

Telomere Dysfunction in Pediatric Patients with Differences/Disorders of Sexual Development

 , ,
, , 

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Declaration of Ethics
2.2. Pediatric Patients
2.3. Culture of Lymphocytes, Preparation of Metaphases, and Analysis of Karyotypes after G-Banding (GTG-Banding)
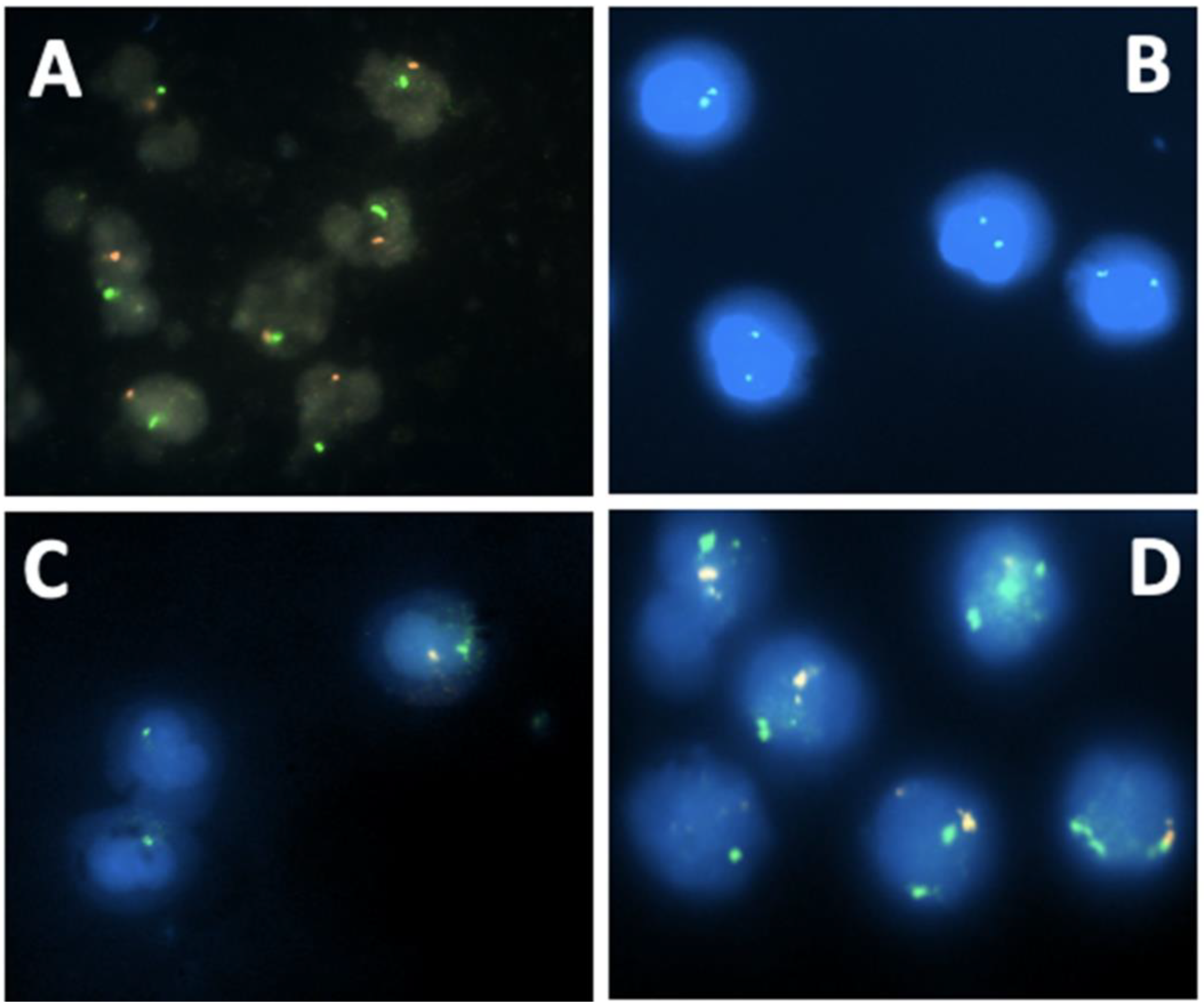
2.4. Detection of SRY by Fluorescence In Situ Hybridization (FISH)
2.5. Staining of Telomere and Centromere Sequences
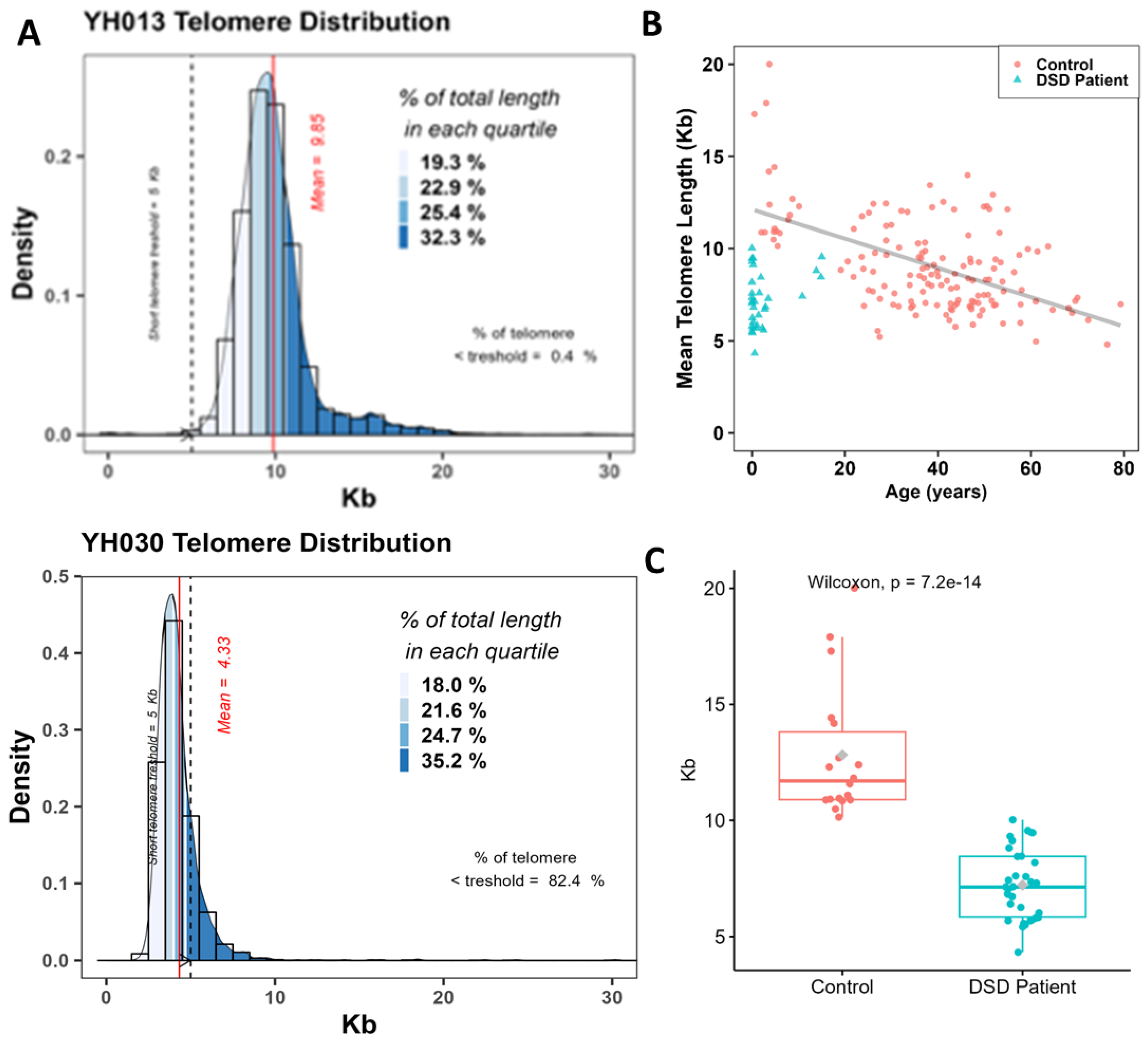
2.6. Telomeres Length Analysis
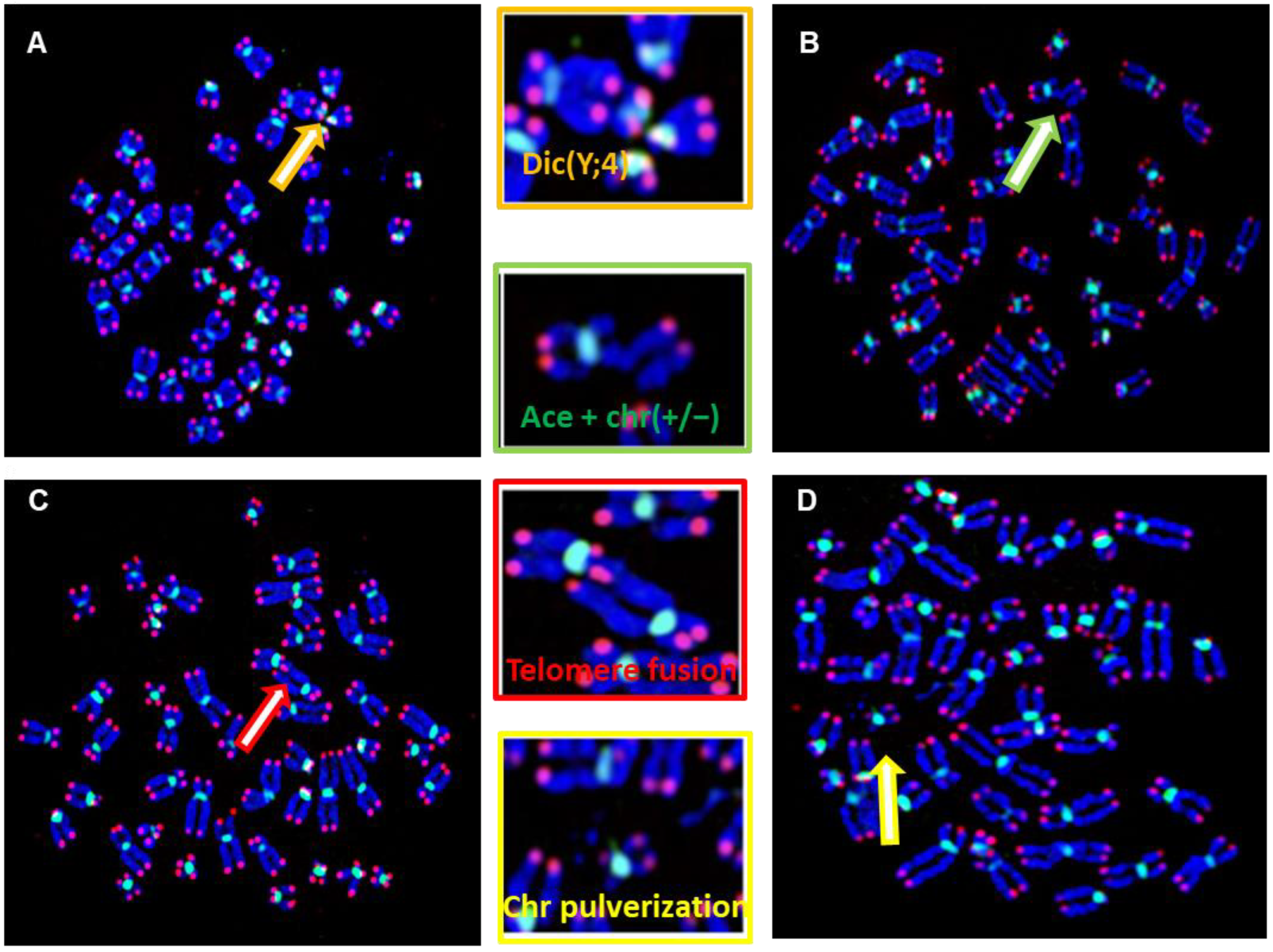
2.7. Scoring of Telomere Aberrations
2.8. Multicolor FISH (M-FISH Technique)
2.9. Statistical Analysis
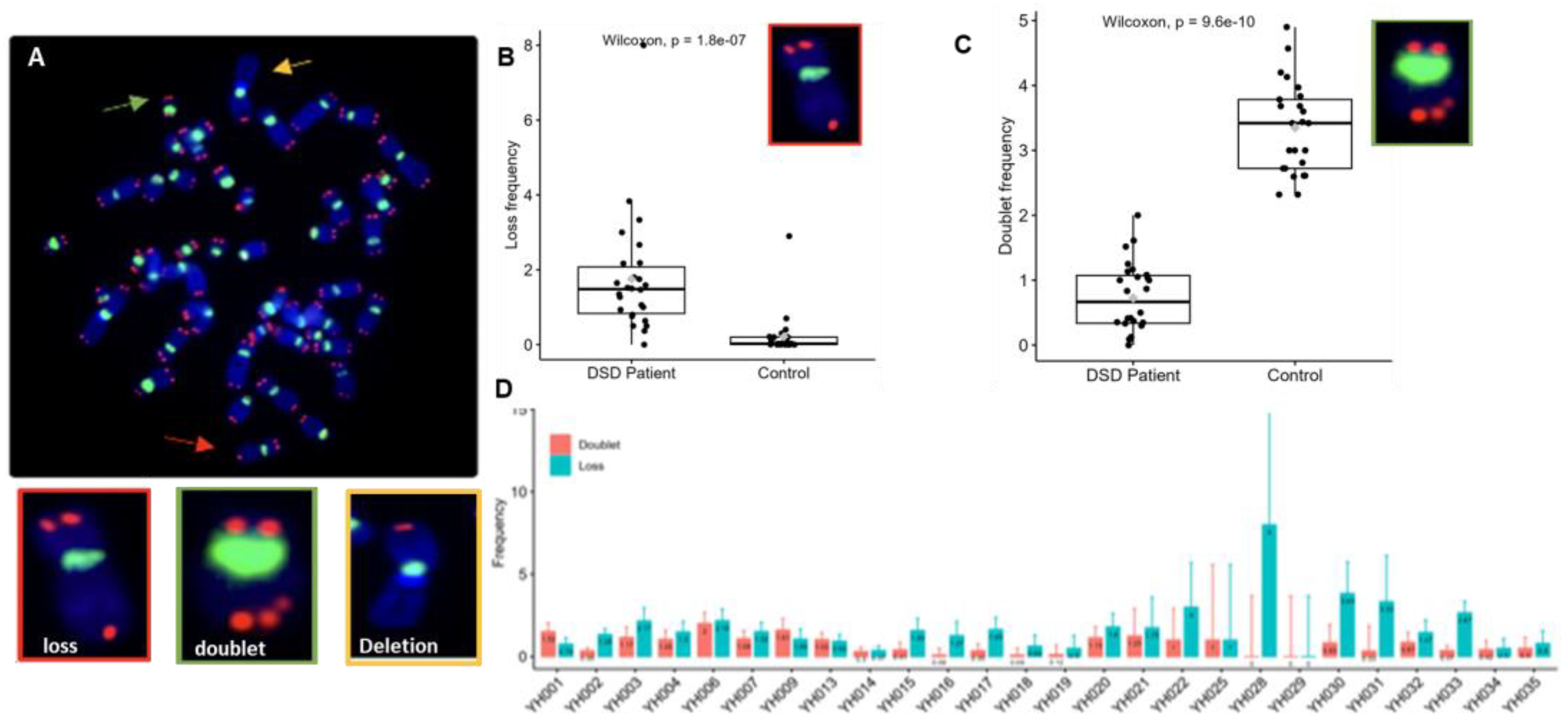
3. Results
3.1. Clinical Profile of DSD Patients
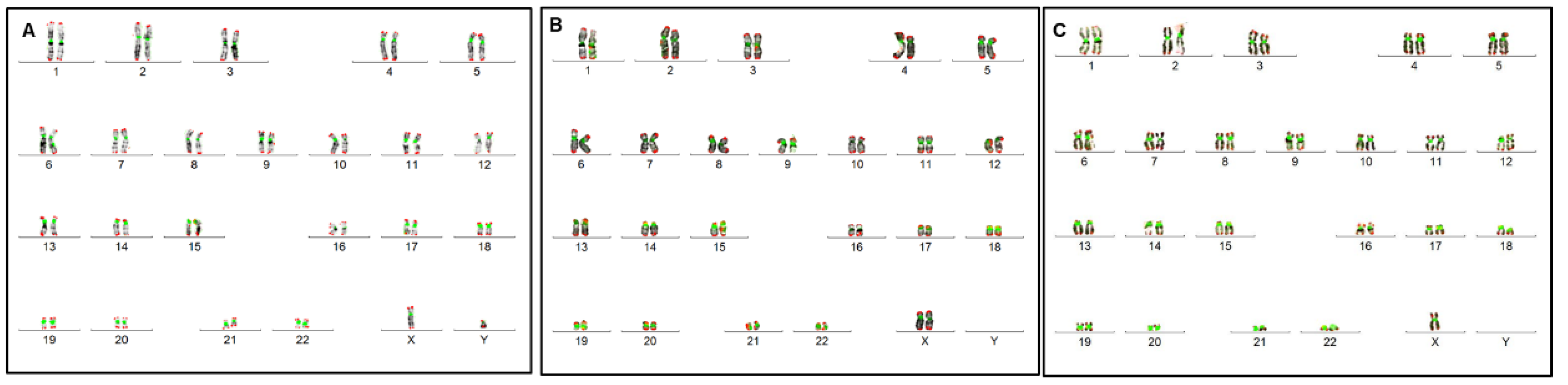
3.2. Conventional and Molecular Cytogenetic Investigations
3.3. Quantification of Telomere Length of DSD Patients
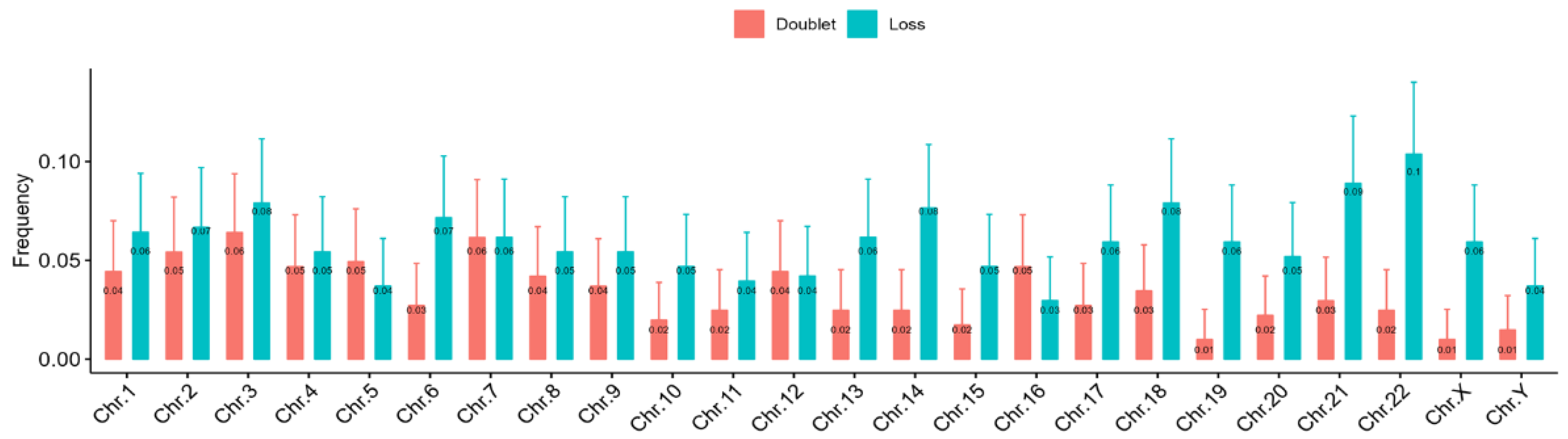
3.4. Telomere Dysfunction of DSDs Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, P.A.; Houk, C.P.; Ahmed, S.F.; Hughes, I.A. International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 2006, 118, e488–e500. [Google Scholar] [CrossRef]
- Bashamboo, A.; McElreavey, K. Consanguinity and disorders of sex development. Hum. Hered. 2014, 77, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, H.; Kylin, H.; Sereda, B.; Bornman, R. High levels of DDT in breast milk: Intake, risk, lactation duration, and involvement of gender. Environ. Pollut. 2012, 170, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Houk, C.P.; Lee, P.A. Consensus Statement on Terminology and Management: Disorders of Sex Development. Sex Dev. 2008, 2, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A. Disorders of sex development: A new definition and classification. