

Linking Periodontitis with Inflammatory Bowel Disease through the Oral–Gut Axis: The Potential Role of Porphyromonas gingivalis
Shandong Key Laboratory of Oral Tissue Regeneration, Shandong Engineering Laboratory for Dental Materials and Oral Tissue Regeneration, Department of Orthodontics, School and Hospital of Stomatology, Cheeloo College of Medicine, Shandong University, No. 44-1 Wenhua Road West, Jinan 250012, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomedicines 2024, 12(3), 685; https://doi.org/10.3390/biomedicines12030685
Submission received: 22 February 2024
/
Revised: 5 March 2024
/
Accepted: 13 March 2024
/
Published: 19 March 2024
(This article belongs to the Section Microbiology in Human Health and Disease)
{kind=link}
Abstract
:Periodontitis and inflammatory bowel disease (IBD) are both chronic inflammatory diseases that are characterized by abnormal host immune responses and microbiota dysbiosis. Emerging evidence implies potential associations between periodontitis and IBD. Porphyromonas gingivalis (P. gingivalis), a primary cause of periodontitis, is thought to play a role in the development of IBD through the oral–gut disease axis. However, the precise mechanisms of its involvement remain enigmatic. In this narrative review, we begin with a discussion of the bidirectional relationship between periodontitis and IBD and the involvement of P. gingivalis in each of the two diseases. Further, we summarize the possible routes by which P. gingivalis links periodontitis and IBD through the oral–gut axis, as well as the underlying mechanisms of its involvement in the pathogenesis of IBD. Collectively, P. gingivalis participates in the progression of IBD through gut dysbiosis, impairment of the intestinal barrier, release of inflammatory mediators, and disturbance of the immune response. The above findings may provide new insights for exploring novel biomarkers and potential therapeutic approaches for IBD.
1. Introduction
Periodontitis represents the inflammatory response of the oral periodontal tissues, including the gingiva, alveolar bone, and periodontal membrane [1]. Swelling and redness of the gingiva are usually the earliest symptoms of periodontitis, which may be followed by attachment loss and alveolar bone resorption. Severe periodontitis can lead to tooth loosening or loss, negatively affecting general health [2,3].
Notably, individuals with periodontitis often suffer from other complications, one of which is inflammatory bowel disease (IBD) [4,5]. IBD is a chronic inflammatory response of the gastrointestinal tract. Clinically, it primarily manifests in two forms: Crohn’s disease (CD) and ulcerative colitis (UC) [6]. CD is a gastrointestinal tract lesion that often presents with parenteral symptoms in the skin, joints, or eyes (common examples include erythema nodosum, pauciarticular large joint arthritis, axial arthropathies, etc.) and is also accompanied by immune dysfunction [7,8]. UC, on the other hand, is an idiopathic, chronic condition affecting the colonic mucosa [9]. To date, the pathogenesis of IBD remains unclear. However, studies have identified various risk factors that contribute to its progression, with the disturbance of the gut microbiota emerging as a potentially important influence [6].
Accumulating epidemiological evidence has shown a significant association between periodontitis and IBD. As mentioned earlier, the microbiota contributes to both periodontitis and IBD. Could there be a connection between the oral and gut microbiota that forms a bridge between periodontitis and IBD? The recent concept of the “oral–gut axis” may hold the answer, suggesting the involvement of oral pathobionts in the pathogenesis of IBD [5,10].
Porphyromonas gingivalis (P. gingivalis) is widely regarded as a key pathogen in the development of periodontitis [11]. With the advancements in microbiology, oral medicine, and related research techniques, the involvement of P. gingivalis in systemic diseases has been discovered [12]. Multiple studies have revealed that P. gingivalis is a risk factor for numerous systemic diseases, such as cardiovascular diseases [13], diabetes [14], and Alzheimer’s disease [15]. Emerging evidence suggests that P. gingivalis is involved in the pathogenesis of IBD through the oral–gut axis [16,17], but the precise mechanisms remain unclear. In this article, we reviewed recent studies examining the intricate interactions between periodontitis and IBD, as well as the evidence for the engagement of P. gingivalis, a keystone pathogen of periodontitis, in the progression of IBD. Further, we summarized the possible routes through which P. gingivalis links the two diseases through the oral–gut axis and the potential mechanisms for its involvement in the pathogenesis of IBD.
2. Role of P. gingivalis as a Keystone Pathogen of Periodontitis
P. gingivalis is a Gram-negative, obligate anaerobic bacteria. It has been detected in 85.75% of subgingival plaque in patients with chronic periodontitis [18]. Researchers have successfully induced periodontitis in rhesus monkeys by implanting P. gingivalis into their subgingival microbiota [19]. As one of the major pathogens in periodontitis, P. gingivalis exhibits a strong correlation between its prevalence and the severity of periodontal disease. Even in low abundance, P. gingivalis has the ability to induce chronic periodontitis by remodeling the commensal bacterial community, resulting in dysbiosis, thus earning its status as a keystone pathogen of periodontitis [20]. Once colonized, P. gingivalis disrupts the host immune defense and promotes inflammation, leading to alterations in the abundance and composition of the subgingival microbiota and ultimately dysbiosis [21].
P. gingivalis is capable of expressing and releasing several virulence factors, including lipopolysaccharides (LPS), Trichoderma, gingipains, tetratricopeptide repeat sequence protein, extracellular polysaccharides, and the hemoglobin uptake system [22,23]. These virulence factors are capable of mediating immune dysfunction in periodontal tissues. This leads to immune cell infiltration and inflammation, ultimately destroying periodontal tissue [23]. The destructed tissues release nutrients, including degraded collagen, sources of amino acids and iron, and heme-containing compounds. These nutrients penetrate the gingival crevice through gingival crevicular fluid (GCF), promoting the growth of periodontitis-associated microorganisms. This, in turn, further exacerbates the dysbiosis associated with periodontitis [24].
In addition, P. gingivalis belongs to the category of intracellular pathogenic bacteria. By invading multiple human cells, P. gingivalis adjusts its expression pattern to evade immune surveillance, allowing it to serve as a reservoir for persistent infection and inflammation induction [25,26]. Moreover, P. gingivalis demonstrates the capacity to interact with host cells, resulting in its long-term colonization within the oral cavity [25].
3. Bidirectional Relationship between Periodontitis and IBD
3.1. Epidemiological Evidence
Three meta-analysis reports integrated observational studies to reveal a potential association between periodontitis and IBD. These studies showed a correlation between periodontitis with CD and UC, with pooled odds ratios ranging from 1.72 to 3.64 for CD and 2.39 to 5.37 for UC [27,28,29]. Furthermore, a study conducted by Schmidt et al. [30] revealed that patients with IBD exhibited more severe periodontitis, characterized by a higher clinical attachment loss (CAL) and gingival bleeding. On the other hand, the prevalence of IBD is elevated in patients with periodontitis. Data from the Women’s Health Initiative observational cohort study revealed an association between poorer oral health and IBD [31]. Another national cohort study conducted in Korea suggested a higher likelihood of UC development among patients with periodontitis, especially among smokers aged ≥65 years. However, the association between periodontitis and CD was not statistically significant in this study [32]. A study from Taiwan showed that patients with periodontitis had a significantly increased likelihood of developing secondary UC [33]. Interestingly, a cohort study including more than 20,000 individuals demonstrated a reduced risk of IBD among patients with periodontitis [34]. This reversed relationship may be influenced by factors such as diet and ethnicity, and it is important to delve deeper into the exact causes.
It must be noted that the existing cohort studies have not been able to explain the exact interaction between periodontitis and IBD. The primary reason for this incomplete understanding is the predominant reliance on cross-sectional data, lacking the support of longitudinal observations to capture the dynamic nature of the association over time. Although longitudinal observations were provided in some of the larger cohort studies, the precise causality between periodontitis and IBD remains unclear [32,33,34,35].
