1. Introduction
Glioblastoma (GB) or grade IV glioma (WHO classification) is an aggressive brain tumor of astrocytic lineage that is inevitably recurrent. It is one of the most common malignant primary brain tumors, with an incidence of 5 per 100,000 [
1]. The median survival time post-diagnosis is approximately 15 months, with a 5-year survival rate of only 5%. The current standard of care is maximally safe surgical resection, followed by radiation and TMZ chemotherapy; however, tumor recurrence occurs in almost all patients, usually 6–9 months after treatment [
2]. The disease outcome has not improved for several decades, and an urgent need for novel therapies exists.
Microtubule targeting agents (MTAs) have long been an important class of anticancer drugs. Microtubules (MTs), the backbone of the cytoskeleton, are involved in multiple cellular processes, including cell migration and mitosis. MTs are polymers consisting of α- and β-tubulin heterodimers and associated proteins. MTAs disrupt polymer dynamics, triggering cell cycle arrest that leads to cell death. Five distinct sites on the β-tubulin subunit have been targeted by MTAs, and an additional binding site has been identified on α-tubulin. MTAs can be categorized into those that inhibit polymerization (tubulin destabilizers, e.g., vinca alkaloids and colchicine) and those that inhibit depolymerization (tubulin stabilizers, e.g., taxanes and epothilones) [
3]. In the clinic, they are utilized as both a first- and second-line standard of care in a variety of solid and hematological malignancies that lack targeted therapies. They are typically administered in combination with other drugs to achieve a greater therapeutic window and are approved for use with radiation, DNA-damaging agents, and immune checkpoint inhibitors.
Despite the success of MTAs as anticancer drugs, their clinical use is limited by poor pharmacokinetics, toxicity, and the development of drug resistance. The high molecular weight and lack of oral bioavailability of MTAs require intravenous administration, resulting in high peak drug concentrations that likely contribute to their associated neuropathic and myeloid toxicities. Their failure to cross the BBB has excluded them from the treatment arsenal for high-grade gliomas (HGGs) and other central nervous system (CNS) cancers, including brain metastases. HGGs have been shown to be sensitive to MTAs in vitro [
4]. Furthermore, glioma tumorigenesis is mediated by neurite-like protrusions, a MT-mediated intercellular process [
5]. Thus, the development of BBB-penetrant MTAs could be a paradigm shift in the treatment of this disease.
The development of resistance to MTAs is mediated by several mechanisms. First, the aberrant expression of β-tubulin isotypes, particularly Class III β-tubulin, is associated with resistance and tumor aggressiveness, forming part of a complex pro-survival pathway that is poorly understood [
6]. A second important resistance mechanism is the overexpression of membrane-bound P-glycoprotein (P-gp) pumps, resulting in drug efflux [
7]. MTAs that bind to the colchicine binding site on β-tubulin are less susceptible to P-gp pump efflux, but to date, they have not been approved as anticancer therapeutics due to toxicities associated with their specific chemical class [
8].
Herein, we describe the identification of a series of small molecules that address many of the shortcomings of known MTAs. To improve the drug-like and physiochemical properties of our molecules, we designed and synthesized a series of analogs optimized for low molecular weight, low polar surface area, and a good CNS multiparametric optimization (MPO) score [
9]. The MPO score uses six calculated properties of the molecule—molecular weight, clog D, clog P, number of hydrogen bond donors (HBDs), pKa and topological polar surface area. These properties for each molecule are transformed into a score from 0 to 1. Scores are added to arrive at an MPO score ranging from 0 to 6 for each molecule. One of these novel molecules, RGN3067, has excellent in vitro early absorption, distribution, metabolism, and elimination (eADME) properties (including solubility, microsomal stability, and cellular permeability). RGN3067 has activity against human HGG cell lines (including the TMZ-resistant cell line LN-18), achieving nanomolar inhibition in a cell viability assay. Additionally, in mice, oral administration of RGN3067 inhibits tumor growth in an HGG PDX model. RGN3067 shows minimal in vivo toxicity and achieves equal levels in plasma and brain.
The results presented here provide the basis for ongoing and future in vivo studies to further evaluate the potential of orally available, BBB-penetrant MTAs for both HGGs and other CNS cancers that currently have limited therapeutic options.
2. Materials and Methods
2.1. Cell Culture
U87 glioblastoma (ATCC HTB-14; Manassas, VA, USA) cells were grown in DMEM/10% FCS, LN-18 glioblastoma cells (ATCC CRL-2610; Manassas, VA, USA) were grown in DMEM/5% FCS, HMC3 microglial cells (ATCC CRL-3304; Manassas, VA, USA) were grown in EMEM/10% FCS, and MCF-7 mammary carcinoma cells (ATCC HTB-22; Manassas, VA, USA) were grown in RPMI/10% FCS. Patient-derived, low-passage, genetically heterogeneous primary cells GBM12, GBM15, GBM39 and GBM43 (established in Mayo Clinic Hospital) were cultured in serum-free DMEM/F12 (Cytiva HyClone; Logan, UT, USA) with 20 ng/mL EGF, 20 ng/mL FGF, 2% B12, 1% N2, and 1% gentamycin solution. Cells were cultured at 37 °C in 5% CO2.
2.2. Cell Viability Assay
Cells (100 µL/well) were seeded in 96-well tissue culture plates (Greiner Bio-One 650090; Kremsmünster, Austria). The plating density (cells/well) for each cell line was as follows: LN-18 (3000), U87 (4000), and HMC3 (5000). After overnight incubation, compounds were added to the cells (50 µL at 3× concentration, final 0.1% DMSO), and plates were incubated for 72 h. Cell viability was assessed by the addition of 25 µL of alamarBlue for a final concentration of 100 µM final concentration (Resazurin redox reagent, Sigma-Aldrich R7017; St. Louis, MO, USA). After a 4 h incubation, fluorescence was read on a CLARIOstar Plus plate reader (BMG Labtech, Ortenberg, Germany) at EX 570/EM 590 nm.