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, I.A.; Aaronson, A.J. How should we classify intersex disorders? J. Pediatr. Urol. 2010, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Amolo, P.; Laigong, P.; Omar, A.; Drop, S. Etiology and Clinical Presentation of Disorders of Sex Development in Kenyan Children and Adolescents. Int. J. Endocrinol. 2019, 2019, 2985347. [Google Scholar] [CrossRef]
- Guerrero-Fernández, J.; Azcona San Julián, C.; Barreiro Conde, J.; Bermúdez De La Vega, J.A.; Carcavilla Urquí, A.; Castaño González, L.A.; Martos Tello, J.M.; Rodríguez Estévez, A.; Yeste Fernández, D.; Martínez Martínez, L.; et al. Guía de actuación en las anomalías de la diferenciación sexual (ADS)/desarrollo sexual diferente (DSD). An. Pediatría 2018, 89, 315.e1–315.e19. [Google Scholar] [CrossRef]
- Sax, L. How common is lntersex? A response to Anne Fausto-Sterling. J. Sex Res. 2002, 39, 174–178. [Google Scholar] [CrossRef]
- Thyen, U.; Lanz, K.; Holterhus, P.-M.; Hiort, O. Epidemiology and initial management of ambiguous genitalia at birth in Germany. Horm. Res. 2006, 66, 195–203. [Google Scholar] [CrossRef]
- Ganie, Y.; Aldous, C.; Balakrishna, Y.; Wiersma, R. Disorders of sex development in children in KwaZulu-Natal Durban South Africa: 20-year experience in a tertiary centre. J. Pediatr. Endocrinol. Metab. JPEM 2017, 30, 11–18. [Google Scholar] [CrossRef]
- Ameyaw, E.; Asafo-Agyei, S.B.; Hughes, I.A.; Zacharin, M.; Chanoine, J.-P. Incidence of disorders of sexual development in neonates in Ghana: Prospective study. Arch. Dis. Child. 2019, 104, 636–638. [Google Scholar] [CrossRef]