3.2. Microbiological Associations between Periodontitis and IBD
The oral cavity and the gut, being the two ends of the digestive tract, are inhabited by unique microbiota that play a pivotal role in human health and disease development [36]. Emerging evidence has demonstrated a strong correlation between oral microbiota and gut diseases. Changes in the gut and oral microbiota of IBD patients provide additional support for the link between periodontitis and IBD [5,37]. Researchers have discovered an abnormal enrichment of typical oral resident bacteria in the intestinal lumen contents and intestinal mucosal tissues of patients suffering from intestinal disease [38]. Therefore, it is conceivable that the translocation of oral pathobionts to the gut and their ectopic colonization may contribute to intestinal disease [39]. Gevers et al. [40] revealed that patients with intestinal inflammation exhibit a significant enrichment of oral bacteria in the gut, including pathogens associated with periodontitis. Similarly, Atarashi et al. [41] demonstrated that periodontal pathogens can ectopically translocate from the oral cavity to the gut, inducing gut dysbiosis and an intestinal immune response that exacerbates intestinal inflammation, suggesting a unique association between periodontitis and IBD. Meanwhile, individuals with IBD have unique oral characteristics compared to healthy individuals. Specifically, they exhibit dysregulation of the oral microbiome, including salivary, dental plaque, tongue, and buccal mucosal microbiota [42,43], and this oral dysbiosis is further associated with the severity of IBD [44]. Moreover, compared to the older population, a higher abundance of the phylum Bacteroidetes was found in the oral microbiome of the younger population [45], which is one of the characteristics of the oral microbiome in IBD patients [42,43], and this may account for the higher prevalence of IBD in young people. These findings suggest a potential link between periodontitis and IBD through microbial communication.
While the existing epidemiological evidence remains controversial in explaining the interaction between periodontitis and IBD, biological evidence suggests that patients with IBD exhibit significant oral dysbiosis. Furthermore, oral bacteria, particularly periodontitis-related pathogens, contribute to the development of IBD through gut translocation and ectopic colonization. Therefore, it is necessary to conduct more high-quality, standardized cohort studies to reveal the exact relationship between periodontitis and IBD.
4. Potential Involvement of P. gingivalis in the Progression of IBD
Several lines of evidence implicate a potential association between P. gingivalis and IBD. Stein et al. [46] analyzed periodontal pathogens in 147 subgingival plaque samples from CD patients, finding that 62.6% of them contained P. gingivalis. By analyzing the microbial macrogenome, Lee et al. [47] found that the abundance of Porphyromonadaceae in fecal samples from patients with CD was significantly higher compared to control volunteers. In vivo experiments have also provided evidence to support the potential association between P. gingivalis and IBD. Researchers observed a significant increase in inflammatory infiltrating cells within the lamina propria of colonic tissue in P. gingivalis-treated mice compared to PBS controls [48]. Similarly, in the Dextran sulfate sodium (DSS)-induced colitis model, mice treated with P. gingivalis exhibited a more severe clinical presentation, characterized by an increased disease activity index (DAI) score and a shortened colon length. On a histological level, it was observed that the administration of P. gingivalis resulted in severe active inflammation with extensive epithelial loss, marked crypt destruction, and dense cellular infiltrations [16,17,47]. These findings demonstrate that P. gingivalis may aggravate colitis induced by DSS. Collectively, the evidence supports the potential involvement of P. gingivalis in the progression of IBD.
5. Linking PD with IBD: Gut Translocation of P. gingivalis
Given the significant abundance of Porphyromonadaceae detected in fecal samples from IBD patients, which suggests a potential role for P. gingivalis in the pathogenesis of IBD [47], researchers have analyzed the gut translocation pathways of P. gingivalis. Two potential routes have been proposed for the migration of P. gingivalis from the oral cavity to the intestine, possibly contributing to the pathogenesis of IBD.
5.1. Enteral Dissemination
Each day, individuals swallow approximately 600 times and produce around 1.5 L of saliva. This saliva transports enzymes, effector cytokines, oral microorganisms, and various inflammatory cells toward the intestine. However, these microorganisms, cells, and their respective products rarely reach and colonize in a healthy intestine due to the presence of gastric acidity and intestinal barriers [10,49]. Currently, gastric acidity is considered to be the largest barrier to the translocation of oral bacteria to the gut [50,51]. It is estimated that more than 99.9% of swallowed bacteria of oral origin cannot survive in the stomach owing to its acidic environment [52,53]. Conversely, P. gingivalis has the unique ability to tolerate acidic environments and pass through the gastric barrier, offering it a natural advantage for gut translocation [54]. In addition, the number of ingested oral pathobionts must reach a threshold for their successful translocation from the oral cavity to the intestine. Quantitative analysis reveals that patients with severe periodontitis can swallow up to 1012–1013 P. gingivalis per day [55,56,57], which provides favorable prerequisites for its gut translocation. The disruption of the colonization resistance of the gut resident microbiota also serves as a favorable condition for the invasion of oral pathobionts [58]. By establishing the human oral microbiota-associated (HOMA) mouse model, Li et al. [59] demonstrated that P. gingivalis plays a key role in competing for colonization with resident bacteria in the small intestine.
The enteral dissemination of P. gingivalis is further supported by in vivo experiments. Researchers observed a significant inflammatory infiltration in the gut of mice in the treatment group that were orally administered P. gingivalis, compared to those in the control group [48]. Likewise, in the DSS-induced colitis model, mice orally administered P. gingivalis exhibited more severe inflammation at both clinical and histologic levels [16,17,47]. Collectively, these findings demonstrate that P. gingivalis can induce or exacerbate inflammation in the gut through enteral dissemination.
5.2. Hematogenous Dissemination
Studies have revealed that both pathological conditions and routine dental activities, including dental treatment, brushing, flossing, and daily chewing, may lead to mechanical damage to the oral cavity. This damage allows oral bacteria and their toxins to spread into the circulation, causing systematic chronic inflammation [60,61,62,63]. An essential clue is the effect of LPS on neutrophils in the circulation. In the context of periodontitis, LPS can induce changes in the neutrophil phenotype and enhance the inflammatory response in the circulation [64]. However, the detailed mechanism behind the altered neutrophil status and circulation hyperinflammation in periodontitis remains controversial. Whether these changes are due to endotoxemia caused by the entry of LPS into the circulation or due to LPS-related immune training of infiltrating neutrophils in the oral microenvironment needs to be further verified [65].
Furthermore, P. gingivalis may achieve hematogenous migration by invading and surviving in dendritic cells (DCs) and macrophages. Specifically, P. gingivalis enters macrophages via complement receptor 3 (CR3) in cholesterol-rich lipid rafts, which is mediated by FimA fimbriae [66,67]. To survive in DCs, the Mfa1 fimbriae of P. gingivalis interact with the C-type lectin DC-specific ICAM-3 grabbing non-integrin (DC-SIGN), facilitating its invasion into these cells [68,69]. Another pathogenic study similarly characterized the role played by different species of fimbriae in the invasion of P. gingivalis into DCs [70]. Mfa1 fimbriae inhibit autophagic destruction by targeting the DC-SIGN-TLR2 axis for more favorable intracellular survival. Conversely, FimA fimbriae act as Toll-like receptor (TLR) 2 agonists to promote DC autophagy. By regulating the expression of both fimbriae, P. gingivalis achieves immune evasion. Nevertheless, the abovementioned ideas are primarily based on the results of molecular mechanism studies. Further in vivo experiments are imperative to verify the hematogenous dissemination of P. gingivalis.
6. Mechanisms of P. gingivalis Involving in IBD
Noteworthy progress has been made in the research exploring the role of P. gingivalis in the progression of IBD. This section presents an overview of the potential mechanisms of P. gingivalis’s involvement in IBD.
6.1. Gut Dysbiosis
Accumulating evidence indicates that gut dysbiosis plays a crucial role in the progression of IBD. Compared to healthy populations, patients with IBD typically exhibit reduced diversity in their gut microbiota, accompanied by a lower proportion of Firmicutes and an increased proportion of Bacteroidetes and Actinobacteria [71,72]. In addition, certain metabolites derived from gut microbiota, including short-chain fatty acids (SCFAs) and bile acids (BAs), are also believed to be associated with the pathogenesis of IBD [73].
After oral administration of P. gingivalis, mice are found to have a significant reduction in the bacterial diversity of their gut microbiota, along with an increased ratio of Bacteroidetes to Firmicutes [48,74,75]. These changes align with the characteristics observed in patients with IBD. Nakajima et al. [74] found that at the genus level, Prevotella was significantly increased in the intestine of mice following P. gingivalis administration. This increase in Prevotella is believed to drive chronic intestinal inflammation and exacerbate chemically induced colitis [76,77].