Short-term cultures of patient-derived cells GBM12, GBM15, GBM39 and GBM43 (used with permission Mayo Foundation) were seeded (1000 cells/well) in solid white Corning 384-well plates (Corning, NY, USA) and cultured overnight. Cells were treated with DMSO or RGN3067 in a 13-point dose response with a 1:3 serial dilution from an initial concentration of 100 μM. After 144 h, cell viability was measured using CellTiter-Glo (Promega, Madison, WI, USA). Data were normalized to DMSO control, and absolute IC50 values were calculated using GraphPad Prism 10 (San Diego, CA, USA).
2.3. Cell Cycle Analysis
Cells (400,000) were seeded into T25 tissue culture flasks and treated with compound for 24 h prior to harvesting with TrypLE (Thermo Fisher Scientific 12605036; Waltham, MA, USA). Cells were washed in ice-cold PBS, resuspended in 1 mL of ice-cold 70% ethanol, and fixed overnight at −20 °C. Cells were centrifuged and resuspended in PBS containing RNAse A at a final concentration of 0.5 mg/mL (MilliporeSigma 10109142001; Burlington, MA, USA) and propidium iodide at a final concentration of 0.04 mg/mL (Sigma-Aldrich P4170, St. Louis, MO, USA) for 1 h at 37 °C. Nuclear staining was analyzed using a BD FACSCanto II flow cytometer (BD Biosciences; Franklin Lakes, NJ, USA) to determine cell cycle distribution. Cell cycle analysis was performed using the BD FACSDiva software v8.0.
2.4. Immunofluorescent Staining of Cellular β-Tubulin
U87 cells (4000/well) were plated in black 96-well plates (Perkin Elmer 6055302; Waltham, MA, USA) and grown overnight. Cells were treated with compounds for 24 h, fixed with 4% paraformaldehyde for 20 min, and permeabilized with FoxP3 permeabilization buffer (eBioscience/Thermo Fisher Scientific 00-5523-00; Waltham, MA, USA) for 10 min at RT. Cells were incubated with an anti-TUBB3 (Tuj1) antibody (1:1000; STEMCELL Technologies 60052; Vancouver, BC, Canada) labeled with Hoechst 33342 dye (1:2000; Thermo Fisher Scientific H3570; Waltham, MA, USA). Images (40×) were taken with an automated high-content imaging microscope (Operetta, Perkin Elmer; Waltham, MA, USA).
2.5. Tubulin Polymerization Assay
Tubulin polymerization was evaluated by monitoring the incorporation of a fluorescent reporter into MTs during polymerization. The tubulin polymerization assay kit (Cytoskeleton, Inc. BK011P; Denver, CO, USA) was used according to the manufacturer’s instructions. Briefly, the tubulin reaction mix was kept on ice until immediately prior to use. The compound (5 µL/well) was added to a half-area 96-well plate (Corning Costar 3686; Corning, NY, USA) and incubated for 1 min in the CLARIOstar Plus plate reader (BMG Labtech; Ortenberg, Germany) at 37 °C before the addition of 50 µL of the tubulin reaction mix/well. Fluorescence was read every min for 160 min at EX 350/EM 435 nm.
2.6. Colchicine Competitive Binding Assay
Porcine tubulin (20 µM, Cytoskeleton, Inc., T240; Denver, CO, USA) was incubated with compounds for 45 min at 37 °C in 80 mM PIPES pH 6.9, 2 mM MgCl2, 0.5 mM EGTA, 0.2 mM GTP prior to the addition of colchicine for a 10 µM final concentration (Selleckchem S2284; Houston, TX, USA). After a 45 min incubation, the fluorescence of the colchicine–tubulin complex was measured at EX 380/EM 435 nm with the CLARIOstar Plus plate reader (BMG Labtech; Ortenberg, Germany).
2.7. Colchicine-Binding Site Cellular Assay
N,N′-ethylene-bis (iodoacetamide) (EBI) cross-links cysteines 239 and 354 in the colchicine binding pocket of β-tubulin, the formation of which can be inhibited by colchicine binding site molecules. A total of 1.5 × 106 MCF-7 cells were seeded overnight in a T25 flask and pre-incubated with compounds for 2 h, then treated with EBI (Abcam ab144980; Cambridge, MA, USA) for 1.5 h at 37 °C. Cells were washed with PBS and lysed for 30 min at 4 °C in RIPA buffer supplemented with a protease inhibitor cocktail (Sigma-Aldrich P8340; St. Louis, MO, USA). Lysates were collected by centrifugation (12,000× g, 4 °C, 30 min) and stored at −80 °C until detection of the EBI: β-tubulin adduct by Western blot. Briefly, 20 µg of each sample was loaded on a 10% Mini-Protean TGX SDS gel (Bio-Rad 4561034; Hercules, CA, USA). Proteins were transferred to nitrocellulose membranes (iBlot dry blotting system, Invitrogen; Waltham, MA, USA) and blocked for 1 h post-transfer (Intercept blocking buffer, Li-COR Biosciences 927-80001; Lincoln, NE, USA). Primary antibodies were incubated overnight at 4 °C (mouse anti-β-tubulin, Sigma-Aldrich T4026, St. Louis, MO, USA; rabbit anti-GAPDH, Cell Signaling Technology 14C10, Danvers, MA, USA) in blocking buffer/0.2% Tween 20. Membranes were washed twice (PBS/0.1% Tween 20) and incubated for 1 h at RT with secondary antibodies (IRDye 680 anti-mouse, Li-COR Biosciences 926-68072; IRDye 800 anti-rabbit, Li-COR Bioscience 926-32211; Lincoln, NE, USA) in blocking buffer/0.1% Tween 20. After three washes, membranes were dried for 2 h prior to imaging on the Odyssey CLx Imager (Li-COR Biosciences; Lincoln, NE, USA). Band intensities were quantified by densitometric analysis using Fiji/ImageJ software (NIH) v2.3.0.