- Alkhzouz, C.; Bucerzan, S.; Miclaus, M.; Mirea, A.-M.; Miclea, D. 46,XX DSD: Developmental, Clinical and Genetic Aspects. Diagnostics 2021, 11, 1379. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Wallace, M.A.; Hofman, L.; Thuline, H.C.; Dorche, C.; Lyon, I.C.; Dobbins, R.H.; Kling, S.; Fujieda, K.; Suwa, S. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 1988, 81, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kim, J. Disorders of sex development. Korean J. Urol. 2012, 53, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, A.B.; Batista, R.L.; Costa, E.M.F.; Finlayson, C.; Sircili, M.H.P.; Dénes, F.T.; Domenice, S.; Mendonca, B.B. Management of 46,XY Differences/Disorders of Sex Development (DSD) Throughout Life. Endocr. Rev. 2019, 40, 1547–1572. [Google Scholar] [CrossRef] [PubMed]
- Mazen, I.; Hiort, O.; Bassiouny, R.; El Gammal, M. Differential Diagnosis of Disorders of Sex Development in Egypt. Horm. Res. 2008, 70, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Pyle, L.C.; Nathanson, K.L. A Practical Guide for Evaluating Gonadal Germ Cell Tumor Predisposition in Differences of Sex Development. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Ayers, K.L.; Bouty, A.; Robevska, G.; van den Bergen, J.A.; Juniarto, A.Z.; Listyasari, N.A.; Sinclair, A.H.; Faradz, S.M.H. Variants in congenital hypogonadotrophic hypogonadism genes identified in an Indonesian cohort of 46,XY under-virilised boys. Hum. Genomics 2017, 11, 1. [Google Scholar] [CrossRef]
- Viswanathan, V.; Eugster, E.A. Etiology and Treatment of Hypogonadism in Adolescents. Pediatr. Clin. N. Am. 2011, 58, 1181–1200. [Google Scholar] [CrossRef]
- Ganmore, I.; Smooha, G.; Izraeli, S. Constitutional aneuploidy and cancer predisposition. Hum. Mol. Genet. 2009, 18, R84–R93. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zöller, B.; Sundquist, J.; Sundquist, K. Risk of solid tumors and hematological malignancy in persons with Turner and Klinefelter syndromes: A national cohort study. Int. J. Cancer 2016, 139, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.L.; Chetty, T.; Jorgensen, A.; Mitchell, R.T. Disorders of Sex Development—Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation. Int. J. Mol. Sci. 2020, 21, 2282. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, E. Congenital Aneuploidy in Klinefelter Syndrome with B-Cell Acute Lymphoblastic Leukemia Might Be Associated with Chromosomal Instability and Reduced Telomere Length. Cancers 2022, 14, 2316. [Google Scholar] [CrossRef]
- Reish, O.; Brosh, N.; Gobazov, R.; Rosenblat, M.; Libman, V.; Mashevich, M. Sporadic aneuploidy in PHA-stimulated lymphocytes of Turner’s syndrome patients. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2006, 14, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Reish, O.; Regev, M.; Kanesky, A.; Girafi, S.; Mashevich, M. Sporadic Aneuploidy in PHA-Stimulated Lymphocytes of Trisomies 21, 18, and 13. Cytogenet. Genome Res. 2011, 133, 184–189. [Google Scholar] [CrossRef]
- Zhong, Q.; Layman, L.C. Genetic considerations in the patient with Turner syndrome--45,X with or without mosaicism. Fertil. Steril. 2012, 98, 775–779. [Google Scholar] [CrossRef]
- Callén, E.; Surrallés, J. Telomere dysfunction in genome instability syndromes. Mutat. Res. Mutat. Res. 2004, 567, 85–104. [Google Scholar] [CrossRef]
- Gadji, M.; Mathur, S.; Bélanger, B.; Jangamreddy, J.R.; Lamoureux, J.; Tsanaclis, A.M.C.; Fortin, D.; Drouin, R.; Mai, S. Three-Dimensional Nuclear Telomere Profiling as a Biomarker for Recurrence in Oligodendrogliomas: A Pilot Study. Int. J. Mol. Sci. 2020, 21, 8539. [Google Scholar] [CrossRef]