The intestinal microbiota is integral to the metabolism of SCFAs and Bas, playing an important role in maintaining the intestinal barrier integrity and immune homeostasis within the host. SCFAs are thought to play a potential anti-inflammatory role in intestinal inflammation, thereby maintaining intestinal barrier integrity [78,79]. In the presence of gut microbiota, primary BAs are converted into secondary BAs, including lithocholic acid and deoxycholic acid. These secondary BAs are involved in immune regulation and exhibit anti-inflammatory activities [80,81]. In the intestine, Ruminocobaceae, Lachnospiraceae, and Marvinbryantia promote the production of SCFAs [82,83,84], and Clostridium and Eubacterium are involved in the conversion of primary BAs to secondary BAs [85]. The relative abundance of the bacterial species mentioned above was reduced in P. gingivalis-treated mice, potentially contributing to the development of intestinal and systemic inflammation [48].
6.2. Impairment of Intestinal Barrier
As a protective umbrella for the intestine, the intestinal epithelial barrier protects against the invasion of various bacteria and the penetration of toxins. The monolayer of intestinal epithelial cells acts as a physical barrier by forming a tight seal with transmembrane protein complexes. These complexes consist of adhesive molecules such as tight junctions, adherent junctions, and desmosomes. Several studies have shown that the administration of P. gingivalis may impair the integrity of the intestinal epithelium. For instance, the administration of P. gingivalis to C57BL/6N mice resulted in a significant decrease in the expression levels of the tight junction proteins ZO-1 and occludin in the small intestine, while no such changes were observed in the large intestine [74,75]. Similarly, Tsuzuno et al. [17] revealed that in a DSS-induced colitis model, treatment with P. gingivalis exacerbated intestinal inflammation in mice. Further investigations demonstrated that P. gingivalis disrupts intestinal barrier function by decreasing the levels of ZO-1 in epithelial cells.
In addition, gut dysbiosis leads to the impairment of the intestinal barrier. Normally, symbiotic bacteria form a microbial barrier on the surface of the mucosal epithelium to resist the invasion of pathogenic microorganisms through colonization resistance and immune response modulation. Gut dysbiosis can result in the disruption of the intestinal microbial barrier function, which increases intestinal permeability and facilitates the invasion of conditioned pathogens. This, in turn, contributes to the inflammatory response in the colon [73]. Furthermore, gut dysbiosis leads to a decrease in intestinal antimicrobial peptides, thereby compromising the intestinal barrier function [86].
6.3. Release of Inflammatory Mediators
6.3.1. LPS
LPS is an important component of the cell wall in Gram-negative bacteria. Due to its virulence and the ability to cause unwanted host inflammation, LPS is known as an endotoxin [11]. The LPS of P. gingivalis acts as a strong pathogenic agent in periodontal tissues. The virulence of LPS is determined by its lipid A component. By responding to the LPS lipid A component of P. gingivalis, host cells generate an inflammatory response in the gingival tissue, creating a favorable environment for pathogens and eventually leading to the progression of periodontal disease [87]. Based on different lipid A structures, LPS can act as an agonist of TLR2 or as an antagonist and/or agonist of TLR4 activation [88,89,90,91,92], causing a series of inflammatory responses.
Previously, it was suggested that P. gingivalis LPS may be involved in the development of intestinal inflammation by inducing a semi-Th2-like response. In a study conducted by Jotwani et al. [93], monocyte-derived DCs (MDDCs) were pulsed with LPS of different oral pathogens. Their findings revealed that, compared to Escherichia coli (E.coli), P. gingivalis LPS induced T cells to produce lower levels of Th1 cytokines and higher levels of Th2 cytokines. An in vivo experiment by Pulendran et al. [94] also supported this conclusion. They demonstrated that P. gingivalis LPS-induced T cell responses were characterized by significantly higher levels of IL-5, IL-10, and IL-13 but lower levels of IFN-γ. Conversely, recent studies have proposed that P. gingivalis-derived LPS may have certain benefits for colitis [47,95]. Seo et al. [95] found that the administration of P. gingivalis LPS attenuated the epithelial damage and lymphocyte infiltration caused by DSS treatment; the DAI score was significantly lower in P. gingivalis-treated mice, suggesting a possible ameliorative activity of P. gingivalis LPS in colitis. Nevertheless, due to limited evidence, whether LPS plays a positive or negative role in IBD remains to be fully elucidated.
6.3.2. Gingipains
Gingipains are a family of cysteine proteases. They can be divided into arginine-specific gingipains (Rgp, including RgpA and RgpB) and lysine-specific gingipains (Kpg) [11,96]. Gingipains are a major virulence factor of P. gingivalis, accounting for 85% of the extracellular protein hydrolytic activity of P. gingivalis [97]. They cause dysregulated immune responses and inflammation by activating host matrix metalloproteinases, inactivating immunosuppressive agents, degrading immune factors, and cleaving immune cell receptors [98,99,100,101].
P. gingivalis secretes gingipains, which evade the intrinsic immune response by selectively inactivating proinflammatory factors produced by DCs and simultaneously sparing those molecules that enhance their survival. Abdi et al. revealed that P. gingivalis significantly reduces the level of IL-12 produced by DCs. Additionally, compared to the W50-NIDCR strain of P. gingivalis, the W50-ATCC strain, which proved to secrete fewer gingipains, was less potent at reducing IL-12 production [102].
P. gingivalis-derived gingipains are also involved in the destruction of the intestinal barrier, including the mucus layer and intestinal epithelial. As part of the intestinal barrier, the mucus layer plays a crucial role in intestinal homeostasis. Reduced mucus secretion due to decreased levels of goblet cells is a hallmark of human IBD, and mice deficient in the major mucin protein MUC2 develop spontaneous colitis [103,104,105]. Post et al. [106] demonstrated that P. gingivalis RgpB can cleave MUC2 mucin in specific sites, resulting in mucus polymer lysis. This process disrupts the intestinal barrier [107,108,109] and contributes to the pathogenesis of IBD [110,111]. Moreover, P. gingivalis-derived gingipains may be responsible for the impairment of the intestinal epithelial barrier. Tsuzuno et al. [17] reported that, in contrast to wild-type strains, the gingipain-deficient P. gingivalis ATCC33277 mutant failed to reduce ZO-1 protein levels, suggesting that gingipains may be involved in the degradation of ZO-1. However, since the two strains are not isogenic and have different fundamental virulence factors [112,113], further experiments are required to confirm the involvement of gingipains in the disruption of the intestinal epithelium.
6.4. Disturbance of Immune Response
6.4.1. Induction of Proinflammatory Cytokines
Inflammatory infiltration in the intestine is also an important pathological change caused by oral administration of P. gingivalis. Administration of P. gingivalis induces elevated mRNA expression levels of proinflammatory cytokines, including TNF-α, IFN-γ, and IL-6, in the intestine [48,74,75]. Similarly, P. gingivalis has been demonstrated to upregulate the expression of IL-6, TNF-α, and IL1-β and exacerbate colitis in DSS-induced mice [17,47]. TNF-α and IFN-γ are typical Th1 cytokines. TNF-α, a pleiotropic cytokine, has been found to be associated with IBD [114,115]. Some studies have observed the overexpression of intestinal TNF-α in patients with CD [116,117,118], leading to the utilization of anti-TNF-α antibody therapy to treat patients with IBD [119,120]. In the intestinal mucosal tissues of IBD patients, the expression level of IFN-γ is highly upregulated, attributed to its immunomodulatory or epithelial effects [121,122]. IL-6 is a key immunomodulator. It acts synergistically with TGF-β to induce the expression of the transcription factor ROR-γ t and promote the differentiation of T helper (Th) 17 cells [123,124]. The aberrant proliferation of Th17 cells and their secretion of a large number of specific cytokines play an important role in the pathogenesis of IBD [125,126]. An elevated level of IL-6 has been found in patients with IBD [127,128]. The inhibition of IL-6 activity by blocking receptors or cytokines with monoclonal antibodies has proved effective in animal models of colitis and in small therapeutic trials for CD [129,130]. IL-1β has the ability to induce proinflammatory innate lymphoid cells and Th17 cells [131]. It also acts as a Th1 skewing factor for the generation of Th1/Th17 cells, which accumulate in the intestine of IBD patients [132,133,134,135,136]. Taken together, P. gingivalis has a colitogenic potency, capable of inducing the expression of intestinal proinflammatory cytokines. However, the exact mechanism underlying the induction of these proinflammatory factors in the intestine remains elusive and warrants further discussion.