2.8. Reversibility Assay
U87 cells were treated with increasing concentrations (up to 10 μM) of RGN3067 and the control compounds colchicine (Selleckchem S2284; Houston, TX, USA) and sabizabulin (MedChem Express HY-120599; Monmouth Junction, NJ, USA) and cultured for 6 h. Compounds were either washed out or left in the medium. Cells were then cultured for an additional 72 h and cell viability was examined using CellTiter-Glo (Promega, Madison, WI, USA).
2.9. Compound Kinetic Solubility
Kinetic solubility was determined in phosphate buffer. The compound (4 µL, 10 mM DMSO) was added to the buffer (396 μL 100 mM phosphate buffer, final 1% DMSO) and shaken at 1000 rpm at RT for 1 h. Samples were filtered (Millipore MSSLBPC10; Burlington, MA, USA), the primary filtrate was discarded, and the subsequent filtrate was collected. The supernatant solution was diluted 10× with DMSO. Test samples (10 µL) and working standard curve samples were added to the stop solution (100 μL, containing internal standard) and water/0.1% FA (100 μL). Samples were analyzed by LC-MS/MS. Albendazole, propranolol, and 4,5-diphenylimidazole were used as positive controls.
2.10. Compound Microsomal Stability
Compounds (5 µL, 20 mM DMSO) were added to 195 µL of methanol to obtain a 500 µM spiking solution. Spiking solution (1.5 μL) and liver microsomes (18.75 μL, 20 mg/mL) were added to Buffer C (479.75 μL) on ice. Plates were pre-incubated at 37 °C for 5 min. At 0 min, spiking/microsome solution (30 μL) was added to the precipitation solution (400 μL) and NADPH stock solution (6 mM in Buffer C, 0.1 M PBS, pH 7.4) on ice. At additional time points, spiking/microsome solution (180 μL) was added to the assay plates, and the reaction initiated with the NADPH stock solution (90 μL, 6 mM). Plates were sealed and incubated at 37 °C. At 5, 15, 30 and 60 min time points, aliquots of the microsomal incubation system (45 μL) were removed and added to precipitation solution (400 μL). Supernatants were evaluated by LC-MS/MS. Ketanserin was used as a positive control. All reactions were performed in duplicate.
2.11. Plasma Protein Binding Studies
Compounds (5 µM/0.2% DMSO) were tested in dialysis chambers. Blank dialysis buffer was added to the receiver side (100 μL), and plasma (100 μL) with test and reference compounds was added to the donor side of the chamber and incubated at 37 °C on an orbital shaker. Plasma was precipitated at 0 and 5 h. Aliquots from donor and receiver sides of the dialysis apparatus (25 μL) were added to sample preparation plates and mixed with aliquots of the opposite matrix (25 μL, blank buffer to plasma and vice versa). Samples were quenched with stop solution (400 μL) containing internal standard, vortexed at 600 rpm for 10 min, and centrifuged at 4000 rpm for 10 min. Supernatants were analyzed by LC-MS. Quinidine and warfarin were used as positive controls.
2.12. Cellular Permeability of RGN3067
Cellular permeability was tested using the MDR1-MDCK (multidrug-resistant-1, Madin–Darby canine kidney) cell line. Donor solution was added to a 96-well plate (2 μL, 2 mM compound; 6 μL DMSO in 1992 μL of Hanks’ balanced salt solution [HBSS] buffer) and shaken at 1000 rpm for 10 min (final 0.4% DMSO). Transepithelial electrical resistance (TEER) values were used as a quality control check for monolayer integrity. At 3–5 days post-seeding, each MDCK II cell monolayer was tested to ensure the TEER value was ≥ 83 Ω⋅cm2. The compound donor working solution was centrifuged (4000 rpm for 5 min) to remove any precipitated compound prior to loading donor chambers. Apical and basolateral plates were warmed at 37 °C for about 5 min prior to the initiation of transport (37 °C, 90 min).
Luciferin marker Lucifer Yellow (150 μL, 50 μM Lucifer Yellow in HBSS, pH 7.4) was added to the apical compartment, and transfer buffer (600 μL) was added to the receiver plate (basolateral compartment) and incubated at 37 °C for 60 min. Apical Lucifer Yellow concentrations for LYT0 and LYT60 were measured by a fluorometer (EX 485/EM 535 nm). Donor and receiver samples were analyzed by LC-MS. Metoprolol, enalapril, and quinidine were used as positive controls. The efflux ratio was calculated as the ratio of apparent permeability (basal-to-apical) and apparent permeability (apical-to-basal).