- Ferlin, A.; Rampazzo, E.; Rocca, M.S.; Keppel, S.; Frigo, A.C.; De Rossi, A.; Foresta, C. In young men sperm telomere length is related to sperm number and parental age. Hum. Reprod. 2013, 28, 3370–3376. [Google Scholar] [CrossRef]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- M’kacher, R.; Bennaceur-Griscelli, A.; Girinsky, T.; Koscielny, S.; Delhommeau, F.; Dossou, J.; Violot, D.; Leclercq, E.; Courtier, M.H.; Béron-Gaillard, N.; et al. Telomere Shortening and Associated Chromosomal Instability in Peripheral Blood Lymphocytes of Patients With Hodgkin’s Lymphoma Prior to Any Treatment Are Predictive of Second Cancers. Int. J. Radiat. Oncol. 2007, 68, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Girinsky, T.; M’Kacher, R.; Lessard, N.; Koscielny, S.; Elfassy, E.; Raoux, F.; Carde, P.; Santos, M.D.; Margainaud, J.-P.; Sabatier, L.; et al. Prospective Coronary Heart Disease Screening in Asymptomatic Hodgkin Lymphoma Patients Using Coronary Computed Tomography Angiography: Results and Risk Factor Analysis. Int. J. Radiat. Oncol. 2014, 89, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; Yao, X.; Rosiene, J.; Tian, H.; Behr, J.; Bosco, N.; Takai, K.K.; de Lange, T.; Imieliński, M. Structural variant evolution after telomere crisis. Nat. Commun. 2021, 12, 2093. [Google Scholar] [CrossRef] [PubMed]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- M’kacher, R.; Colicchio, B.; Borie, C.; Junker, S.; Marquet, V.; Heidingsfelder, L.; Soehnlen, K.; Najar, W.; Hempel, W.M.; Oudrhiri, N.; et al. Telomere and Centromere Staining Followed by M-FISH Improves Diagnosis of Chromosomal Instability and Its Clinical Utility. Genes 2020, 11, 475. [Google Scholar] [CrossRef]
- M’kacher, R.; Colicchio, B.; Marquet, V.; Borie, C.; Najar, W.; Hempel, W.M.; Heidingsfelder, L.; Oudrhiri, N.; Jawhari, M.A.; Wilhelm-Murer, N.; et al. Telomere aberrations, including telomere loss, doublets, and extreme shortening, are increased in patients with infertility. Fertil. Steril. 2021, 115, 164–173. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomere dysfunction and chromosome instability. Mutat. Res. Mol. Mech. Mutagen. 2012, 730, 28–36. [Google Scholar] [CrossRef]