6.4.2. Upregulation of Th17/Treg Ratio
IBD is a non-specific mucosal inflammatory disease. Intrinsically, it is a CD4+ T cell-mediated disturbance in the balance between the microbial ecosystem and the host immune system, with a key role played by the equilibrium between Th17 cells and Treg cells [6]. Both Th17 and Treg cells are differentiated from naive CD4+ cells.
Th17 cells promote tissue inflammation, while Treg cells suppress autoimmunity in IBD; they are functionally related through differentiation and suppression [137]. Th17 cells and Treg cells share a common signaling pathway mediated by TGF-β. In the presence of IL-6 or IL-21, naive CD4+T cells differentiate into Th17 cells, while in the absence of proinflammatory cytokines, naive CD4+T cells differentiate into Treg cells [138]. The disruption of the Th17/Treg balance can lead to the development of various autoimmune diseases, including IBD.
Jia et al. [139] found that the cell fragments of P. gingivalis, obtained by ultrasonic treatment, upregulated the expression of the Th17-associated transcription factor RoRγt and enhanced the production of the proinflammatory cytokines IL-17 and IL-6 through the TLR4 pathway. Conversely, they downregulated the expression of the Treg transcription factor Foxp3 and suppressed the production of anti-inflammatory factors TGF-β and IL-10. Further in vivo experiments showed that P. gingivalis-stimulated CD4+ T cells exacerbated DSS-induced colitis by increasing the Th17/Treg ratio in colonic and lamina propria lymphocytes, which was achieved through the JAK-STAT signaling pathway.
However, the detailed mechanisms underlying how P. gingivalis affects Th17/Treg homeostasis and ultimately leads to the development of IBD still need to be investigated. Abnormalities in the Th17/Treg differentiation ratio in the context of IBD are attributed to various factors, including the regulation of inflammatory cytokine expression, influence on BAs release, and induction of intestinal flora dysbiosis [137]. Whether P. gingivalis is involved in one or more of these mechanisms warrants further investigation.
The identified mechanisms by which Pg is involved in IBD are depicted in Figure 1.
7. Perspectives
Periodontitis and IBD, both complex chronic inflammatory diseases characterized by abnormal host immune responses and microbiota dysbiosis, exhibit a high risk of coexistence and are potentially related. Recently, epidemiological, translational medicine and basic science studies have begun to elucidate the oral–gut association behind periodontitis and IBD [35].
In this article, we focus on the role of P. gingivalis in linking periodontitis and IBD through the oral–gut axis. We summarize the routes of P. gingivalis gut translocation and outline the possible mechanisms of its involvement in the pathogenesis of IBD, which contributes to the search for novel biomarkers and potential therapeutic treatments for IBD. A recent study reported that KYT-36, as a highly selective inhibitor peptide against gingipains, may have promising applications in the treatment of IBD and periodontitis [140]. Furthermore, Jia et al. [139] found that Lactobacillus rhamnosus GG (LGG) could decrease the IL-17+Th17 ratio and increase the CD25+Foxp3+Treg ratio through the TLR2 pathway, reversing the increased Th17/Treg ratio caused by P. gingivalis and alleviating DSS-induced colitis. This provides a theoretical basis for subsequent investigations on the treatment of inflammatory diseases.
In addition to gut translocation and ectopic colonization of oral pathobionts, oral-derived immune cells can also be involved in the development of intestinal inflammation through the oral–gut axis. During periodontal inflammation, oral draining lymph nodes produce Th17 cells that recognize oral pathobionts. Oral pathobiont-reactive Th17 cells express intestinal homing molecules, such as CCR9 and α4β7. When oral-derived Th17 cells reach the intestine, they can be activated by translocated oral pathobionts and promote the development of colitis [37]. Given that oral pathobionts, such as P. gingivalis, upregulate IL1-β expression [17] and are involved in regulating Th17 differentiation [139], it is likely that microbial and immune pathways work synergistically to exacerbate intestinal inflammation.
To better elucidate the correlation between periodontitis and IBD, further clinical evidence is required to assess the importance of the oral–intestinal axis in the development of intestinal inflammation. Additionally, more research efforts are needed to explore the microbiological and immunological associations between periodontitis and IBD, thus providing new targets for the diagnosis and treatment of periodontitis and IBD.
Author Contributions
Conceptualization, methodology, validation, project administration, J.Z. and Q.F.; formal analysis, investigation, writing—original draft preparation, writing—review and editing, visualization, Y.L. and X.H.; supervision, Q.F.; funding acquisition, Q.F. All authors have read and agreed to the published version of the manuscript.
Funding
We sincerely thank the foundation support of the National Natural Science Foundation of China (No. 82270980, 82071122), the National Young Scientist Support Foundation (2019), Excellent Young Scientist Foundation of Shandong Province (No. ZR2021JQ29), Taishan Young Scientist Project of Shandong Province (2019), Periodontitis innovation team of Jinan City (2021GXRC021), Major In-novation Projects in Shandong Province (No. 2021SFGC0502), Oral Microbiome Innovation Team of Shandong Province (No. 2020KJK001), Shandong Province Key Research, Development Program (No. 2021ZDSYS18), Natural Science Foundation of Shandong Province (ZR2021QH340) and the Horizontal Scientific Research Fund of Shandong University (1350022003).
Acknowledgments
We sincerely thank all colleagues in our laboratory for all of their kind advice and support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef]
- Slots, J. Periodontitis: Facts, fallacies and the future. Periodontol. 2000 2017, 75, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Chapple, I.L.C.; Mealey, B.L.; Van Dyke, T.E.; Bartold, P.M.; Dommisch, H.; Eickholz, P.; Geisinger, M.L.; Genco, R.J.; Glogauer, M.; Goldstein, M.; et al. Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: Consensus report of workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S74–S84. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhu, T.; Liu, F.; Zhuang, Z.; Zhao, L. Co-pathogens in Periodontitis and Inflammatory Bowel Disease. Front. Med. 2021, 8, 723719. [Google Scholar] [CrossRef] [PubMed]
- Read, E.; Curtis, M.A.; Neves, J.F. The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef]
- Byrd, K.M.; Gulati, A.S. The “Gum-Gut” Axis in Inflammatory Bowel Diseases: A Hypothesis-Driven Review of Associations and Advances. Front. Immunol. 2021, 12, 620124. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, W.; Wang, H.; Liang, S. Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Adv. Protein Chem. Struct. Biol. 2020, 120, 45–84. [Google Scholar] [PubMed]
