
Menopause-Associated Depression: Impact of Oxidative Stress and Neuroinflammation on the Central Nervous System—A Review


 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hormonal Changes during Perimenopause
3. Monoamine Hypothesis
4. Perimenopausal Depression and BDNF Deficiency
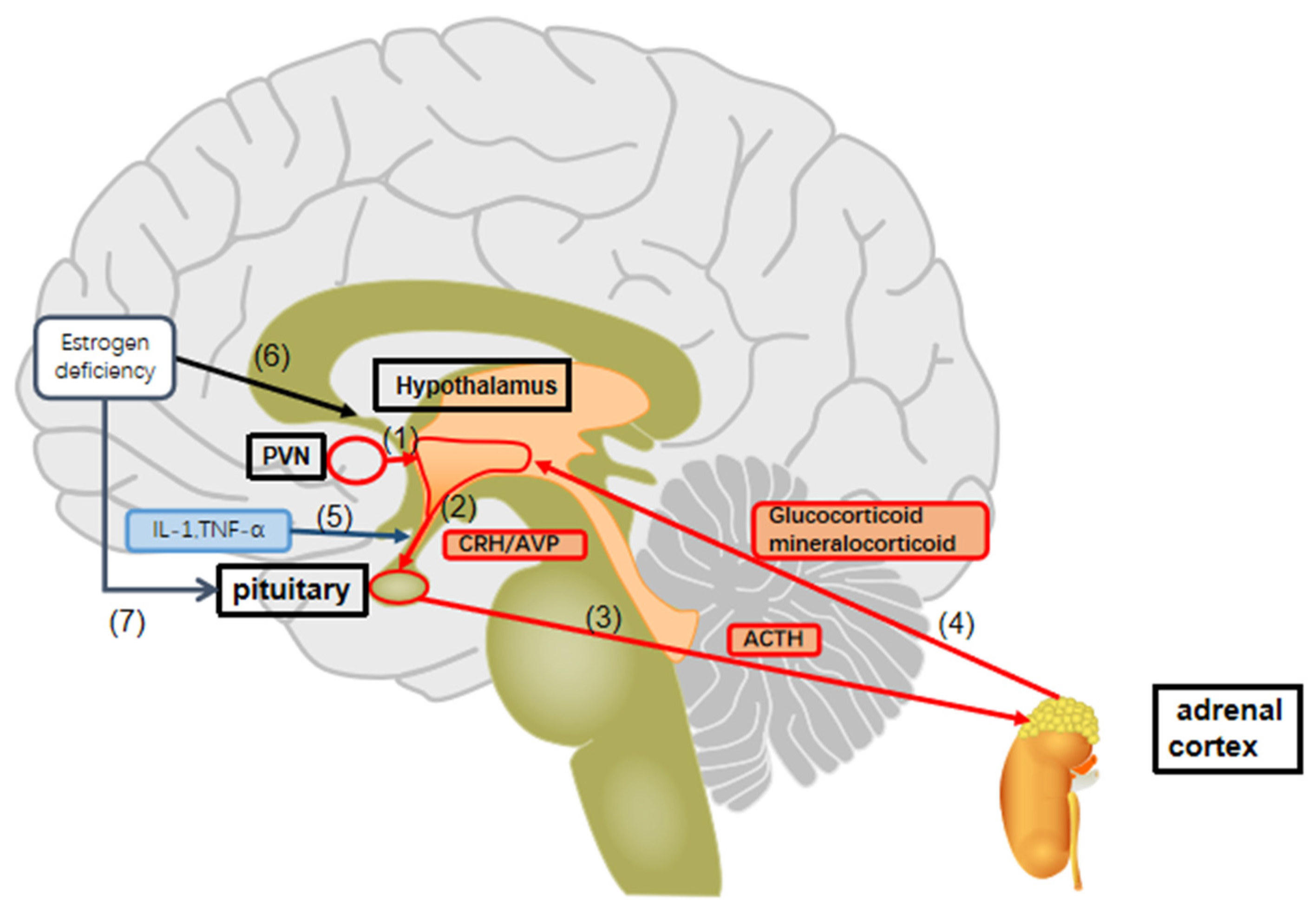
5. Perimenopausal Depression and HPA Axis Dysregulation
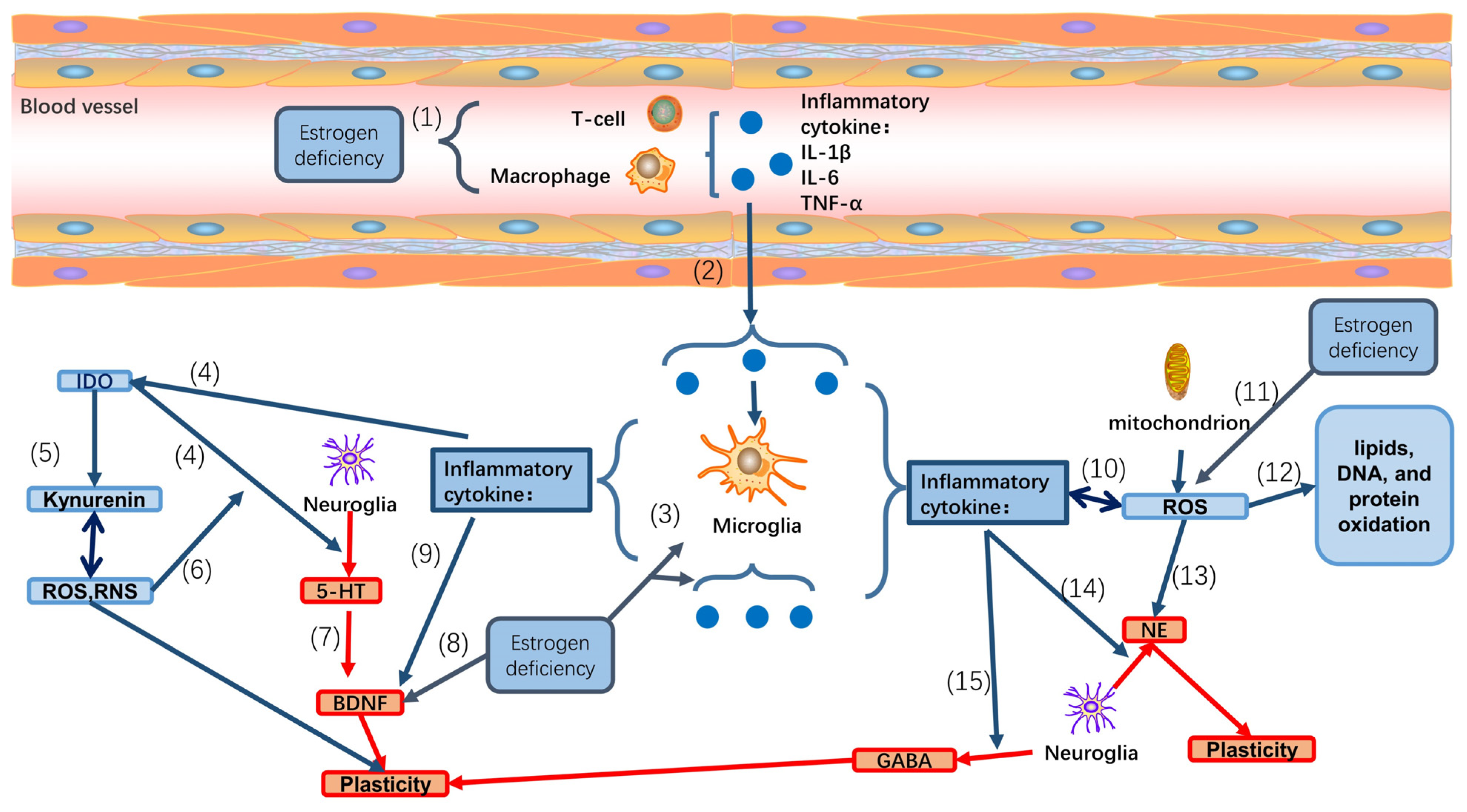
6. Effect of Neuroinflammation in Perimenopausal Depression
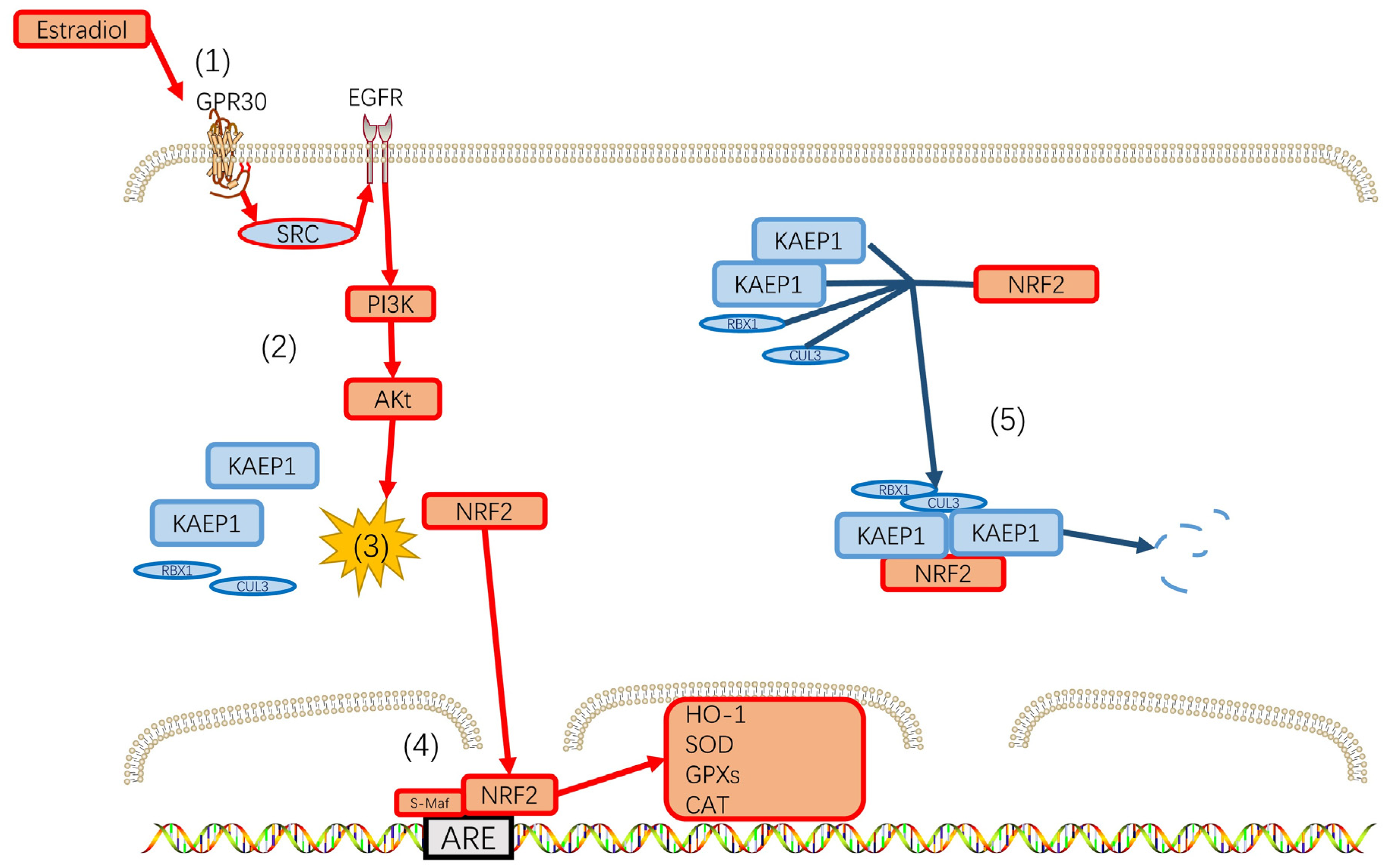
7. Perimenopausal Depression and Reactive Oxygen Species (ROS)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| 4-HNE | 4-hydroxynenal |
| 5-HIAA | 5-hydroxyindoleacetic acid |
| 5-HT2A | 5-hydroxytryptamine receptor 2A |
| ACEI | Angiotensin-converting enzyme inhibitors |
| ACTH | Adrenocorticotrophin |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate |
| ANP | Atrial natiuretic peptide |
| ARBs | Angiotensin receptor blockers |
| AREs | antioxidant response elements |
| ARNIs | Angiotensin receptor-neprilysin inhibitor |
| AVP | Arginine vasopressin |
| BAP | biological antioxidant potential |
| BDNF | Brain-derived neurotrophic factor |
| BH4 | Tetrahydrobiopterin |
| BNP | B-type natiuretic peptide |
| CCL2 | CC-chemokine ligand 2 |
| cGMP | Cyclic GMP |
| CNP | C-type natiuretic peptide |
| CNS | Central nervous system |
| CO | carbon monoxide |
| CRH | Corticotrophin-releasing hormone |
| CXCL1 | CXC-chemokine ligand 1 |
| d-ROMs | derivatives of reactive oxygen metabolites |
| EAE | experimental autoimmune encephalomyelitis |
| ER | estrogen receptor |
| ETC | electron transport chain |
| FSH | Follicle Stimulating Hormone |
| GABA | gamma-aminobutyric acid |
| GAD | glutamic acid decarboxylase |
| GC | guanylyl cyclase |
| Gpxs | glutathione peroxidases |
| GR | Glucocorticoid receptor |
| HO-1 | Heme oxygenase 1 |
| HPA | Hypothalamic-pituitary-adrenal |
| HSD | hydroxysteroid dehydrogenase |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-γ | interferon-gamma |
| IL | Interleukin |
| IL-1RA | interleukin-1 receptor antagonist |
| IL-1β | Interleukin-1beta |
| InhA | inhibin A |
| InhB | inhibin B |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LPS | lipopolysaccharide |
| LTP | long-term potentiation |
| MAO-A | Monoamine oxidase-A |
| MAOIs | Monoamine oxidase inhibitors |
| MAPK | mitogen-activated protein kinase |
| MDA | Malonaldehyde |
| MDD | Major Depressive Disorder |
| MR | Mineralocorticoid receptor |
| mTORC1 | Mammalian target of rapamycin complex-1 |
| NE | norepinephrine |
| NMDA | N-methyl-D-aspartate |
| NO | nitric oxide |
| Nps | natiuretic peptides |
| Nrf2 | nuclear respiratory factor 2 |
| O2− | oxide ion |
| ox LDL | oxidized lipoproteins |
| PFC | Prefrontal cortex |
| PLCγ1 | phospholipase Cγ1 |
| POMC | Pro-opiomelanocortin mRNA |
| PVN | Paraventricular nucleus |
| RAAS | renin–angiotensin–aldosterone system |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SAR | superoxide anion radicals |
| SERM | Selective estrogen receptor modulators |
| sMaf | small musculoaponeurotic fibrosarcoma |
| SNRIs | Serotonin/norepinephrine reuptake inhibitors |
| SSRIs | Selective serotonin reuptake inhibitors |
| TAK1 | TGF-β activated kinase 1 |
| TCAs | Tricyclic antidepressants |
| TNF-α | Tumor necrosis factor-alpha |
| TrkB | Tyrosine kinase receptor B |
References
- Gotlib, I.H.; Joormann, J. Cognition and depression: Current status and future directions. Annu. Rev. Clin. Psychol. 2010, 6, 285–312. [Google Scholar] [CrossRef]
- Bakunina, N.; Pariante, C.M.; Zunszain, P.A. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015, 144, 365–373. [Google Scholar] [CrossRef]
- Gorlova, A.; Svirin, E.; Pavlov, D.; Cespuglio, R.; Proshin, A.; Schroeter, C.A.; Lesch, K.P.; Strekalova, T. Understanding the Role of Oxidative Stress, Neuroinflammation and Abnormal Myelination in Excessive Aggression Associated with Depression: Recent Input from Mechanistic Studies. Int. J. Mol. Sci. 2023, 24, 915. [Google Scholar] [CrossRef]
- Lai, J.Y.; Ho, J.X.; Kow, A.S.F.; Liang, G.; Tham, C.L.; Ho, Y.C.; Lee, M.T. Interferon therapy and its association with depressive disorders—A review. Front. Immunol. 2023, 14, 1048592. [Google Scholar] [CrossRef]
- Moderie, C.; Nunez, N.; Fielding, A.; Comai, S.; Gobbi, G. Sex Differences in Responses to Antidepressant Augmentations in Treatment-Resistant Depression. Int. J. Neuropsychopharmacol. 2022, 25, 479–488. [Google Scholar] [CrossRef]