2.13. Pharmacokinetics of RGN3067
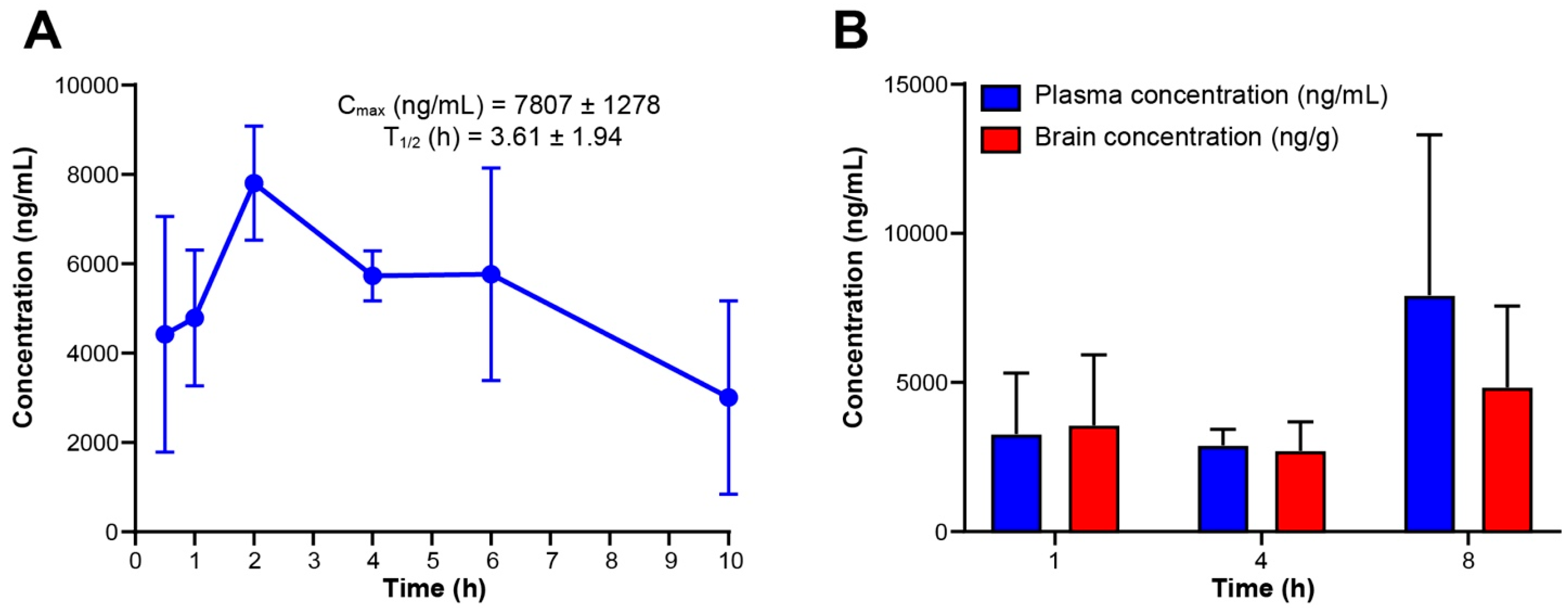
Pharmacokinetic studies were carried out by DMPK at WuXi AppTec, Inc. (Cranbury, NJ, USA). The pharmacokinetics of RGN3067 were evaluated in three male CD-1 mice. RGN3067 formulated as a 10 mg/mL solution (20% DMSO, 80% PEG400: PVP 95:5) was administered by oral gavage at a dose of 100 mg/kg. Plasma samples were drawn at 0.5, 1, 2, 4, 6, 10 and 24 h. Samples were analyzed by LC-MS/MS. A calibration curve was established using Waters ACQUITY UPLC BEH C18 column and formic acid in water and formic acid in acetonitrile as mobile phase. Tolbutamide and labetalol were used as internal standards for LC-MS/MS analysis. Compound was detected using electrospray ionization (ESI) positive detection. Pharmacokinetic parameters were calculated using the PO-noncompartmental linear model in Phoenix WinNonlin software v8.3 (Certara, Princeton, NJ, USA).
2.14. Brain Pharmacokinetics of RGN3067
Brain pharmacokinetic studies were carried out by DMPK at WuXi AppTec, Inc. (Cranbury, NJ, USA). The brain penetrance of RGN3067 was evaluated in three male CD-1 mice. RGN3067 was formulated as a 10 mg/mL solution (20% DMSO, 80% PEG400: PVP 95:5) and administered by oral gavage at a dose of 100 mg/kg. Plasma and brain samples were collected at 1, 4, and 8 h. Samples were analyzed by LC-MS/MS. A calibration curve was established using Waters ACQUITY UPLC BEH C18 column and formic acid in water and formic acid in acetonitrile as mobile phase. Tolbutamide and labetalol were used as internal standards for LC-MS/MS analysis. Compound was detected using electrospray ionization (ESI) positive detection. Pharmacokinetic parameters were calculated using the PO-noncompartmental linear model in Phoenix WinNonlin software v8.3 (Certara, Princeton, NJ, USA).
2.15. Maximum Tolerated Dose Study with RGN3067
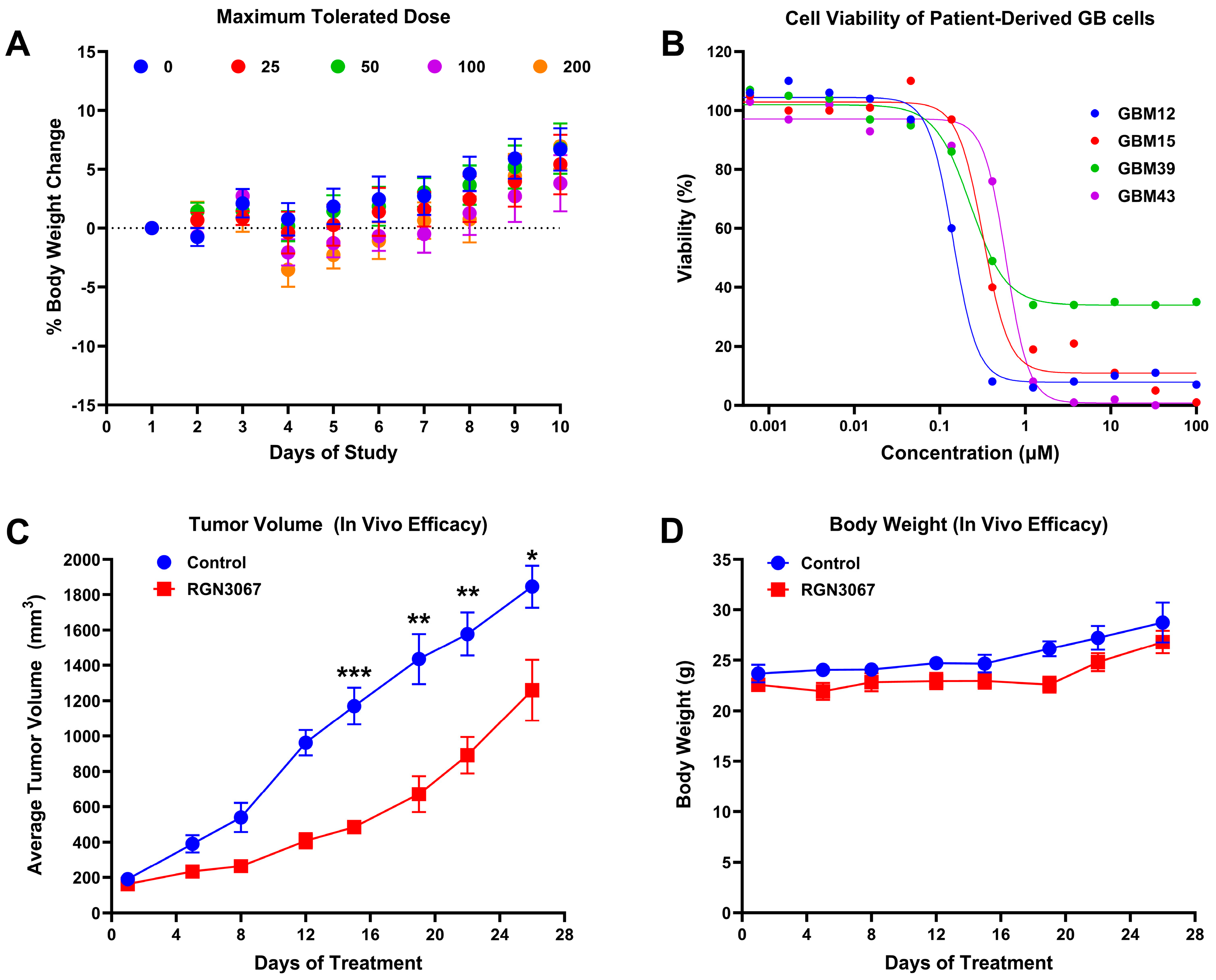
RGN3067 was administered to athymic female mice (4–6 weeks old) twice daily for 5 days by oral gavage at doses of 25, 50, 100 or 200 mg/kg (BID; 6 mice per group, including vehicle control). One mouse from each group was euthanized for PK analysis at 6 h post-initial dose. The weight and physical states of the animals were monitored during the 5 days of treatment and for 5 days following drug administration. The maximum tolerated dose was defined as the dose at which none of the mice within the cohort lost more than 20% body weight during the 10-day study and was used for subsequent efficacy testing.
2.16. The Efficacy of RGN3067 in a PDX Model of GB
Animal protocols were approved by the Animal Care and Use Committee (ACUC) of the institutes and CRO labs. All studies were performed in compliance with the guidelines. GBM12 cells (5 × 10