- Zheng, S.; Yu, S.; Fan, X.; Zhang, Y.; Sun, Y.; Lin, L.; Wang, H.; Pan, Y.; Li, C. Porphyromonas gingivalis survival skills: Immune evasion. J. Periodontal Res. 2021, 56, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Kuramitsu, H.K.; Kang, I.C.; Qi, M. Interactions of Porphyromonas gingivalis with host cells: Implications for cardiovascular diseases. J. Periodontol. 2003, 74, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Montevecchi, M.; Valeriani, L.; Gatto, M.R.; D’Alessandro, G.; Piana, G. Subgingival pathogens in chronic periodontitis patients affected by type 2 diabetes mellitus: A retrospective case-control study. J. Periodontal Implant. Sci. 2021, 51, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Zhang, C.; Yu, N.; Lu, Z.; Zhang, S.; Li, Y.; Li, Q.; Liu, J.; Liu, D.; et al. Porphyromonas gingivalis exacerbates ulcerative colitis via Porphyromonas gingivalis peptidylarginine deiminase. Int. J. Oral. Sci. 2021, 13, 31. [Google Scholar] [CrossRef]
- Tsuzuno, T.; Takahashi, N.; Yamada-Hara, M.; Yokoji-Takeuchi, M.; Sulijaya, B.; Aoki-Nonaka, Y.; Matsugishi, A.; Katakura, K.; Tabeta, K.; Yamazaki, K. Ingestion of Porphyromonas gingivalis exacerbates colitis via intestinal epithelial barrier disruption in mice. J. Periodontal Res. 2021, 56, 275–288. [Google Scholar] [CrossRef]
- How, K.Y.; Song, K.P.; Chan, K.G. Porphyromonas gingivalis: An Overview of Periodontopathic Pathogen below the Gum Line. Front. Microbiol. 2016, 7, 53. [Google Scholar] [CrossRef]
- Holt, S.C.; Ebersole, J.; Felton, J.; Brunsvold, M.; Kornman, K.S. Implantation of Bacteroides gingivalis in nonhuman primates initiates progression of periodontitis. Science 1988, 239, 55–57. [Google Scholar] [CrossRef]
- Olsen, I.; Lambris, J.D.; Hajishengallis, G. Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J. Oral. Microbiol. 2017, 9, 1340085. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Gao, Z.Y.; Sun, C.T.; Wen, H.; Gao, B.; Li, S.B.; Tong, Q. The potential role of P. gingivalis in gastrointestinal cancer: A mini review. Infect. Agent. Cancer 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.P.; Hutcherson, J.A.; Wang, Y.; Nowakowska, Z.M.; Potempa, J.; Yoder-Himes, D.R.; Scott, D.A.; Whiteley, M.; Lamont, R.J. Genes Contributing to Porphyromonas gingivalis Fitness in Abscess and Epithelial Cell Colonization Environments. Front. Cell Infect. Microbiol. 2017, 7, 378. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Plaza, K.; Kalinska, M.; Bochenska, O.; Meyer-Hoffert, U.; Wu, Z.; Fischer, J.; Falkowski, K.; Sasiadek, L.; Bielecka, E.; Potempa, B.; et al. Gingipains of Porphyromonas gingivalis Affect the Stability and Function of Serine Protease Inhibitor of Kazal-type 6 (SPINK6), a Tissue Inhibitor of Human Kallikreins. J. Biol. Chem. 2016, 291, 18753–18764. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.J.; Dashper, S.G.; Slakeski, N.; Chen, Y.Y.; Reynolds, E.C. Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol. Oral. Microbiol. 2016, 31, 365–378. [Google Scholar] [CrossRef] [PubMed]
- She, Y.Y.; Kong, X.B.; Ge, Y.P.; Liu, Z.Y.; Chen, J.Y.; Jiang, J.W.; Jiang, H.B.; Fang, S.L. Periodontitis and inflammatory bowel disease: A meta-analysis. BMC Oral Health 2020, 20, 67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiao, D.; Chen, R.; Zhu, F.; Gong, J.; Yan, F. The Association between Periodontitis and Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. Biomed. Res. Int. 2021, 2021, 6692420. [Google Scholar] [CrossRef]
- Domokos, Z.; Uhrin, E.; Szabó, B.; Czumbel, M.L.; Dembrovszky, F.; Kerémi, B.; Varga, G.; Hegyi, P.; Hermann, P.; Németh, O. Patients with inflammatory bowel disease have a higher chance of developing periodontitis: A systematic review and meta-analysis. Front. Med. 2022, 9, 1020126. [Google Scholar] [CrossRef]
- Schmidt, J.; Weigert, M.; Leuschner, C.; Hartmann, H.; Raddatz, D.; Haak, R.; Mausberg, R.F.; Kottmann, T.; Schmalz, G.; Ziebolz, D. Active matrix metalloproteinase-8 and periodontal bacteria-interlink between periodontitis and inflammatory bowel disease? J. Periodontol. 2018, 89, 699–707. [Google Scholar] [CrossRef]
- Kato, I.; Sun, J.; Larson, J.; Hastert, T.; Abrams, J. History of Inflammatory Bowel Disease and Self-Reported Oral Health: Women’s Health Initiative Observational Study. J. Womens Health 2020, 29, 1032–1040. [Google Scholar] [CrossRef]
- Kang, E.A.; Chun, J.; Kim, J.H.; Han, K.; Soh, H.; Park, S.; Hong, S.W.; Moon, J.M.; Lee, J.; Lee, H.J.; et al. Periodontitis combined with smoking increases risk of the ulcerative colitis: A national cohort study. World J. Gastroenterol. 2020, 26, 5661–5672. [Google Scholar] [CrossRef]
- Lin, C.Y.; Tseng, K.S.; Liu, J.M.; Chuang, H.C.; Lien, C.H.; Chen, Y.C.; Lai, C.Y.; Yu, C.P.; Hsu, R.J. Increased Risk of Ulcerative Colitis in Patients with Periodontal Disease: A Nationwide Population-Based Cohort Study. Int. J. Environ. Res. Public. Health 2018, 15, 2602. [Google Scholar] [CrossRef]
- Yin, W.; Ludvigsson, J.F.; Liu, Z.; Roosaar, A.; Axéll, T.; Ye, W. Inverse Association Between Poor Oral Health and Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2017, 15, 525–531. [Google Scholar] [CrossRef]
- Newman, K.L.; Kamada, N. Pathogenic associations between oral and gastrointestinal diseases. Trends Mol. Med. 2022, 28, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Kamada, N. Periodontal connection with intestinal inflammation: Microbiological and immunological mechanisms. Periodontol. 2000 2022, 89, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Hein, R.; Schmidt, T.M.; Kamada, N. The Bacterial Connection between the Oral Cavity and the Gut Diseases. J. Dent. Res. 2020, 99, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, G.; Cao, H.; Yu, D.; Fang, X.; de Vos, W.M.; Wu, H. Gut dysbacteriosis and intestinal disease: Mechanism and treatment. J. Appl. Microbiol. 2020, 129, 787–805. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic colonization of oral bacteria in the intestine drives T(H)1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef]
- Somineni, H.K.; Weitzner, J.H.; Venkateswaran, S.; Dodd, A.; Prince, J.; Karikaran, A.; Sauer, C.G.; Abramowicz, S.; Zwick, M.E.; Cutler, D.J.; et al. Site- and Taxa-Specific Disease-Associated Oral Microbial Structures Distinguish Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2021, 27, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Said, H.S.; Suda, W.; Nakagome, S.; Chinen, H.; Oshima, K.; Kim, S.; Kimura, R.; Iraha, A.; Ishida, H.; Fujita, J.; et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Elmaghrawy, K.; Fleming, P.; Fitzgerald, K.; Cooper, S.; Dominik, A.; Hussey, S.; Moran, G.P. The Oral Microbiome in Treatment-Naïve Paediatric IBD Patients Exhibits Dysbiosis Related to Disease Severity that Resolves Following Therapy. J. Crohns Colitis 2023, 17, 553–564. [Google Scholar] [CrossRef]