- Hasin, D.S.; Sarvet, A.L.; Meyers, J.L.; Saha, T.D.; Ruan, W.J.; Stohl, M.; Grant, B.F. Epidemiology of Adult DSM-5 Major Depressive Disorder and Its Specifiers in the United States. JAMA Psychiatry 2018, 75, 336–346. [Google Scholar] [CrossRef]
- Metcalf, C.A.; Duffy, K.A.; Page, C.E.; Novick, A.M. Cognitive Problems in Perimenopause: A Review of Recent Evidence. Curr. Psychiatry Rep. 2023, 25, 501–511. [Google Scholar] [CrossRef]
- Bromberger, J.T.; Epperson, C.N. Depression During and After the Perimenopause: Impact of Hormones, Genetics, and Environmental Determinants of Disease. Obstet. Gynecol. Clin. N. Am. 2018, 45, 663–678. [Google Scholar] [CrossRef]
- Han, Y.; Gu, S.; Li, Y.; Qian, X.; Wang, F.; Huang, J.H. Neuroendocrine pathogenesis of perimenopausal depression. Front. Psychiatry 2023, 14, 1162501. [Google Scholar] [CrossRef]
- Boulet, M.J.; Oddens, B.J.; Lehert, P.; Vemer, H.M.; Visser, A. Climacteric and menopause in seven South-east Asian countries. Maturitas 1994, 19, 157–176. [Google Scholar] [CrossRef]
- Makara-Studzińska, M.; Kryś-Noszczyk, K.M.; Wdowiak, A.; Kamińska, M.; Bakalczuk, S.; Bakalczuk, G. Comparison of biopsychosocial functioning of women of different nationalities in the perimenopausal period. Prz. Menopauzalny 2014, 13, 339–343. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, L.; Yan, R.; Dennerstein, L. Menopausal symptoms: Experience of Chinese women. Climacteric 2000, 3, 135–144. [Google Scholar] [CrossRef]
- Abel, K.M.; Freeman, M.P. Optimizing Mental Health for Women: Recognizing and Treating Mood Disorders throughout the Lifespan. J. Clin. Psychiatry 2023, 84, 49041. [Google Scholar] [CrossRef]
- Kulkarni, J. Perimenopausal depression—An under-recognised entity. Aust. Prescr. 2018, 41, 183–185. [Google Scholar] [CrossRef]
- Assaf, A.R.; Bushmakin, A.G.; Joyce, N.; Louie, M.J.; Flores, M.; Moffatt, M. The Relative Burden of Menopausal and Postmenopausal Symptoms versus Other Major Conditions: A Retrospective Analysis of the Medical Expenditure Panel Survey Data. Am. Health Drug Benefits 2017, 10, 311–321. [Google Scholar]
- Sharman Moser, S.; Chodick, G.; Bar-On, S.; Shalev, V. Healthcare Utilization and Prevalence of Symptoms in Women with Menopause: A Real-World Analysis. Int. J. Women’s Health 2020, 12, 445–454. [Google Scholar] [CrossRef]
- Herson, M.; Kulkarni, J. Hormonal Agents for the Treatment of Depression Associated with the Menopause. Drugs Aging 2022, 39, 607–618. [Google Scholar] [CrossRef]
- Maki, P.M.; Kornstein, S.G.; Joffe, H.; Bromberger, J.T.; Freeman, E.W.; Athappilly, G.; Bobo, W.V.; Rubin, L.H.; Koleva, H.K.; Cohen, L.S.; et al. Guidelines for the evaluation and treatment of perimenopausal depression: Summary and recommendations. Menopause 2018, 25, 1069–1085. [Google Scholar] [CrossRef]
- Parry, B.L. Perimenopausal depression. Am. J. Psychiatry 2008, 165, 23–27. [Google Scholar] [CrossRef]
- Garay, R.P.; Charpeaud, T.; Logan, S.; Hannaert, P.; Garay, R.G.; Llorca, P.M.; Shorey, S. Pharmacotherapeutic approaches to treating depression during the perimenopause. Expert Opin. Pharmacother. 2019, 20, 1837–1845. [Google Scholar] [CrossRef]
- Gilmor, M.L.; Owens, M.J.; Nemeroff, C.B. Inhibition of norepinephrine uptake in patients with major depression treated with paroxetine. Am. J. Psychiatry 2002, 159, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Littleton-Kearney, M.T.; Ostrowski, N.L.; Cox, D.A.; Rossberg, M.I.; Hurn, P.D. Selective estrogen receptor modulators: Tissue actions and potential for CNS protection. CNS Drug Rev. 2002, 8, 309–330. [Google Scholar] [CrossRef]
- Hare, D.L.; Toukhsati, S.R.; Johansson, P.; Jaarsma, T. Depression and cardiovascular disease: A clinical review. Eur. Heart J. 2014, 35, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Chung, H.F.; Dobson, A.J.; Pandeya, N.; Giles, G.G.; Bruinsma, F.; Brunner, E.J.; Kuh, D.; Hardy, R.; Avis, N.E.; et al. Age at natural menopause and risk of incident cardiovascular disease: A pooled analysis of individual patient data. Lancet Public Health 2019, 4, e553–e564. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Y.; Sun, Y.; Alolga, R.N.; Xiao, P.; Ma, G. Antihypertensive Drug Use and the Risk of Depression: A Systematic Review and Network Meta-analysis. Front. Pharmacol. 2021, 12, 777987. [Google Scholar] [CrossRef] [PubMed]
