.; Novák, P.; Vaněk, O. High-level expression and purification of soluble form of human natural killer cell receptor NKR-P1 in HEK293S GnTI— Cells. Protein Expr. Purif. 2017, 140, 36–43. [Google Scholar] [CrossRef]
- Herrmann, J.; Berberich, H.; Hartmann, J.; Beyer, S.; Davies, K.; Koch, J. Homo-oligomerization of the Activating Natural Killer Cell Receptor NKp30 Ectodomain Increases Its Binding Affinity for Cellular Ligands. J. Biol. Chem. 2014, 289, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Pechar, M.; Pola, R.; Laga, R.; Braunova, A.; Filippov, S.K.; Bogomolova, A.; Bednarova, L.; Vanek, O.; Ulbrich, K. Coiled coil peptides and polymer-peptide conjugates: Synthesis, self-assembly, characterization and potential in drug delivery systems. Biomacromolecules 2014, 15, 2590–2599. [Google Scholar] [CrossRef]
- Matta, J.; Baratin, M.; Chiche, L.; Forel, J.M.; Cognet, C.; Thomas, G.; Farnarier, C.; Piperoglou, C.; Papazian, L.; Chaussabel, D.; et al. Induction of B7-H6, a ligand for the natural killer cell-activating receptor NKp30, in inflammatory conditions. Blood 2013, 122, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.; Turley, H.; Talks, K.L.; Maxwell, P.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Sowa, T.; Menju, T.; Chen-Yoshikawa, T.F.; Takahashi, K.; Nishikawa, S.; Nakanishi, T.; Shikuma, K.; Motoyama, H.; Hijiya, K.; Aoyama, A.; et al. Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med. 2016, 6, 288–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrich, K.; Šubr, V.; Strohalm, J.; Plocová, D.; Jelínková, M.; Říhová, B. Polymeric Drugs Based on Conjugates of Synthetic and Natural Macromolecules. I. Synthesis and Physico-Chemical Characterisation. J. Control. Release 2000, 64, 63–79. [Google Scholar] [CrossRef]
- Pola, R.; Král, V.; Filippov, S.K.; Kaberov, L.; Etrych, T.; Sieglová, I.; Sedláček, J.; Fábry, M.; Pechar, M. Polymer Cancerostatics Targeted by Recombinant Antibody Fragments to Gd2-Positive Tumor Cells. Biomacromolecules 2019, 20, 412–421. [Google Scholar] [CrossRef]
- Perrier, S.; Takolpuckdee, P.; Westwood, J.; Lewis, D.M. Versatile Chain Transfer Agents for Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerization to Synthesize Functional Polymeric Architectures. Macromolecules 2004, 37, 2709–2717. [Google Scholar] [CrossRef]
- Green, N.M. A Spectrophotometric Assay for Avidin and Biotin Based on Binding of Dyes by Avidin. Biochem. J. 1965, 94, 23C–24C. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.W.; Woolfson, D.N. CCBuilder 2.0: Powerful and accessible coiled-coil modeling. Protein Sci. 2018, 27, 103–111. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalousková, B.; Skořepa, O.; Cmunt, D.; Abreu, C.; Krejčová, K.; Bláha, J.; Sieglová, I.; Král, V.; Fábry, M.; Pola, R.; et al. Tumor Marker B7-H6 Bound to the Coiled Coil Peptide-Polymer Conjugate Enables Targeted Therapy by Activating Human Natural Killer Cells. Biomedicines 2021, 9, 1597. https://doi.org/10.3390/biomedicines9111597
Kalousková B, Skořepa O, Cmunt D, Abreu C, Krejčová K, Bláha J, Sieglová I, Král V, Fábry M, Pola R, et al. Tumor Marker B7-H6 Bound to the Coiled Coil Peptide-Polymer Conjugate Enables Targeted Therapy by Activating Human Natural Killer Cells. Biomedicines. 2021; 9(11):1597. https://doi.org/10.3390/biomedicines9111597
Chicago/Turabian StyleKalousková, Barbora, Ondřej Skořepa, Denis Cmunt, Celeste Abreu, Kateřina Krejčová, Jan Bláha, Irena Sieglová, Vlastimil Král, Milan Fábry, Robert Pola, and et al. 2021. "Tumor Marker B7-H6 Bound to the Coiled Coil Peptide-Polymer Conjugate Enables Targeted Therapy by Activating Human Natural Killer Cells" Biomedicines 9, no. 11: 1597. https://doi.org/10.3390/biomedicines9111597