- Willis, J.R.; Saus, E.; Iraola-Guzmán, S.; Ksiezopolska, E.; Cozzuto, L.; Bejarano, L.A.; Andreu-Somavilla, N.; Alloza-Trabado, M.; Blanco, A.; Puig-Sola, A.; et al. Citizen-science reveals changes in the oral microbiome in Spain through age and lifestyle factors. NPJ Biofilms Microbiomes 2022, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.M.; Lammert, F.; Zimmer, V.; Granzow, M.; Reichert, S.; Schulz, S.; Ocklenburg, C.; Conrads, G. Clinical periodontal and microbiologic parameters in patients with Crohn’s disease with consideration of the CARD15 genotype. J. Periodontol. 2010, 81, 535–545. [Google Scholar] [CrossRef]
- Lee, Y.C.; Liu, C.Y.; Lee, C.L.; Zhang, R.H.; Huang, C.J.; Yen, T.L. The Periodontopathic Pathogen, Porphyromonas gingivalis, Involves a Gut Inflammatory Response and Exacerbates Inflammatory Bowel Disease. Pathogens 2022, 11, 84. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, W.; Dai, K.; Liu, N.; Wang, J.; Lu, X.; Ma, J.; Zhang, M.; Xu, M.; Long, X.; et al. Inflammatory response of gut, spleen, and liver in mice induced by orally administered Porphyromonas gingivalis. J. Oral. Microbiol. 2022, 14, 2088936. [Google Scholar] [CrossRef]
- Humphrey, S.P.; Williamson, R.T. A review of saliva: Normal composition, flow, and function. J. Prosthet. Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef]
- Howden, C.W.; Hunt, R.H. Relationship between gastric secretion and infection. Gut 1987, 28, 96–107. [Google Scholar] [CrossRef]
- Martinsen, T.C.; Bergh, K.; Waldum, H.L. Gastric juice: A barrier against infectious diseases. Basic. Clin. Pharmacol. Toxicol. 2005, 96, 94–102. [Google Scholar] [CrossRef]
- Giannella, R.A.; Broitman, S.A.; Zamcheck, N. Gastric acid barrier to ingested microorganisms in man: Studies in vivo and in vitro. Gut 1972, 13, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.Y.; Pratap, S.; Southerland, J.H.; Farmer-Dixon, C.M.; Lakshmyya, K.; Gangula, P.R. Role of oral and gut microbiome in nitric oxide-mediated colon motility. Nitric Oxide 2018, 73, 81–88. [Google Scholar] [CrossRef]
- He, J.; Huang, W.; Pan, Z.; Cui, H.; Qi, G.; Zhou, X.; Chen, H. Quantitative analysis of microbiota in saliva, supragingival, and subgingival plaque of Chinese adults with chronic periodontitis. Clin. Oral. Investig. 2012, 16, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Boutaga, K.; Savelkoul, P.H.; Winkel, E.G.; van Winkelhoff, A.J. Comparison of subgingival bacterial sampling with oral lavage for detection and quantification of periodontal pathogens by real-time polymerase chain reaction. J. Periodontol. 2007, 78, 79–86. [Google Scholar] [CrossRef]
- Saygun, I.; Nizam, N.; Keskiner, I.; Bal, V.; Kubar, A.; Açıkel, C.; Serdar, M.; Slots, J. Salivary infectious agents and periodontal disease status. J. Periodontal Res. 2011, 46, 235–239. [Google Scholar] [CrossRef]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G., 3rd; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e414. [Google Scholar] [CrossRef]
- Li, B.; Ge, Y.; Cheng, L.; Zeng, B.; Yu, J.; Peng, X.; Zhao, J.; Li, W.; Ren, B.; Li, M.; et al. Oral bacteria colonize and compete with gut microbiota in gnotobiotic mice. Int. J. Oral. Sci. 2019, 11, 10. [Google Scholar] [CrossRef]
- Priyamvara, A.; Dey, A.K.; Bandyopadhyay, D.; Katikineni, V.; Zaghlol, R.; Basyal, B.; Barssoum, K.; Amarin, R.; Bhatt, D.L.; Lavie, C.J. Periodontal Inflammation and the Risk of Cardiovascular Disease. Curr. Atheroscler. Rep. 2020, 22, 28. [Google Scholar] [CrossRef]
- Ambrosio, N.; Marín, M.J.; Laguna, E.; Herrera, D.; Sanz, M.; Figuero, E. Detection and quantification of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans in bacteremia induced by interdental brushing in periodontally healthy and periodontitis patients. Arch. Oral. Biol. 2019, 98, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.; Gonder, U.; Robinson, S.J.; Crean, S.; Singhrao, S.K. Exploring the Association between Alzheimer’s Disease, Oral Health, Microbial Endocrinology and Nutrition. Front. Aging Neurosci. 2017, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Hassanpour, S.; Borenstein, A.; Sima, C.; Oveisi, M.; Scholey, J.; Cherney, D.; Glogauer, M. Distinct Oral Neutrophil Subsets Define Health and Periodontal Disease States. J. Dent. Res. 2016, 95, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Darveau, R.P. Does Oral Endotoxin Contribute to Systemic Inflammation? Front. Oral Health 2022, 3, 911420. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hajishengallis, G. Lipid raft-dependent uptake, signalling and intracellular fate of Porphyromonas gingivalis in mouse macrophages. Cell Microbiol. 2008, 10, 2029–2042. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Shakhatreh, M.A.; James, D.; Liang, S.; Nishiyama, S.; Yoshimura, F.; Demuth, D.R.; Hajishengallis, G. Fimbrial proteins of porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J. Immunol. 2007, 179, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Zeituni, A.E.; McCaig, W.; Scisci, E.; Thanassi, D.G.; Cutler, C.W. The native 67-kilodalton minor fimbria of Porphyromonas gingivalis is a novel glycoprotein with DC-SIGN-targeting motifs. J. Bacteriol. 2010, 192, 4103–4110. [Google Scholar] [CrossRef]
- Zeituni, A.E.; Jotwani, R.; Carrion, J.; Cutler, C.W. Targeting of DC-SIGN on human dendritic cells by minor fimbriated Porphyromonas gingivalis strains elicits a distinct effector T cell response. J. Immunol. 2009, 183, 5694–5704. [Google Scholar] [CrossRef]
- El-Awady, A.R.; Miles, B.; Scisci, E.; Kurago, Z.B.; Palani, C.D.; Arce, R.M.; Waller, J.L.; Genco, C.A.; Slocum, C.; Manning, M.; et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PLoS Pathog. 2015, 10, e1004647. [Google Scholar] [CrossRef]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339.e324. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, Z.; Xu, C.; Kan, S.; Chen, D. Disturbances of the Gut Microbiota and Microbiota-Derived Metabolites in Inflammatory Bowel Disease. Nutrients 2022, 14, 5140. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Arimatsu, K.; Kato, T.; Matsuda, Y.; Minagawa, T.; Takahashi, N.; Ohno, H.; Yamazaki, K. Oral Administration of P. gingivalis Induces Dysbiosis of Gut Microbiota and Impaired Barrier Function Leading to Dissemination of Enterobacteria to the Liver. PLoS ONE 2015, 10, e0134234. [Google Scholar] [CrossRef] [PubMed]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Kanda, N.; Hoashi, T.; Saeki, H. The Defect in Regulatory T Cells in Psoriasis and Therapeutic Approaches. J. Clin. Med. 2021, 10, 3880. [Google Scholar] [CrossRef]
- Gao, Y.; Davis, B.; Zhu, W.; Zheng, N.; Meng, D.; Walker, W.A. Short-chain fatty acid butyrate, a breast milk metabolite, enhances immature intestinal barrier function genes in response to inflammation in vitro and in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G521–G530. [Google Scholar] [CrossRef]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Jia, W.; Rajani, C.; Xu, H.; Zheng, X. Gut microbiota alterations are distinct for primary colorectal cancer and hepatocellular carcinoma. Protein Cell 2021, 12, 374–393. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.T.; Schmidt, A.W.; Venkataraman, A.; Kim, K.S.; Waldron, C.; Schmidt, T.M. Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio 2019, 10, 02566-18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Z.; He, Y.; Li, P.; Zhou, H.; Zeng, N. Regional distribution of Christensenellaceae and its associations with metabolic syndrome based on a population-level analysis. PeerJ 2020, 8, e9591. [Google Scholar] [CrossRef]