- Nassour, J.; Radford, R.; Correia, A.; Fusté, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef]
- M’Kacher, R.; Colicchio, B.; Junker, S.; El Maalouf, E.; Heidingsfelder, L.; Plesch, A.; Dieterlen, A.; Jeandidier, E.; Carde, P.; Voisin, P. High Resolution and Automatable Cytogenetic Biodosimetry Using In Situ Telomere and Centromere Hybridization for the Accurate Detection of DNA Damage: An Overview. Int. J. Mol. Sci. 2023, 24, 5699. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. Iscn 2020: An International System for Human Cytogenomic Nomenclature 2020. Cytogenet. Genome Res. 2020, 160, 7–8. [Google Scholar]
- Riana Bornman, M.S.; Bouwman, H. Environmental pollutants and diseases of sexual development in humans and wildlife in South Africa: Harbingers of impact on overall health? Reprod. Domest. Anim. Zuchthyg. 2012, 47 (Suppl. S4), 327–332. [Google Scholar] [CrossRef]
- Barriga, H.H.A.; Velásquez, F.C.; Carrillo, C.B.; Tacuri, A.P.; Hurtado, M.B.; Loarte, T.V.; Rondón, L.O.; Linares, M.O.; Abuhadba, E.A.R. Variantes en el número de copias y consanguinidad parental en neonatos de altura con anomalías congénitas en Perú. Rev. Fac. Cienc. Médicas 2022, 79, 132. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, L.; Sampaio, D.R.; Paris, F.; Audran, F.; Orsini, M.; Neto, J.B.; Sultan, C. High prevalence of micropenis in 2710 male newborns from an intensive-use pesticide area of Northeastern Brazil. Int. J. Androl. 2012, 35, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Syndrome de Turner Protocole National de Diagnostic et de Soins. Available online: https://www.has-sante.fr/upload/docs/application/pdf/2021-11/pnds_turner_29_10.pdf (accessed on 3 January 2023).