- van der Most, P.J.; Dolga, A.M.; Nijholt, I.M.; Luiten, P.G.; Eisel, U.L. Statins: Mechanisms of neuroprotection. Prog. Neurobiol. 2009, 88, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Taniguti, E.H.; Ferreira, Y.S.; Stupp, I.J.V.; Fraga-Junior, E.B.; Doneda, D.L.; Lopes, L.; Rios-Santos, F.; Lima, E.; Buss, Z.S.; Viola, G.G.; et al. Atorvastatin prevents lipopolysaccharide-induced depressive-like behaviour in mice. Brain Res. Bull. 2019, 146, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Shen, D.; Liu, J.; Pan, J.; Yu, L.; Shi, W.; Deng, L.; Zhu, L.; Yang, F.; Liu, J.; et al. Statin Function as an Anti-inflammation Therapy for Depression in Patients with Coronary Artery Disease by Downregulating Interleukin-1β. J. Cardiovasc. Pharmacol. 2016, 67, 129–135. [Google Scholar] [CrossRef]
- Liu, X.; Lou, X.; Cheng, X.; Meng, Y. Impact of metoprolol treatment on mental status of chronic heart failure patients with neuropsychiatric disorders. Drug Des. Dev. Ther. 2017, 11, 305–312. [Google Scholar] [CrossRef]
- van Melle, J.P.; Verbeek, D.E.; van den Berg, M.P.; Ormel, J.; van der Linde, M.R.; de Jonge, P. Beta-blockers and depression after myocardial infarction: A multicenter prospective study. J. Am. Coll. Cardiol. 2006, 48, 2209–2214. [Google Scholar] [CrossRef]
- McAinsh, J.; Cruickshank, J.M. Beta-blockers and central nervous system side effects. Pharmacol. Ther. 1990, 46, 163–197. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: Rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur. J. Heart Fail. 2013, 15, 1062–1073. [Google Scholar] [CrossRef]
- Breckenridge, A. Angiotensin converting enzyme inhibitors and quality of life. Am. J. Hypertens. 1991, 4, 79S–82S. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.F.; Bulpitt, C.J.; Fletcher, A.E. Angiotensin converting enzyme inhibitors and quality of life: The European trial. J. Hypertens Suppl. 1985, 3, S91–S94. [Google Scholar] [PubMed]
- Malik, J.; Shahid, A.W.; Shah, M.; Rana, G.; Kamal, A.; Naeem, H. Outcome of angiotensin receptor-neprilysin inhibitor on anxiety and depression in heart failure with reduced ejection fraction vs. heart failure with preserved ejection fraction. J. Community Hosp. Intern. Med. Perspect. 2021, 11, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.G.; Holsen, L.M.; Lancaster, K.; Makris, N.; Whitfield-Gabrieli, S.; Remington, A.; Weiss, B.; Buka, S.; Klibanski, A.; Goldstein, J.M. 17β-estradiol differentially regulates stress circuitry activity in healthy and depressed women. Neuropsychopharmacology 2015, 40, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Lascurain, M.B.; Camuñas-Palacín, A.; Thomas, N.; Breadon, C.; Gavrilidis, E.; Hudaib, A.R.; Gurvich, C.; Kulkarni, J. Improvement in depression with oestrogen treatment in women with schizophrenia. Arch. Women’s Ment. Health 2019, 23, 149–154. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Ben Dor, R.; Martinez, P.E.; Guerrieri, G.M.; Harsh, V.L.; Thompson, K.; Koziol, D.E.; Nieman, L.K.; Rubinow, D.R. Effects of Estradiol Withdrawal on Mood in Women with Past Perimenopausal Depression: A Randomized Clinical Trial. JAMA Psychiatry 2015, 72, 714–726. [Google Scholar] [CrossRef]
- Soares, C.N.; Almeida, O.P.; Joffe, H.; Cohen, L.S. Efficacy of estradiol for the treatment of depressive disorders in perimenopausal women: A double-blind, randomized, placebo-controlled trial. Arch. Gen. Psychiatry 2001, 58, 529–534. [Google Scholar] [CrossRef]
- McGee, E.A.; Hsueh, A.J. Initial and cyclic recruitment of ovarian follicles. Endocr. Rev. 2000, 21, 200–214. [Google Scholar] [CrossRef]
- Welt, C.K.; Jimenez, Y.; Sluss, P.M.; Smith, P.C.; Hall, J.E. Control of estradiol secretion in reproductive ageing. Hum. Reprod. 2006, 21, 2189–2193. [Google Scholar] [CrossRef]
- Allshouse, A.; Pavlovic, J.; Santoro, N. Menstrual Cycle Hormone Changes Associated with Reproductive Aging and How They May Relate to Symptoms. Obstet. Gynecol. Clin. N. Am. 2018, 45, 613–628. [Google Scholar] [CrossRef]
- Harlow, S.D.; Gass, M.; Hall, J.E.; Lobo, R.; Maki, P.; Rebar, R.W.; Sherman, S.; Sluss, P.M.; de Villiers, T.J.; Group, S.C. Executive summary of the Stages of Reproductive Aging Workshop + 10: Addressing the unfinished agenda of staging reproductive aging. Menopause 2012, 19, 387–395. [Google Scholar] [CrossRef]