- Jalanka, J.; Cheng, J.; Hiippala, K.; Ritari, J.; Salojärvi, J.; Ruuska, T.; Kalliomäki, M.; Satokari, R. Colonic Mucosal Microbiota and Association of Bacterial Taxa with the Expression of Host Antimicrobial Peptides in Pediatric Ulcerative Colitis. Int. J. Mol. Sci. 2020, 21, 6044. [Google Scholar] [CrossRef]
- Herath, T.D.; Wang, Y.; Seneviratne, C.J.; Darveau, R.P.; Wang, C.Y.; Jin, L. The expression and regulation of matrix metalloproteinase-3 is critically modulated by Porphyromonas gingivalis lipopolysaccharide with heterogeneous lipid A structures in human gingival fibroblasts. BMC Microbiol. 2013, 13, 73. [Google Scholar] [CrossRef]
- Herath, T.D.; Darveau, R.P.; Seneviratne, C.J.; Wang, C.Y.; Wang, Y.; Jin, L. Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS ONE 2013, 8, e58496. [Google Scholar] [CrossRef]
- Hirschfeld, M.; Ma, Y.; Weis, J.H.; Vogel, S.N.; Weis, J.J. Cutting edge. repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J. Immunol. 2000, 165, 618–622. [Google Scholar] [CrossRef]
- Hirschfeld, M.; Weis, J.J.; Toshchakov, V.; Salkowski, C.A.; Cody, M.J.; Ward, D.C.; Qureshi, N.; Michalek, S.M.; Vogel, S.N. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 2001, 69, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P.; Pham, T.T.; Lemley, K.; Reife, R.A.; Bainbridge, B.W.; Coats, S.R.; Howald, W.N.; Way, S.S.; Hajjar, A.M. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect. Immun. 2004, 72, 5041–5051. [Google Scholar] [CrossRef]
- Triantafilou, M.; Gamper, F.G.; Lepper, P.M.; Mouratis, M.A.; Schumann, C.; Harokopakis, E.; Schifferle, R.E.; Hajishengallis, G.; Triantafilou, K. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol. 2007, 9, 2030–2039. [Google Scholar] [CrossRef]
- Jotwani, R.; Pulendran, B.; Agrawal, S.; Cutler, C.W. Human dendritic cells respond to Porphyromonas gingivalis LPS by promoting a Th2 effector response in vitro. Eur. J. Immunol. 2003, 33, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Kumar, P.; Cutler, C.W.; Mohamadzadeh, M.; Van Dyke, T.; Banchereau, J. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J. Immunol. 2001, 167, 5067–5076. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Oh, S.J.; Ahn, J.S.; Shin, Y.Y.; Yang, J.W.; Kim, H.S. Implication of Porphyromonas gingivalis in colitis and homeostasis of intestinal epithelium. Lab. Anim. Res. 2019, 35, 26. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, N.; Belibasakis, G.N. Porphyromonas gingivalis: An invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 2012, 333, 1–9. [Google Scholar] [CrossRef] [PubMed]
- de Diego, I.; Veillard, F.; Sztukowska, M.N.; Guevara, T.; Potempa, B.; Pomowski, A.; Huntington, J.A.; Potempa, J.; Gomis-Rüth, F.X. Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 2014, 289, 32291–32302. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nguyen, K.A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol. 2000 2010, 54, 15–44. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Travis, J.; Potempa, J. The biphasic virulence activities of gingipains: Activation and inactivation of host proteins. Curr. Protein Pept. Sci. 2003, 4, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Potempa, J.; Banbula, A.; Travis, J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol. 2000 2000, 24, 153–192. [Google Scholar] [CrossRef]
- Potempa, M.; Potempa, J.; Okroj, M.; Popadiak, K.; Eick, S.; Nguyen, K.A.; Riesbeck, K.; Blom, A.M. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J. Immunol. 2008, 181, 5537–5544. [Google Scholar] [CrossRef]
- Abdi, K.; Chen, T.; Klein, B.A.; Tai, A.K.; Coursen, J.; Liu, X.; Skinner, J.; Periasamy, S.; Choi, Y.; Kessler, B.M.; et al. Mechanisms by which Porphyromonas gingivalis evades innate immunity. PLoS ONE 2017, 12, e0182164. [Google Scholar] [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- van der Post, S.; Subramani, D.B.; Bäckström, M.; Johansson, M.E.V.; Vester-Christensen, M.B.; Mandel, U.; Bennett, E.P.; Clausen, H.; Dahlén, G.; Sroka, A.; et al. Site-specific O-glycosylation on the MUC2 mucin protein inhibits cleavage by the Porphyromonas gingivalis secreted cysteine protease (RgpB). J. Biol. Chem. 2013, 288, 14636–14646. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.S.; Kissoon-Singh, V.; Gibson, D.L.; Ma, C.; Montero, M.; Sham, H.P.; Ryz, N.; Huang, T.; Velcich, A.; Finlay, B.B.; et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010, 6, e1000902. [Google Scholar] [CrossRef]
- Wenzel, U.A.; Magnusson, M.K.; Rydström, A.; Jonstrand, C.; Hengst, J.; Johansson, M.E.; Velcich, A.; Öhman, L.; Strid, H.; Sjövall, H.; et al. Spontaneous colitis in Muc2-deficient mice reflects clinical and cellular features of active ulcerative colitis. PLoS ONE 2014, 9, e100217. [Google Scholar] [CrossRef]
- Sheng, Y.H.; Hasnain, S.Z.; Florin, T.H.; McGuckin, M.A. Mucins in inflammatory bowel diseases and colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Kimura, R.K.; Nakamura, T.; Kawabata, S.; Hamada, S. Distribution and molecular characterization of Porphyromonas gingivalis carrying a new type of fimA gene. J. Clin. Microbiol. 2000, 38, 1909–1914. [Google Scholar] [CrossRef]
- Nagano, K.; Hasegawa, Y.; Abiko, Y.; Yoshida, Y.; Murakami, Y.; Yoshimura, F. Porphyromonas gingivalis FimA fimbriae: Fimbrial assembly by fimA alone in the fim gene cluster and differential antigenicity among fimA genotypes. PLoS ONE 2012, 7, e43722. [Google Scholar] [CrossRef] [PubMed]
- Marafini, I.; Monteleone, I.; Di Fusco, D.; Cupi, M.L.; Paoluzi, O.A.; Colantoni, A.; Ortenzi, A.; Izzo, R.; Vita, S.; De Luca, E.; et al. TNF-α Producing Innate Lymphoid Cells (ILCs) Are Increased in Active Celiac Disease and Contribute to Promote Intestinal Atrophy in Mice. PLoS ONE 2015, 10, e0126291. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; Braegger, C.P.; Walker-Smith, J.A.; MacDonald, T.T. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut 1993, 34, 1705–1709. [Google Scholar] [CrossRef]
- Paredes, J.M.; Moreno, N.; Latorre, P.; Ripollés, T.; Martinez, M.J.; Vizuete, J.; Moreno-Osset, E. Clinical Impact of Sonographic Transmural Healing After Anti-TNF Antibody Treatment in Patients with Crohn’s Disease. Dig. Dis. Sci. 2019, 64, 2600–2606. [Google Scholar] [CrossRef]
- Braegger, C.P.; Nicholls, S.; Murch, S.H.; Stephens, S.; MacDonald, T.T. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet 1992, 339, 89–91. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Reibetanz, J.; Germer, C.T. Anti-TNF antibodies in prevention and treatment of postoperative recurrence of Crohn’s disease. Chirurg 2015, 86, 498. [Google Scholar] [CrossRef]