- Ennazk, L.; Mghari, G.E.; Ansari, N.E. Le DSD mosaïque (45,X0, 46,XY), un sous-type peu connu du syndrome de Turner. Ann. Endocrinol. 2016, 77, 469. [Google Scholar] [CrossRef]
- Bojesen, A.; Juul, S.; Gravholt, C.H. Prenatal and postnatal prevalence of Klinefelter syndrome: A national registry study. J. Clin. Endocrinol. Metab. 2003, 88, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Rochira, V.; Pasquali, D.; Balercia, G.; Jannini, E.A.; Ferlin, A.; Balercia, G.; Bonomi, M.; Calogero, A.; Corona, G.; et al. Klinefelter syndrome (KS): Genetics, clinical phenotype and hypogonadism. J. Endocrinol. Investig. 2017, 40, 123–134. [Google Scholar] [CrossRef]
- Achermann, J.C.; Domenice, S.; Bachega, T.A.S.S.; Nishi, M.Y.; Mendonca, B.B. Disorders of sex development: Effect of molecular diagnostics. Nat. Rev. Endocrinol. 2015, 11, 478–488. [Google Scholar] [CrossRef]
- DesGroseilliers, M.; Beaulieu Bergeron, M.; Brochu, P.; Lemyre, E.; Lemieux, N. Phenotypic variability in isodicentric Y patients: Study of nine cases. Clin. Genet. 2006, 70, 145–150. [Google Scholar] [CrossRef]
- Abdelmoula, N.B.; Amouri, A. [Dicentric Y chromosome]. Ann. Biol. Clin. 2005, 63, 363–375. [Google Scholar]
- Roubin, M.; de Grouchy, J.; Chauveau, P.; Rappaport, R.; Pellerin, D. [Dicentric Y chromosome in a male pseudohermaphrodite 45,X/46,X, dic (Y)/47, XYY]. Ann. Genet. 1977, 20, 185–189. [Google Scholar] [PubMed]
- Alexander, D.S.; Soudek, D.; Laraya, P. Unstable dicentric iso(Yq) chromosome in a pseudohermaphrodite. Am. J. Med. Genet. 1978, 1, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Reshmi, S.C.; Miller, J.L.; Deplewski, D.; Close, C.; Henderson, L.J.; Littlejohn, E.; Schwartz, S.; Waggoner, D.J. Evidence of a mechanism for isodicentric chromosome Y formation in a 45,X/46,X,idic(Y)(p11.31)/46,X,del(Y)(p11.31) mosaic karyotype. Eur. J. Med. Genet. 2011, 54, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Ponzio, G.; DeMarchi, M.; Carbonara, A.; Godano, A.; Massara, F. Dicentric Y chromosome in a patient with gonadal dysgenesis and seminoma. Hum. Genet. 1981, 58, 282–284. [Google Scholar] [CrossRef]
- Ernst, A.; Jones, D.T.W.; Maass, K.K.; Rode, A.; Deeg, K.I.; Jebaraj, B.M.C.; Korshunov, A.; Hovestadt, V.; Tainsky, M.A.; Pajtler, K.W.; et al. Telomere dysfunction and chromothripsis. Int. J. Cancer 2016, 138, 2905–2914. [Google Scholar] [CrossRef]
- Vasilopoulos, E.; Fragkiadaki, P.; Kalliora, C.; Fragou, D.; Docea, A.O.; Vakonaki, E.; Tsoukalas, D.; Calina, D.; Buga, A.M.; Georgiadis, G.; et al. The association of female and male infertility with telomere length (Review). Int. J. Mol. Med. 2019, 44, 375–389. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | No. of Patients |
|---|---|
| DSDs | 35 |
| Assigned sex | |
| Male | 17 |
| Female | 13 |
| ND | 5 |
| Age (years) | |
| At diagnosis | 2.6 |
| At analysis | 3.5 |
| Type | |
| Clitoral hypertrophy | 8 |
| Micropenis + cryptorchidism | 5 |
| Isolated hypospadias | 4 |
| Isolated cryptorchidism | 2 |
| Isolated micropenis | 1 |
| Hypogonadism + gynecomastia | 1 |
| Short stature + Turner syndrome | 1 |
| Other DSDs with diverse congenital malformations | 12 |
| Cytogenetic Profile | No. of Patients |
|---|---|
| Nb of analyzed DSDs patients | 35 |
| Conventional cytogenetics | 35 |
| Molecular cytogenetic (SRY) | 35 |
| Karyotype results | |
| 46,XY | 18 |
| 46,XX | 14 |
| 46,XX[12]/45,X[5] | 1 |
| 45,X[28]/46,XY[20] | 1 |
| nuc ish(DXZ1x2,SRYx1)[85/200] | 1 |
| Structural chromosome aberrations | 8 |
| ID | Assigned SEXE | Age Month | Reason for Consultation | FISH (SRY) | Karyotypes | Telomere Length (kb) | Telomere Loss/Cell | Telomere Doublet/Cell | Additional Chromosome Aberrations |
|---|---|---|---|---|---|---|---|---|---|
| YH001 | M | 0 | Micropenis and cryptorchidism | XY (SRY+) | 46,XY | 9.31 | 0.78 | 1.52 | |
| YH002 | M | 178 | Micropenis and cryptorchidism) | XY (SRY+) | 46,XY | 8.5 | 1.35 | 0.35 | |