- Freeman, E.W.; Sammel, M.D.; Lin, H.; Nelson, D.B. Associations of hormones and menopausal status with depressed mood in women with no history of depression. Arch. Gen. Psychiatry 2006, 63, 375–382. [Google Scholar] [CrossRef]
- Gordon, J.L.; Girdler, S.S.; Meltzer-Brody, S.E.; Stika, C.S.; Thurston, R.C.; Clark, C.T.; Prairie, B.A.; Moses-Kolko, E.; Joffe, H.; Wisner, K.L. Ovarian hormone fluctuation, neurosteroids, and HPA axis dysregulation in perimenopausal depression: A novel heuristic model. Am. J. Psychiatry 2015, 172, 227–236. [Google Scholar] [CrossRef]
- Chandankhede, M.; Gupta, M.; Pakhmode, S. Assessment of Psychological Status and Oxidative Stress in Postmenopausal Women: A Cross-Sectional Study. J. Menopausal. Med. 2021, 27, 155–161. [Google Scholar] [CrossRef]
- Sanchez-Rodriguez, M.A.; Castrejon-Delgado, L.; Zacarias-Flores, M.; Arronte-Rosales, A.; Mendoza-Nunez, V.M. Quality of life among post-menopausal women due to oxidative stress boosted by dysthymia and anxiety. BMC Women’s Health 2017, 17, 1. [Google Scholar] [CrossRef]
- Metcalf, C.A.; Johnson, R.L.; Duffy, K.A.; Freeman, E.W.; Sammel, M.D.; Epperson, C.N. Depressed, stressed, and inflamed: C-reactive protein linked with depression symptoms in midlife women with both childhood and current life stress. Stress Health 2023. [Google Scholar] [CrossRef]
- Zainal, N.H.; Newman, M.G. Prospective network analysis of proinflammatory proteins, lipid markers, and depression components in midlife community women. Psychol. Med. 2023, 53, 5267–5278. [Google Scholar] [CrossRef]
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61, 7–11. [Google Scholar]
- Kasper, S.; Hamon, M. Beyond the monoaminergic hypothesis: Agomelatine, a new antidepressant with an innovative mechanism of action. World J. Biol. Psychiatry 2009, 10, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The serotonin theory of depression: A systematic umbrella review of the evidence. Mol. Psychiatry 2023, 28, 3243–3256. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hernandez, O.T.; Martinez-Mota, L.; Herrera-Perez, J.J.; Jimenez-Rubio, G. Role of Estradiol in the Expression of Genes Involved in Serotonin Neurotransmission: Implications for Female Depression. Curr. Neuropharmacol. 2019, 17, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Sumner, B.E.; Fink, G. The density of 5-hydoxytryptamine2A receptors in forebrain is increased at pro-oestrus in intact female rats. Neurosci. Lett. 1997, 234, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Holschneider, D.P.; Kumazawa, T.; Chen, K.; Shih, J.C. Tissue-specific effects of estrogen on monoamine oxidase A and B in the rat. Life Sci. 1998, 63, 155–160. [Google Scholar] [CrossRef]
- Rekkas, P.V.; Wilson, A.A.; Lee, V.W.; Yogalingam, P.; Sacher, J.; Rusjan, P.; Houle, S.; Stewart, D.E.; Kolla, N.J.; Kish, S.; et al. Greater monoamine oxidase a binding in perimenopausal age as measured with carbon 11-labeled harmine positron emission tomography. JAMA Psychiatry 2014, 71, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Rajewska, J.; Rybakowski, J.K. Depression in premenopausal women: Gonadal hormones and serotonergic system assessed by D-fenfluramine challenge test. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 705–709. [Google Scholar] [CrossRef]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- De Vincenti, A.P.; Rios, A.S.; Paratcha, G.; Ledda, F. Mechanisms That Modulate and Diversify BDNF Functions: Implications for Hippocampal Synaptic Plasticity. Front. Cell. Neurosci. 2019, 13, 135. [Google Scholar] [CrossRef]
- Bohm-Levine, N.; Goldberg, A.R.; Mariani, M.; Frankfurt, M.; Thornton, J. Reducing luteinizing hormone levels after ovariectomy improves spatial memory: Possible role of brain-derived neurotrophic factor. Horm. Behav. 2020, 118, 104590. [Google Scholar] [CrossRef]
- Fan, J.; Li, B.; Ge, T.; Zhang, Z.; Lv, J.; Zhao, J.; Wang, P.; Liu, W.; Wang, X.; Mlyniec, K.; et al. Berberine produces antidepressant-like effects in ovariectomized mice. Sci. Rep. 2017, 7, 1310. [Google Scholar] [CrossRef]
- Scharfman, H.E.; MacLusky, N.J. Estrogen and brain-derived neurotrophic factor (BDNF) in hippocampus: Complexity of steroid hormone-growth factor interactions in the adult CNS. Front. Neuroendocr. 2006, 27, 415–435. [Google Scholar] [CrossRef]