6) were injected subcutaneously into athymic nude female mice aged 4–6 weeks. Subcutaneous (SC) tumor size was measured using an external caliper. The greatest longitudinal diameter (length) and the greatest transverse diameter (width) were determined while mice were scruffed and conscious. Tumor volume was calculated by the modified ellipsoidal formula: V = ½ (Length × Width
2) [
10]. RGN3067 solution was prepared for each mouse. RGN3067 (9.91 mg) was dissolved in DMSO (17 µL) by vortexing for 30–60 s. Approximately 69 μL of PEG400: PVP (95:5, Sigma-Aldrich; St. Louis, MO, USA) solution was added and vortexed.
After tumors reached 100–150 mm3, mice were dosed with RGN3067 solution (43 µL, 100 mg/kg) or vehicle control twice daily by oral gavage (total dose was 200 mg/kg). Mice were dosed using cycles of 5 days on/2 days off, for a total of four cycles. Mice were euthanized once the tumor reached 2000 mm3.
2.17. Metabolite Profiling of RGN3067 in Hepatocytes
Metabolite profiling was carried out by WuXi DMPK (Nanjing, China). RGN3067 (10 μM) was profiled in CD-1 mice (BioIVT, Westbury, NY, USA), SD rats (BioIVT), beagle dogs (BioIVT), cynomolgus monkeys (Research Institute for Liver Diseases [RILD], Shanghai, China), and human cryopreserved hepatocytes (BioIVT). The positive control was 7-ethoxycoumarin (30 μM). Compounds were added to hepatocytes (400 µL, 1 × 106/mL), and reactions were carried out at 37 °C in 5% CO2/saturated humidity for 120 min. Reactions were stopped by the addition of ice-cold ACN (400 μL). Samples were shaken at 300 rpm for 10 min, then centrifuged at 13,000× g for 10 min. Supernatants were evaporated under a stream of N2. Residue from the RGN3067 incubation was reconstituted with ACN: H2O (200 μL, v:v, 3:7), followed by centrifugation at 14,000 rpm for 10 min, and the supernatant was used for qualitative and semi-quantitative analysis using LC-UV/HRMS. A Shimadzu UFLC-20A with autosampler was used for chromatographic separation. Detection and structure were determined using a Thermo Q-Extractive mass spectrometer in full MS/MS2 scan mode and Xcalibur Data Acquisition and Interpretation Software v4.2.
2.18. Data Processing
All statistical analyses were performed using GraphPad Prism v10 (San Diego, CA, USA). Absolute IC50 values were determined by a nonlinear regression model. Tests to determine significance are detailed in the relevant figure and table legends. Briefly, the Shapiro–Wilk test was used for testing the normality of the data. Unpaired t-tests or one-way ANOVAs were used to determine the statistical significance of treatment conditions. Asterisks denote significance (* p < 0.05; ** p < 0.01; *** p < 0.001, **** p < 0.0001).