| YH003 | F | 3 | Prader Type I | XX (SRY−) | 46,XX | 9.15 | 2.17 | 1.17 | |
| YH004 | M | 0 | ND | XY (SRY+) | 46,XY | 7.66 | 1.5 | 1.05 | DNA fragmentation Dicentric with interstitial telomeres |
| YH005 | M | 4 | Clitoral hypertrophy | XX (SRY−) | 46,XX | 6.32 | NA | NA | |
| YH006 | M | 33 | ND | XY (SRY+) | 46,XY | 7.57 | 2.19 | 2 | |
| YH007 | M | 19 | Hypospadias | XY (SRY+) | 46,XY | 8.09 | 1.52 | 1.08 | |
| YH008 | M | 120 | Micropenis and cryptorchidism | 28% XY/57% X (SRY+) | 45,X[28]/46,XY[20] | 6.09 | NA | NA | |
| YH009 | M | 7 | Hypospadias | ish(DXZ1x2,SRYx1)[85/200] (SRY+) | 46,XY[115]/47,XXY[85] | 8.21 | 1.06 | 1.71 | |
| YH010 | F | 42 | Clitoral hypertrophy | XX (SRY−) | 46,XX | 7.29 | 0 | 0 | |
| YH011 | M | 1 | ND | XY (SRY+) | 46,XY | 5.42 | 0 | 0 | |
| YH012 | M | 166 | Hypogonadism and gynecomastia | XY (SRY+) | 46,XY | 8.12 | 0 | 0 | |
| YH013 | F | 3 | Clitoral hypertrophy | XY (SRY+) | 46,XY | 9.85 | 0.94 | 1.03 | |
| YH014 | F | 0 | Clitoral hypertrophy | XX (SRY−) | 46,XX | 9.47 | 0.37 | 0.3 | Chromosomal breakage 46,XX,del(15)(q10q26) |
| YH015 | F | 180 | Statural retardation and Turner syndrome | 71% XX/29% X (SRY−) | 46,XX[12]/45,X[5] | 9.55 | 1.59 | 0.42 | |
| YH016 | ND | ND | XX (SRY−) | 46,XX | 5.56 | 1.28 | 0.09 | chromatid fusion of ch 8 and 10 | |
| YH017 | M | 13 | ND | XY (SRY+) | 46,XY | 5.83 | 1.65 | 0.35 | chromatid fusion of ch 15 and 4 chromatid fusion of ch 15 and 9 |
| YH018 | ND | 0 | ND | XY (SRY+) | 46,XY | 6.71 | 0.6363636364 | 0.09090909091 | |
| YH019 | ND | 0 | ND | XX (SRY−) | 46,XX | 8.18 | 0.5 | 0.125 | |
| YH020 | M | 130 | Hypospadias | XY (SRY+) | 46,XY | 7.42 | 1.8 | 1.13 | Chromosome 2 breakage |
| YH021 | ND | 0 | ND | XY (SRY+) | 46,XY | 5.5 | 1.75 | 1.25 | |
| YH022 | M | 27 | Micropenis and cryptorchidism | XY (SRY+) | 46,XY | 5.7 | 3 | 1 | |
| YH023 | F | 23 | Clitoral hypertrophy | XX (SRY−) | 46,XX | 6.34 | NA | NA | |
| YH024 | M | 11 | Micropenis and cryptorchidism | XY (SRY+) | 46,XY | 5.67 | NA | NA | |
| YH025 | F | 17 | external Genitalia Anomaly | XX (SRY−) | 46,XX | 6.39 | 1 | 1 | |
| YH026 | F | 0 | Clitoral hypertrophy | XX (SRY−) | 46,XX | 7.02 | NA | NA | |
| YH027 | F | 0 | XX (SRY−) | 46,XX | 5.8 | NA | NA | ||
| YH028 | ND | 0 | Polymalformation | XY (SRY+) | 46,XY | 6.42 | 8 | 0 | |
| YH029 | M | 1 | ND | XY (SRY+) | 46,XY | 6.25 | NA | NA | |
| YH030 | M | 7 | Ovotestis | XX (SRY−) | 46,XX | 4.33 | 3.83 | 0.8 | Acentric chromosome |
| YH031 | F | 30 | Prader Type II | XY (SRY+) | 46,XY | 5.57 | 3.333 | 0.33 | |
| YH032 | M | 1 | hypospadias | XY (SRY+) | 46,XY | 7.14 | 1.47 | 0.87 | Chromosome breakage |
| YH033 | F | 35 | ND | XX (SRY−) | 46,XX | 6.84 | 2.67 | 0.374 | DNA Fragmentation |
| YH034 | F | 3 | ND | XX (SRY−) | 46,XX | 6.01 | 0.5 | 0.42 | |
| YH035 | F | 2 | Micro penis | XY (SRY+) | 46,XY | 5.68 | 0.8 | 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Younoussa, H.; Gadji, M.; Soumboundou, M.; Colicchio, B.; Said, A.; Ndoye, N.A.; Junker, S.; Plesch, A.; Heidingsfelder, L.; Diagne, N.R.; et al. Telomere Dysfunction in Pediatric Patients with Differences/Disorders of Sexual Development. Biomedicines 2024, 12, 565. https://doi.org/10.3390/biomedicines12030565
Younoussa H, Gadji M, Soumboundou M, Colicchio B, Said A, Ndoye NA, Junker S, Plesch A, Heidingsfelder L, Diagne NR, et al. Telomere Dysfunction in Pediatric Patients with Differences/Disorders of Sexual Development. Biomedicines. 2024; 12(3):565. https://doi.org/10.3390/biomedicines12030565
Chicago/Turabian StyleYounoussa, Haifaou, Macoura Gadji, Mamadou Soumboundou, Bruno Colicchio, Ahmed Said, Ndeye Aby Ndoye, Steffen Junker, Andreas Plesch, Leonhard Heidingsfelder, Ndeye Rama Diagne, and et al. 2024. "Telomere Dysfunction in Pediatric Patients with Differences/Disorders of Sexual Development" Biomedicines 12, no. 3: 565. https://doi.org/10.3390/biomedicines12030565