- Ishizuka, Y.; Kakiya, N.; Witters, L.A.; Oshiro, N.; Shirao, T.; Nawa, H.; Takei, N. AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signaling in neurons. J. Neurochem. 2013, 127, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Lee, J.G.; Seo, M.K.; Lee, C.H.; Cho, H.Y.; Lee, B.J.; Seol, W.; Kim, Y.H. Differential effects of antidepressant drugs on mTOR signalling in rat hippocampal neurons. Int. J. Neuro-Psychopharmacol. 2014, 17, 1831–1846. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Abelaira, H.M.; Reus, G.Z.; Neotti, M.V.; Quevedo, J. The role of mTOR in depression and antidepressant responses. Life Sci. 2014, 101, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR Signaling in Tissue Regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef]
- Gassen, N.C.; Rein, T. Is There a Role of Autophagy in Depression and Antidepressant Action? Front. Psychiatry 2019, 10, 337. [Google Scholar] [CrossRef]
- Fukumoto, K.; Fogaca, M.V.; Liu, R.J.; Duman, C.H.; Li, X.Y.; Chaki, S.; Duman, R.S. Medial PFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant-like effects of 5-HT(1A) receptor stimulation. Neuropsychopharmacology 2020, 45, 1725–1734. [Google Scholar] [CrossRef]
- Naert, G.; Ixart, G.; Maurice, T.; Tapia-Arancibia, L.; Givalois, L. Brain-derived neurotrophic factor and hypothalamic-pituitary-adrenal axis adaptation processes in a depressive-like state induced by chronic restraint stress. Mol. Cell. Neurosci. 2011, 46, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.T.; Wang, B.; Xiao, Z.Y.; You, Y.; Tian, S.W. Apelin-13 Upregulates BDNF Against Chronic Stress-induced Depression-like Phenotypes by Ameliorating HPA Axis and Hippocampal Glucocorticoid Receptor Dysfunctions. Neuroscience 2018, 390, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Wu, H.; Li, Z.; Li, C.; Chen, D.; Zong, J.; Liu, Z.; Wei, S.; Peng, W. Jie-Yu-He-Huan Capsule Ameliorates Anxiety-Like Behaviours in Rats Exposed to Chronic Restraint Stress via the cAMP/PKA/CREB/BDNF Signalling Pathway. Oxidative Med. Cell. Longev. 2021, 2021, 1703981. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ren, L.; Zhang, C. Relationship between depression and inflammatory factors and brain-derived neurotrophic factor in patients with perimenopause syndrome. Exp. Ther. Med. 2018, 15, 4436–4440. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.Y.; Wang, Y.W.; Zhou, F.L.; Ma, X.C.; Yan, R.Z.; Zhang, L.; Wang, Q.L.; Yu, X. Association between MKP-1, BDNF, and Gonadal Hormones with Depression on Perimenopausal Women. J. Women’s Health 2016, 25, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M., Jr.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry 2017, 22, 527–536. [Google Scholar] [CrossRef]
- Mikulska, J.; Juszczyk, G.; Gawronska-Grzywacz, M.; Herbet, M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. [Google Scholar] [CrossRef]
- Labad, J.; Soria, V.; Salvat-Pujol, N.; Segalas, C.; Real, E.; Urretavizcaya, M.; de Arriba-Arnau, A.; Ferrer, A.; Crespo, J.M.; Jimenez-Murcia, S.; et al. Hypothalamic-pituitary-adrenal axis activity in the comorbidity between obsessive-compulsive disorder and major depression. Psychoneuroendocrinology 2018, 93, 20–28. [Google Scholar] [CrossRef]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef]
- Goncharova, N.D. Stress responsiveness of the hypothalamic-pituitary-adrenal axis: Age-related features of the vasopressinergic regulation. Front. Endocrinol. 2013, 4, 26. [Google Scholar] [CrossRef]
- Zhe, D.; Fang, H.; Yuxiu, S. Expressions of hippocampal mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) in the single-prolonged stress-rats. Acta Histochem. Cytochem. 2008, 41, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.Y.; Zhao, J.; Rahman, M.; St-Cyr, S.; McGowan, P.O.; Kim, J.C. Hippocampus-Anterior Hypothalamic Circuit Modulates Stress-Induced Endocrine and Behavioral Response. Front. Neural Circuits 2022, 16, 894722. [Google Scholar] [CrossRef] [PubMed]
- Gold, P.W.; Chrousos, G.P. Organization of the stress system and its dysregulation in melancholic and atypical depression: High vs low CRH/NE states. Mol. Psychiatry 2002, 7, 254–275. [Google Scholar] [CrossRef] [PubMed]
- Jarva, J.A.; Oinonen, K.A. Do oral contraceptives act as mood stabilizers? Evidence of positive affect stabilization. Arch. Women’s Ment. Health 2007, 10, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Zanardi, R.; Rossini, D.; Magri, L.; Malaguti, A.; Colombo, C.; Smeraldi, E. Response to SSRIs and role of the hormonal therapy in post-menopausal depression. Eur. Neuropsychopharmacol. 2007, 17, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Young, E.A.; Altemus, M.; Parkison, V.; Shastry, S. Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology 2001, 25, 881–891. [Google Scholar] [CrossRef]