4. Discussion
Despite the development of new treatments for GB over the last few decades, the disease continues to have a poor prognosis, and these therapies have not impacted the median survival of 15 months after diagnosis [
23]. Thus, innovative therapeutic strategies are urgently needed to delay recurrence and extend the survival of patients with GB tumors. Evidence suggests that gliomas are particularly sensitive to MTAs, and preclinical studies by this group and others have shown that tubulin-targeting therapies can be effective in treating brain cancers. Many MTAs, such as taxanes, vinca alkaloids, and epothilones, show potent anticancer activities in preclinical studies and clinical trials [
4,
24], but their effectiveness is often limited by poor aqueous solubility, multidrug resistance, inability to cross the BBB, and drug-induced cytotoxicity, such as peripheral neuropathy [
25,
26,
27,
28,
29]. Besides the taxane and vinca alkaloid binding sites, the colchicine binding site of tubulin has been examined extensively. While colchicine itself has had limited clinical success due to its high toxicity and low therapeutic index [
30], colchicine site inhibitors are attractive molecules due to several advantages they possess over taxane- and vinca-binding site compounds, including selective toxicity against tumor vasculature and an insensitivity to P-gp efflux pump-mediated multidrug resistance [
31,
32,
33].
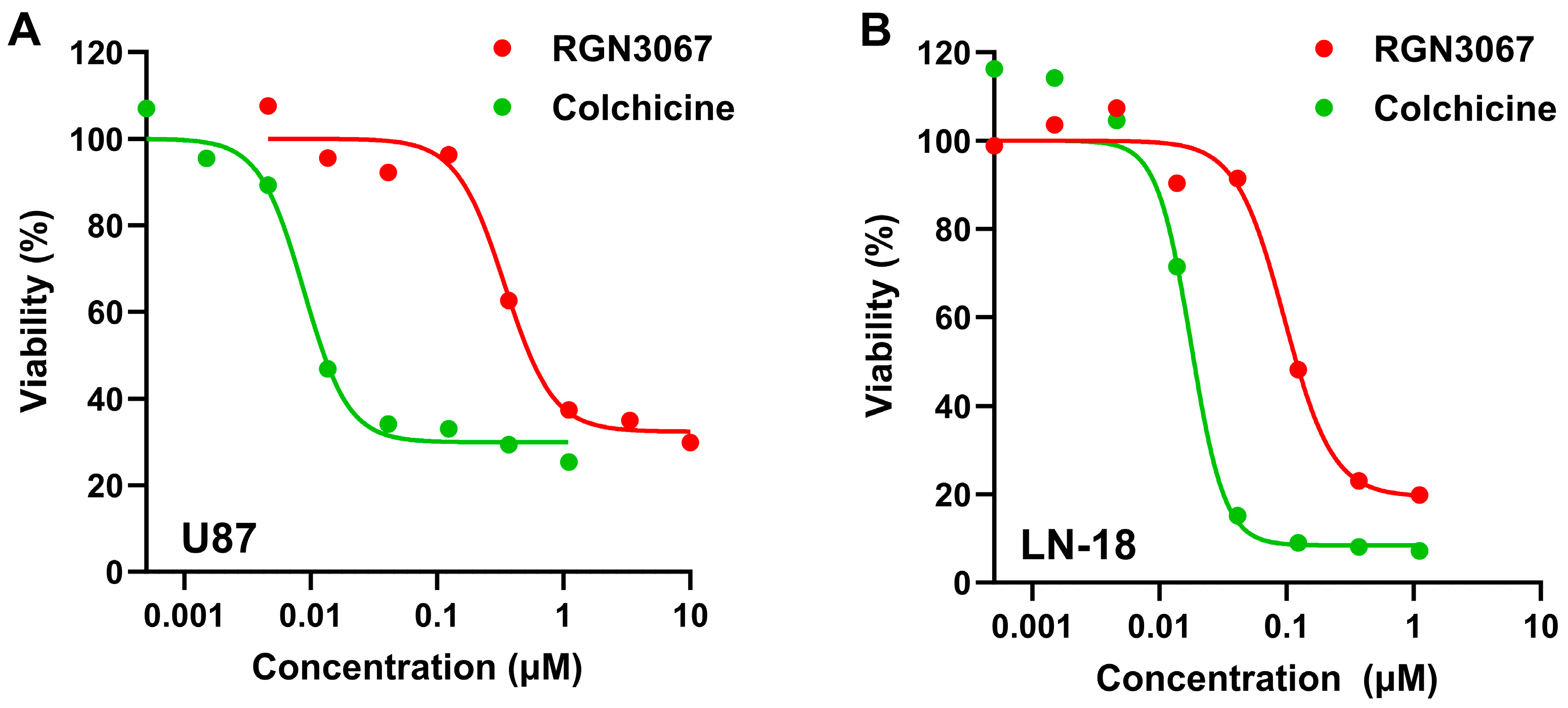
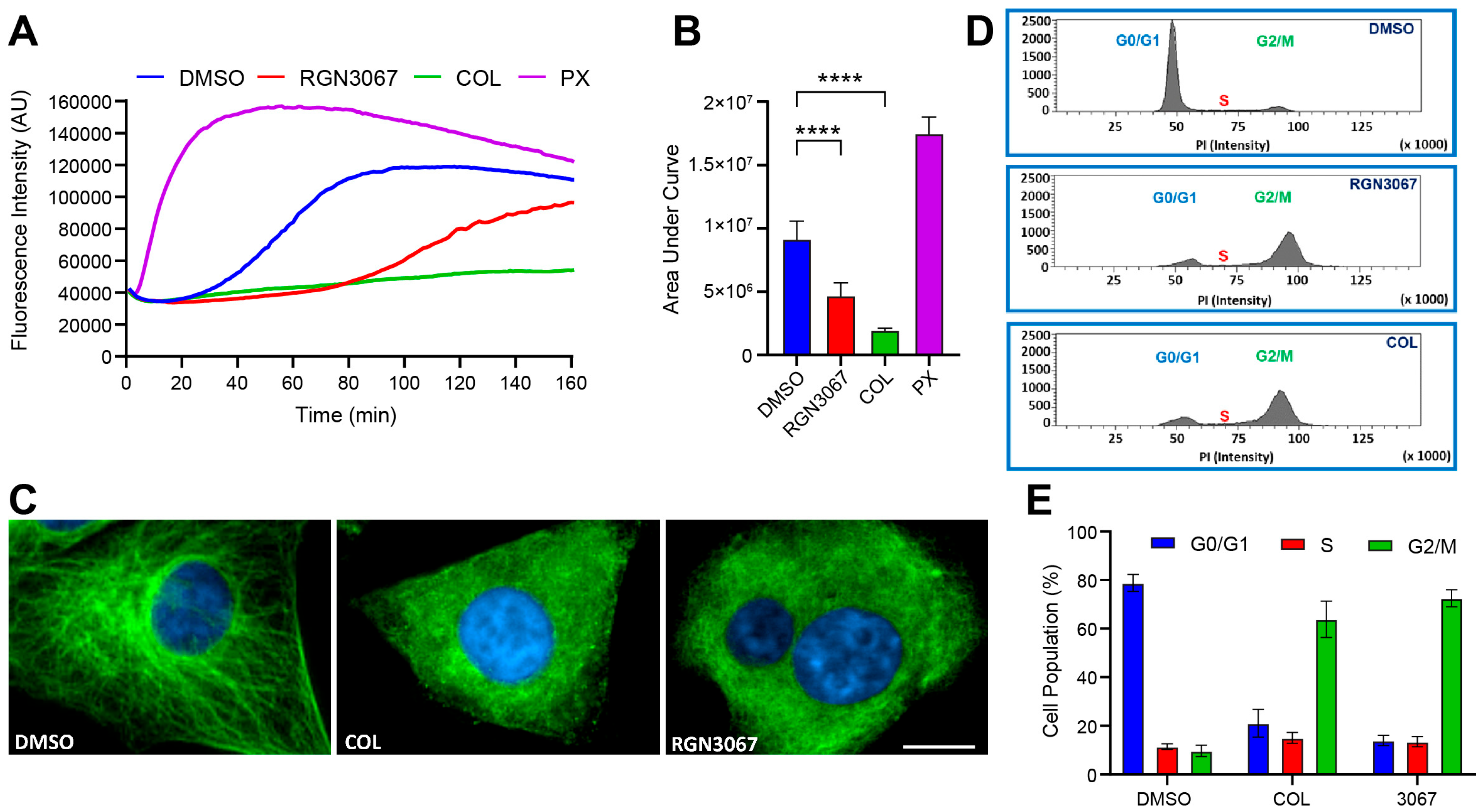
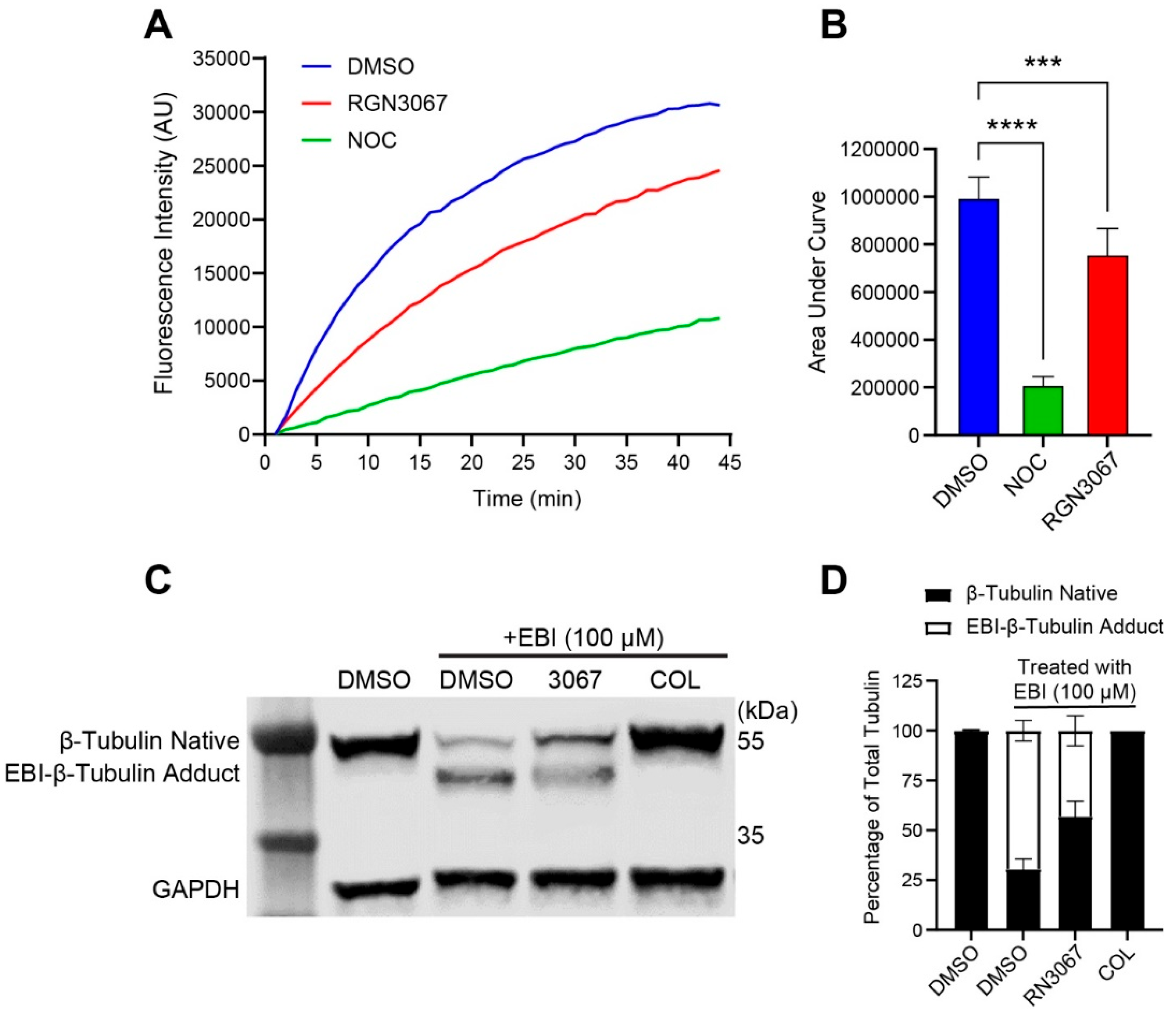
This study describes the development of RGN3067, a MTA that was designed to cross the BBB using CNS MPO parameters. We showed that RGN3067 inhibits the polymerization of tubulin in vitro and binds to the colchicine binding pocket of β-tubulin. In line with other tubulin destabilizers, we found that RGN3067 induces cell cycle arrest in the G2/M phase and inhibits the growth of human GB cancer cell lines (including the TMZ-resistant cell line LN-18) and patient-derived GB cell lines, with nanomolar potency. The effectiveness of RGN3067 in the TMZ-resistant line LN-18 is worth noting, as TMZ resistance remains a significant barrier to the successful treatment of patients with HGG [
34]. Only 50% percent of GB patients are responsive to TMZ, and the vast majority of responsive patients eventually develop TMZ resistance. This resistance is most commonly due to the overexpression of O6-methylguanine methyltransferase (MGMT), which is frequently correlated with the loss of epigenetic silencing of the
MGMT promoter. LN-18 cells replicate the loss of epigenetic silencing and show constitutively high levels of MGMT activity [
35]. Due to the significance of TMZ resistance in the clinic, future in vivo efficacy studies will include TMZ-resistant tumors.