- Langer, V.; Vivi, E.; Regensburger, D.; Winkler, T.H.; Waldner, M.J.; Rath, T.; Schmid, B.; Skottke, L.; Lee, S.; Jeon, N.L.; et al. IFN-γ drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J. Clin. Investig. 2019, 129, 4691–4707. [Google Scholar] [CrossRef]
- Beaurepaire, C.; Smyth, D.; McKay, D.M. Interferon-gamma regulation of intestinal epithelial permeability. J. Interferon Cytokine Res. 2009, 29, 133–144. [Google Scholar] [CrossRef]
- Zhao, L.; Qiu, D.K.; Ma, X. Th17 cells: The emerging reciprocal partner of regulatory T cells in the liver. J. Dig. Dis. 2010, 11, 126–133. [Google Scholar] [CrossRef]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Jiang, P.; Zheng, C.; Xiang, Y.; Malik, S.; Su, D.; Xu, G.; Zhang, M. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor. Rev. 2022, 69, 28–42. [Google Scholar] [CrossRef]
- Daley, T.; Smith, A.D. Predicting the molecular complexity of sequencing libraries. Nat. Methods 2013, 10, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Amoussina, L.; Schenk, M.; Yu, J.; Brüstle, A.; Weigmann, B.; Atreya, R.; Wirtz, S.; Becker, C.; Hoffman, A.; et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J. Clin. Investig. 2008, 118, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Waetzig, G.H.; Butzin, S.; Ziolkiewicz, M.; Al-Massad, N.; Thieme, F.; Lövgren, U.; Rasmussen, B.B.; Reinheimer, T.M.; Seegert, D.; et al. Evaluation of interleukin-6 and its soluble receptor components sIL-6R and sgp130 as markers of inflammation in inflammatory bowel diseases. Int. J. Colorectal Dis. 2018, 33, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996, discussion 947. [Google Scholar] [CrossRef]
- Danese, S.; Vermeire, S.; Hellstern, P.; Panaccione, R.; Rogler, G.; Fraser, G.; Kohn, A.; Desreumaux, P.; Leong, R.W.; Comer, G.M.; et al. Randomised trial and open-label extension study of an anti-interleukin-6 antibody in Crohn’s disease (ANDANTE I and II). Gut 2019, 68, 40–48. [Google Scholar] [CrossRef]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef]
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. [Google Scholar] [CrossRef] [PubMed]
- Bsat, M.; Chapuy, L.; Rubio, M.; Wassef, R.; Richard, C.; Schwenter, F.; Loungnarath, R.; Soucy, G.; Mehta, H.; Sarfati, M. Differential Pathogenic Th17 Profile in Mesenteric Lymph Nodes of Crohn’s Disease and Ulcerative Colitis Patients. Front. Immunol. 2019, 10, 1177. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Filì, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef]
- Calderón-Gómez, E.; Bassolas-Molina, H.; Mora-Buch, R.; Dotti, I.; Planell, N.; Esteller, M.; Gallego, M.; Martí, M.; Garcia-Martín, C.; Martínez-Torró, C.; et al. Commensal-Specific CD4(+) Cells from Patients with Crohn’s Disease Have a T-Helper 17 Inflammatory Profile. Gastroenterology 2016, 151, 489–500.e483. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, A.N.; West, N.R.; Stubbington, M.J.T.; Wendt, E.; Suijker, K.I.M.; Datsi, A.; This, S.; Danne, C.; Campion, S.; Duncan, S.H.; et al. Circulating and Tissue-Resident CD4(+) T Cells with Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered during Inflammation. Gastroenterology 2017, 153, 1320–1337.e1316. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.B.; Luo, M.M.; Chen, Z.Y.; He, B.H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 8813558. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Jia, L.; Wu, R.; Han, N.; Fu, J.; Luo, Z.; Guo, L.; Su, Y.; Du, J.; Liu, Y. Porphyromonas gingivalis and Lactobacillus rhamnosus GG regulate the Th17/Treg balance in colitis via TLR4 and TLR2. Clin. Transl. Immunol. 2020, 9, e1213. [Google Scholar] [CrossRef]
- Guevara, T.; Rodríguez-Banqueri, A.; Lasica, A.M.; Ksiazek, M.; Potempa, B.A.; Potempa, J.; Gomis-Rüth, F.X. Structural determinants of inhibition of Porphyromonas gingivalis gingipain K by KYT-36, a potent, selective, and bioavailable peptidase inhibitor. Sci. Rep. 2019, 9, 4935. [Google Scholar] [CrossRef]
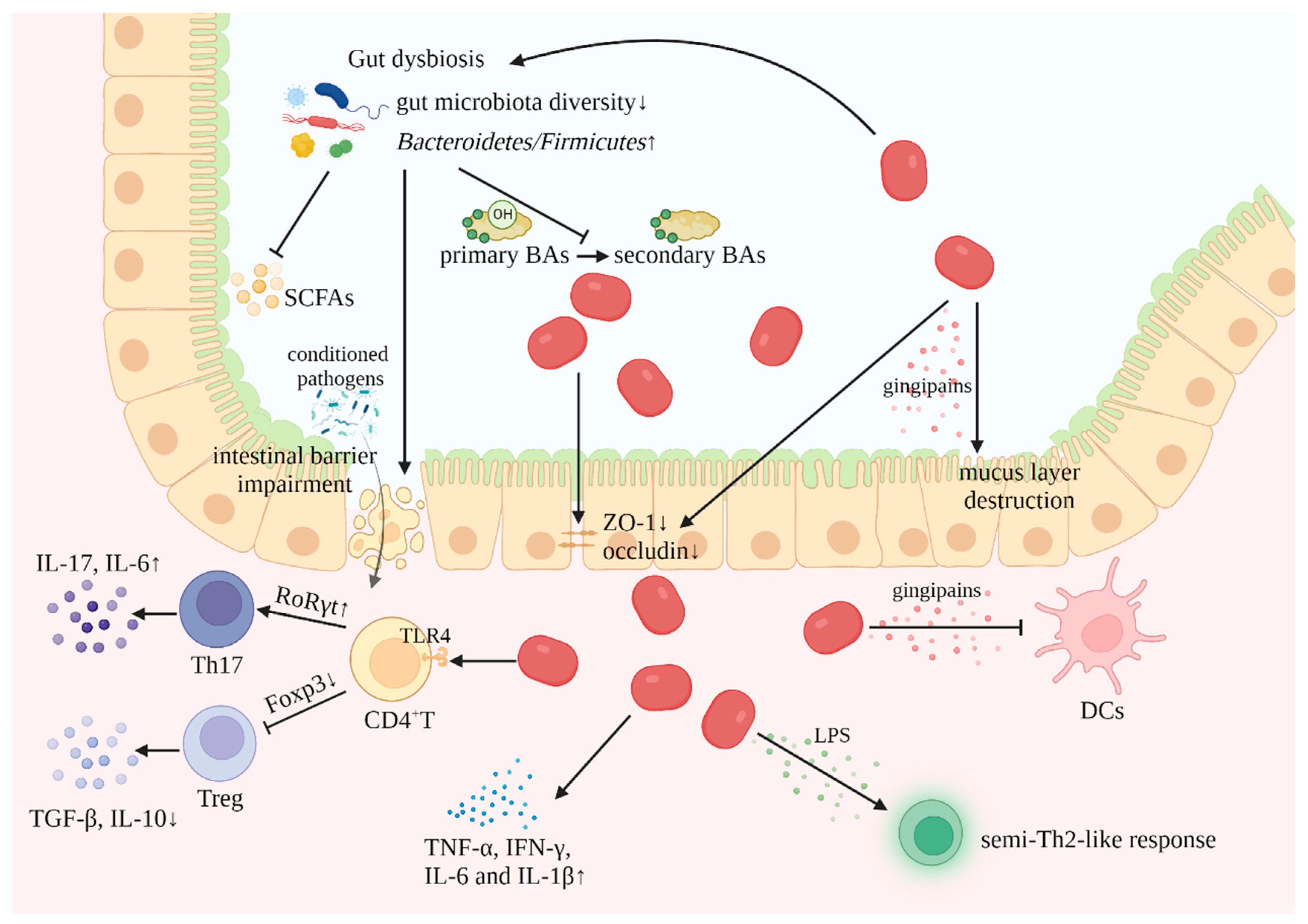
Figure 1.
Mechanisms of P. gingivalis involvement in IBD (created with BioRender.com). (1) Gut dysbiosis: The reduced diversity of gut microbiota, accompanied by the increased ratio of Bacteroidetes to Firmicutes, leads to decreased production of SCFAs and inhibits the conversion of BAs. (2) Impairment of intestinal barrier: P. gingivalis decreases the expression level of the tight junction proteins ZO-1 and occludin, disrupting the epithelial barrier; meanwhile, gut dysbiosis contributes to the impairment of the intestinal barrier, increasing the invasion of conditioned pathogens. (3) Release of inflammatory mediators: P. gingivalis-derived LPS induces a semi-Th2-like response; in addition, P. gingivalis secretes gingipains that evade the intrinsic immune response by inhibiting DCs and causing the destruction of the intestinal barrier. (4) Disturbance of immune response: P. gingivalis induces the expression of proinflammatory cytokines; in addition, it upregulates the Th17/Treg ratio by regulating the transcription factors RoRγt and Foxp3. (SCFAs, short-chain fatty acids; BAs, bile acids; LPS, lipopolysaccharides; DCs, dendritic cells).
Figure 1.
Mechanisms of P. gingivalis involvement in IBD (created with BioRender.com). (1) Gut dysbiosis: The reduced diversity of gut microbiota, accompanied by the increased ratio of Bacteroidetes to Firmicutes, leads to decreased production of SCFAs and inhibits the conversion of BAs. (2) Impairment of intestinal barrier: P. gingivalis decreases the expression level of the tight junction proteins ZO-1 and occludin, disrupting the epithelial barrier; meanwhile, gut dysbiosis contributes to the impairment of the intestinal barrier, increasing the invasion of conditioned pathogens. (3) Release of inflammatory mediators: P. gingivalis-derived LPS induces a semi-Th2-like response; in addition, P. gingivalis secretes gingipains that evade the intrinsic immune response by inhibiting DCs and causing the destruction of the intestinal barrier. (4) Disturbance of immune response: P. gingivalis induces the expression of proinflammatory cytokines; in addition, it upregulates the Th17/Treg ratio by regulating the transcription factors RoRγt and Foxp3. (SCFAs, short-chain fatty acids; BAs, bile acids; LPS, lipopolysaccharides; DCs, dendritic cells).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, X.; Li, Y.; Zhang, J.; Feng, Q. Linking Periodontitis with Inflammatory Bowel Disease through the Oral–Gut Axis: The Potential Role of Porphyromonas gingivalis. Biomedicines 2024, 12, 685. https://doi.org/10.3390/biomedicines12030685
AMA Style
Huang X, Li Y, Zhang J, Feng Q. Linking Periodontitis with Inflammatory Bowel Disease through the Oral–Gut Axis: The Potential Role of Porphyromonas gingivalis. Biomedicines. 2024; 12(3):685. https://doi.org/10.3390/biomedicines12030685
Chicago/Turabian StyleHuang, Xinyi, Yilin Li, Jun Zhang, and Qiang Feng. 2024. "Linking Periodontitis with Inflammatory Bowel Disease through the Oral–Gut Axis: The Potential Role of Porphyromonas gingivalis" Biomedicines 12, no. 3: 685. https://doi.org/10.3390/biomedicines12030685
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.