- Gordon, J.L.; Eisenlohr-Moul, T.A.; Rubinow, D.R.; Schrubbe, L.; Girdler, S.S. Naturally Occurring Changes in Estradiol Concentrations in the Menopause Transition Predict Morning Cortisol and Negative Mood in Perimenopausal Depression. Clin. Psychol. Sci. 2016, 4, 919–935. [Google Scholar] [CrossRef]
- McCarthy, M.; Raval, A.P. The peri-menopause in a woman’s life: A systemic inflammatory phase that enables later neurodegenerative disease. J. Neuroinflamm. 2020, 17, 317. [Google Scholar] [CrossRef]
- Mishra, A.; Brinton, R.D. Inflammation: Bridging Age, Menopause and APOEepsilon4 Genotype to Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 312. [Google Scholar] [CrossRef]
- Wang, Y.; Mishra, A.; Brinton, R.D. Transitions in metabolic and immune systems from pre-menopause to post-menopause: Implications for age-associated neurodegenerative diseases. F1000Research 2020, 9, 68. [Google Scholar] [CrossRef]
- Huang, W.Y.; Hsin, I.L.; Chen, D.R.; Chang, C.C.; Kor, C.T.; Chen, T.Y.; Wu, H.M. Circulating interleukin-8 and tumor necrosis factor-alpha are associated with hot flashes in healthy postmenopausal women. PLoS ONE 2017, 12, e0184011. [Google Scholar] [CrossRef]
- Malutan, A.M.; Dan, M.; Nicolae, C.; Carmen, M. Proinflammatory and anti-inflammatory cytokine changes related to menopause. Prz. Menopauzalny 2014, 13, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Pfeilschifter, J.; Koditz, R.; Pfohl, M.; Schatz, H. Changes in proinflammatory cytokine activity after menopause. Endocr. Rev. 2002, 23, 90–119. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2015, 6, 635. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Raison, C.L.; Borisov, A.S.; Majer, M.; Drake, D.F.; Pagnoni, G.; Woolwine, B.J.; Vogt, G.J.; Massung, B.; Miller, A.H. Activation of central nervous system inflammatory pathways by interferon-alpha: Relationship to monoamines and depression. Biol. Psychiatry 2009, 65, 296–303. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Avital, A.; Goshen, I.; Kamsler, A.; Segal, M.; Iverfeldt, K.; Richter-Levin, G.; Yirmiya, R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 2003, 13, 826–834. [Google Scholar] [CrossRef]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Peng, W.H.; Kan, H.W.; Wu, C.C.; Wang, D.W.; Ho, Y.C. Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain-Focusing on AMPA Receptor. Biomedicines 2022, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- Viviani, B.; Boraso, M. Cytokines and neuronal channels: A molecular basis for age-related decline of neuronal function? Exp. Gerontol. 2011, 46, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Min, S.S.; Quan, H.Y.; Ma, J.; Han, J.S.; Jeon, B.H.; Seol, G.H. Chronic brain inflammation impairs two forms of long-term potentiation in the rat hippocampal CA1 area. Neurosci. Lett. 2009, 456, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Nistico, R.; Mango, D.; Mandolesi, G.; Piccinin, S.; Berretta, N.; Pignatelli, M.; Feligioni, M.; Musella, A.; Gentile, A.; Mori, F.; et al. Inflammation subverts hippocampal synaptic plasticity in experimental multiple sclerosis. PLoS ONE 2013, 8, e54666. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.; Herrera, A.J.; Cano, J.; Machado, A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J. Neurochem. 2002, 81, 150–157. [Google Scholar] [CrossRef]
- Wichers, M.C.; Maes, M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J. Psychiatry Neurosci. 2004, 29, 11–17. [Google Scholar]
- Sirivelu, M.P.; MohanKumar, P.S.; MohanKumar, S.M. Differential effects of systemic interleukin-1beta on gene expression in brainstem noradrenergic nuclei. Life Sci. 2012, 90, 77–81. [Google Scholar] [CrossRef]
- Tong, L.; Balazs, R.; Soiampornkul, R.; Thangnipon, W.; Cotman, C.W. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol. Aging 2008, 29, 1380–1393. [Google Scholar] [CrossRef]
- Tong, L.; Prieto, G.A.; Kramar, E.A.; Smith, E.D.; Cribbs, D.H.; Lynch, G.; Cotman, C.W. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1beta via p38 mitogen-activated protein kinase. J. Neurosci. 2012, 32, 17714–17724. [Google Scholar] [CrossRef]