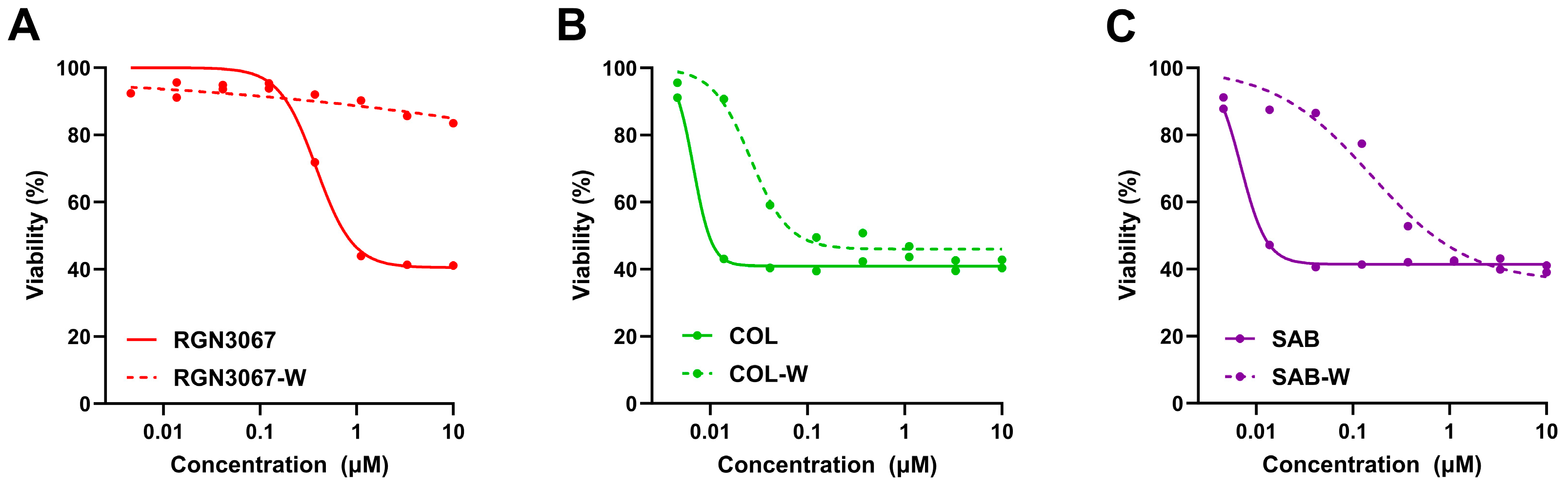
RGN3067 penetrates the rodent brain effectively, showing that a favorable CNS MPO score and the lack of P-gp pump efflux in the MDR1-MDCK model are accurate predictors of BBB penetrance. Equal amounts of RGN3067 were observed in the brain and plasma after oral administration. Furthermore, RGN3067 decreased the rate of tumor growth in a GB PDX model relative to control and caused no discernible toxicities in mice. A contributing factor to the low toxicity of RGN3067 in the PDX model may be its reversible binding to tubulin. Reversibility experiments showed that RGN3067 has a similar profile to sabizabulin, a potent tubulin destabilizer with a good safety profile in patients [
20]. However, unlike RGN3067, SAB is not BBB-penetrant with a brain-to-plasma ratio between 5% and 9% in mice [
36]. In contrast, the activity of colchicine, a classical MTA with a narrow therapeutic window, was nearly irreversible. These findings are notable from a therapeutic standpoint, as the low toxicity of RGN3067 makes it a promising candidate for the treatment of GB.
To facilitate the preclinical development of RGN3067, it was extensively profiled in eADME assays. The solubility and plasma binding of RGN3067 predict good exposure in vivo. The observed protein binding in human plasma is similar to that observed in rodents, and the human liver microsomal stability exceeds the stability observed in rodents. Metabolite ID studies in hepatocytes from multiple species indicate a consistent pattern of metabolism, increasing the likelihood that future animal studies will accurately predict human exposure and risk levels in the clinic for this class of molecules.
In summary, the design of RGN3067 addresses the limitations in physicochemical properties that have prevented previous MTAs from clinical success for the treatment of GB and other brain cancers. Our family of small-molecule tubulin destabilizers have been optimized chemically to cross the BBB, with the goal of developing a new generation of MTAs to treat glioblastoma and other CNS cancers. We show that our proof-of-concept molecules penetrate the brains of mice and have activity against glioma cells both in vitro and in a GB PDX model.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