- Payne, L.C.; Krueger, J.M. Interactions of cytokines with the hypothalamus-pituitary axis. J. Immunother. 1992, 12, 171–173. [Google Scholar] [CrossRef]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: A mechanism for the generation of glucocorticoid resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, X.; Zhou, Y.; Zheng, Q.; Chen, Z.; Zhang, H.; Sun, Z.; Xu, G.; Hu, G. Hypothalamus-pituitary-adrenal axis imbalance and inflammation contribute to sex differences in separation- and restraint-induced depression. Horm. Behav. 2020, 122, 104741. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Hiew, L.F.; Poon, C.H.; You, H.Z.; Lim, L.W. TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases. Cells 2021, 10, 1382. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tan, Q.; Zhao, Z.; Niu, G.; Li, S.; Li, W.; Song, C.; Leng, H. Distinct pathological changes of osteochondral units in early OVX-OA involving TGF-β signaling. Front. Endocrinol. 2022, 13, 1074176. [Google Scholar] [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- Petrik, D.; Lagace, D.C.; Eisch, A.J. The neurogenesis hypothesis of affective and anxiety disorders: Are we mistaking the scaffolding for the building? Neuropharmacology 2012, 62, 21–34. [Google Scholar] [CrossRef]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef]
- Wang, Y.; Symes, A.J. Smad3 deficiency reduces neurogenesis in adult mice. J. Mol. Neurosci. 2010, 41, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Nicks, K.M.; Fowler, T.W.; Akel, N.S.; Perrien, D.S.; Suva, L.J.; Gaddy, D. Bone turnover across the menopause transition: The role of gonadal inhibins. Ann. N. Y. Acad. Sci. 2010, 1192, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Cardoso, A.; Vale, N. Oxidative Stress in Depression: The Link with the Stress Response, Neuroinflammation, Serotonin, Neurogenesis and Synaptic Plasticity. Antioxidants 2023, 12, 470. [Google Scholar] [CrossRef]
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discov. Today 2020, 25, 1270–1276. [Google Scholar] [CrossRef]
- Doser, R.L.; Hoerndli, F.J. Regulation of neuronal excitability by reactive oxygen species and calcium signaling: Insights into brain aging. Curr. Res. Neurobiol. 2021, 2, 100012. [Google Scholar] [CrossRef]
- Insel, K.C.; Moore, I.M.; Vidrine, A.N.; Montgomery, D.W. Biomarkers for cognitive aging part II: Oxidative stress, cognitive assessments, and medication adherence. Biol. Res. Nurs. 2012, 14, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sarandol, A.; Sarandol, E.; Eker, S.S.; Erdinc, S.; Vatansever, E.; Kirli, S. Major depressive disorder is accompanied with oxidative stress: Short-term antidepressant treatment does not alter oxidative-antioxidative systems. Hum. Psychopharmacol. 2007, 22, 67–73. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Eichwald, T.; da Silva, L.B.; Staats Pires, A.C.; Niero, L.; Schnorrenberger, E.; Filho, C.C.; Espindola, G.; Huang, W.L.; Guillemin, G.J.; Abdenur, J.E.; et al. Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor. Antioxidants 2023, 12, 1037. [Google Scholar] [CrossRef]
- Cook, I.; Wang, T.; Leyh, T.S. Tetrahydrobiopterin regulates monoamine neurotransmitter sulfonation. Proc. Natl. Acad. Sci. USA 2017, 114, E5317–E5324. [Google Scholar] [CrossRef]
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10, 1603. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Grinberg, Y.Y.; Dibbern, M.E.; Levasseur, V.A.; Kraig, R.P. Insulin-like growth factor-1 abrogates microglial oxidative stress and TNF-alpha responses to spreading depression. J. Neurochem. 2013, 126, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Smith, A.N.; Shaughness, M.; Collier, S.; Hopkins, D.; Byrnes, K.R. Therapeutic targeting of microglia mediated oxidative stress after neurotrauma. Front. Med. 2022, 9, 1034692. [Google Scholar] [CrossRef]
- Hassamal, S. Chronic stress, neuroinflammation, and depression: An overview of pathophysiological mechanisms and emerging anti-inflammatories. Front. Psychiatry 2023, 14, 1130989. [Google Scholar] [CrossRef]
- Wang, J.; Dore, S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 2007, 130, 1643–1652. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Ufer, C.; Kuhn, H.; Borchert, A. Molecular biology of glutathione peroxidase 4: From genomic structure to developmental expression and neural function. Biol. Chem. 2007, 388, 1007–1017. [Google Scholar] [CrossRef]
- Buday, K.; Conrad, M. Emerging roles for non-selenium containing ER-resident glutathione peroxidases in cell signaling and disease. Biol. Chem. 2021, 402, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Bayo Jimenez, M.T.; Frenis, K.; Hahad, O.; Steven, S.; Cohen, G.; Cuadrado, A.; Munzel, T.; Daiber, A. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic. Biol. Med. 2022, 187, 72–91. [Google Scholar] [CrossRef] [PubMed]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation through the SIRT1/Nrf2 Pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, M.; Li, H.; Cai, L.; Yang, L. Rice Protein Exerts Anti-Inflammatory Effect in Growing and Adult Rats via Suppressing NF-kappaB Pathway. Int. J. Mol. Sci. 2019, 20, 6164. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Jo, M.G.; Kim, M.O. 17-beta Estradiol Rescued Immature Rat Brain against Glutamate-Induced Oxidative Stress and Neurodegeneration via Regulating Nrf2/HO-1 and MAP-Kinase Signaling Pathway. Antioxidants 2021, 10, 892. [Google Scholar] [CrossRef]
- Samy, D.M.; Mostafa, D.K.; Saleh, S.R.; Hassaan, P.S.; Zeitoun, T.M.; Ammar, G.A.G.; Elsokkary, N.H. Carnosic Acid Mitigates Depression-Like Behavior in Ovariectomized Mice via Activation of Nrf2/HO-1 Pathway. Mol. Neurobiol. 2023, 60, 610–628. [Google Scholar] [CrossRef]
- Marathe, N.; Rangaswami, H.; Zhuang, S.; Boss, G.R.; Pilz, R.B. Pro-survival effects of 17β-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J. Biol. Chem. 2012, 287, 978–988. [Google Scholar] [CrossRef]
- Xu, Y.; Pan, J.; Chen, L.; Zhang, C.; Sun, J.; Li, J.; Nguyen, L.; Nair, N.; Zhang, H.; O’Donnell, J.M. Phosphodiesterase-2 inhibitor reverses corticosterone-induced neurotoxicity and related behavioural changes via cGMP/PKG dependent pathway. Int. J. Neuropsychopharmacol. 2013, 16, 835–847. [Google Scholar] [CrossRef]
- Masood, A.; Nadeem, A.; Mustafa, S.J.; O’Donnell, J.M. Reversal of oxidative stress-induced anxiety by inhibition of phosphodiesterase-2 in mice. J. Pharmacol. Exp. Ther. 2008, 326, 369–379. [Google Scholar] [CrossRef]
- Stratton, R.C.; Squires, P.E.; Green, A.K. 17Beta-estradiol elevates cGMP and, via plasma membrane recruitment of protein kinase GIalpha, stimulates Ca2+ efflux from rat hepatocytes. J. Biol. Chem. 2010, 285, 27201–27212. [Google Scholar] [CrossRef]
- Cui, R.; Iso, H.; Yamagishi, K.; Ohira, T.; Tanigawa, T.; Kitamura, A.; Kiyama, M.; Imano, H.; Konishi, M.; Shimamoto, T. Relationship of urinary cGMP excretion with aging and menopausal status in a general population. J. Atheroscler. Thromb. 2009, 16, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Huang, Y.; Jin, S.L.; Frith, S.A.; Suvarna, N.; Conti, M.; O’Donnell, J.M. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology 2002, 27, 587–595. [Google Scholar] [CrossRef]
- Ishikawa, A.; Matsushita, H.; Shimizu, S.; Morita, N.; Hanai, R.; Sugiyama, S.; Watanabe, K.; Wakatsuki, A. Impact of Menopause and the Menstrual Cycle on Oxidative Stress in Japanese Women. J. Clin. Med. 2023, 12, 829. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Neri, S.; Sciacchitano, S.; Pino, L.D.; Costa, M.P.; Marchese, G.; Celotta, G.; Cassibba, N.; Pennisi, G.; Caschetto, S. Behaviour of some indicators of oxidative stress in postmenopausal and fertile women. Maturitas 2006, 53, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Matteo, M.; Rollo, T.; De Rosario, F.; Greco, P.; Vendemiale, G.; Serviddio, G. Sex hormones modulate circulating antioxidant enzymes: Impact of estrogen therapy. Redox Biol. 2013, 1, 340–346. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, G.; Kow, A.S.F.; Yusof, R.; Tham, C.L.; Ho, Y.-C.; Lee, M.T. Menopause-Associated Depression: Impact of Oxidative Stress and Neuroinflammation on the Central Nervous System—A Review. Biomedicines 2024, 12, 184. https://doi.org/10.3390/biomedicines12010184
Liang G, Kow ASF, Yusof R, Tham CL, Ho Y-C, Lee MT. Menopause-Associated Depression: Impact of Oxidative Stress and Neuroinflammation on the Central Nervous System—A Review. Biomedicines. 2024; 12(1):184. https://doi.org/10.3390/biomedicines12010184
Chicago/Turabian StyleLiang, Gengfan, Audrey Siew Foong Kow, Rohana Yusof, Chau Ling Tham, Yu-Cheng Ho, and Ming Tatt Lee. 2024. "Menopause-Associated Depression: Impact of Oxidative Stress and Neuroinflammation on the Central Nervous System—A Review" Biomedicines 12, no. 1: 184. https://doi.org/10.3390/biomedicines12010184