
From Pediatric to Adult Brain Cancer: Exploring Histone H3 Mutations in Australian Brain Cancer Patients

 , , , , , , and add
Show full author list
, , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. DIPG Cell Culture
2.3. Cell-Free Conditioned Media
2.4. Primers, Probes and Synthetic gBlock DNA
2.5. H3.3-K27M, H3.3-G34R and H3.1-K27M ddPCR Assays
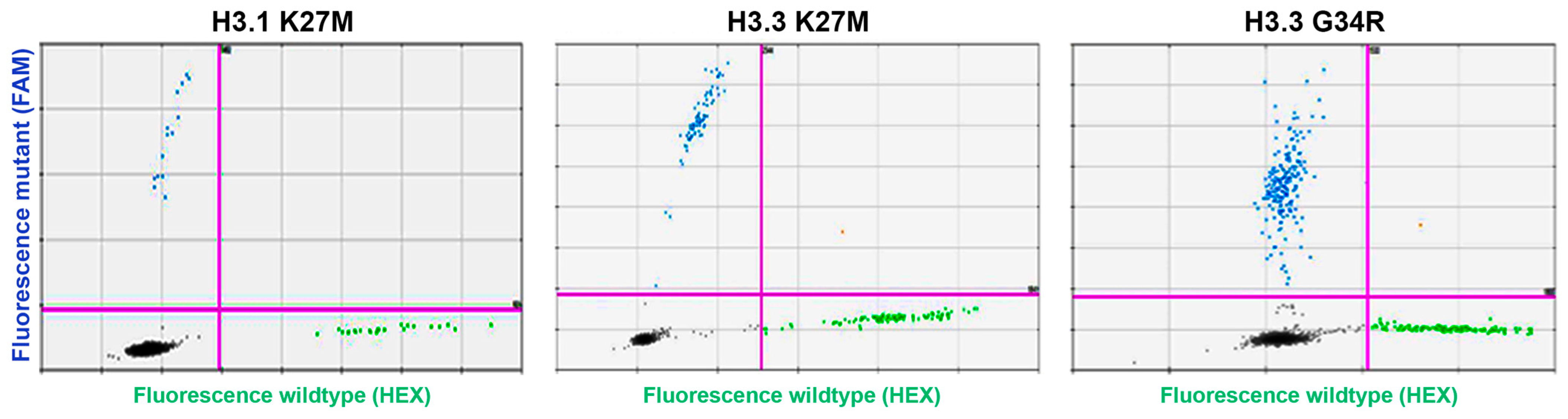
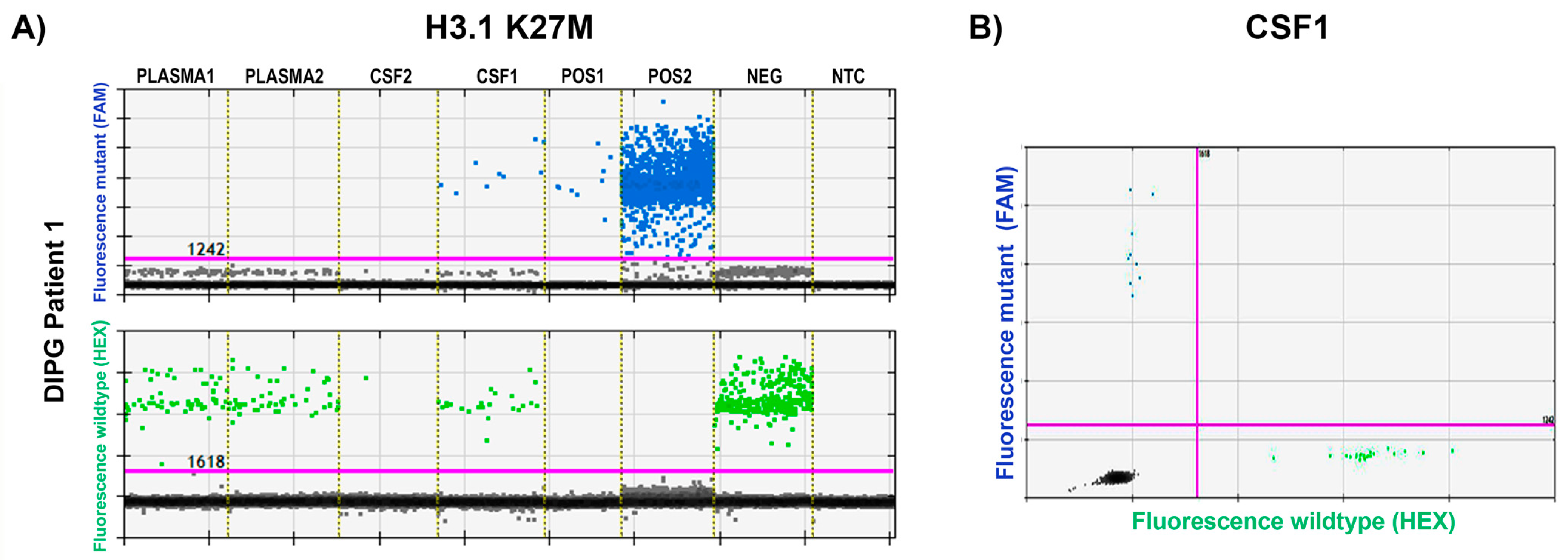
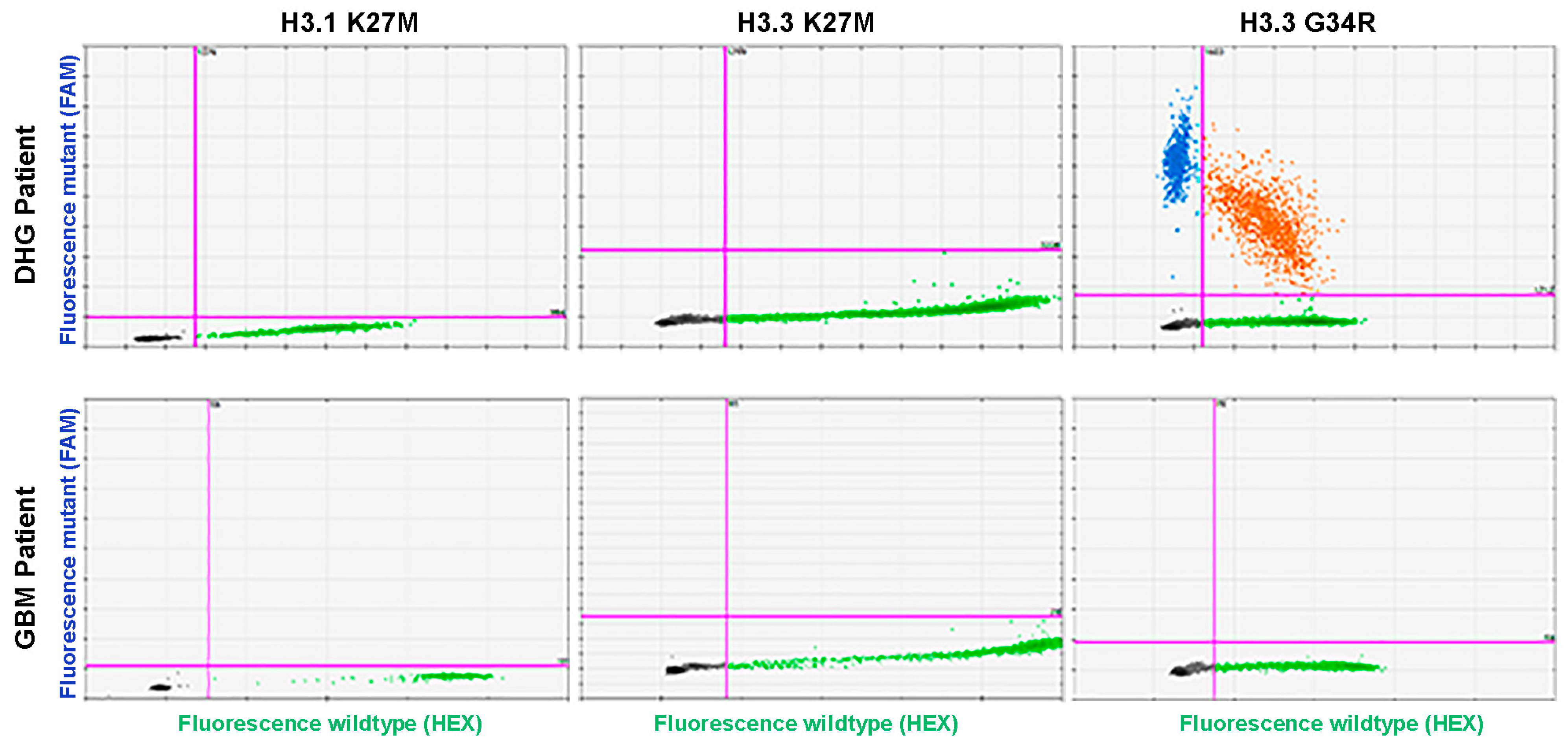
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crowell, C.; Mata-Mbemba, D.; Bennett, J.; Matheson, K.; Mackley, M.; Perreault, S.; Erker, C. Systematic review of diffuse hemispheric glioma, H3 G34-mutant: Outcomes and associated clinical factors. Neurooncol. Adv. 2022, 4, vdac133. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.G.; Ngo, T.N.M.; Le, H.T.; Dunn, I.F. The prognostic significance of HIST1H3B/C and H3F3A K27M mutations in diffuse midline gliomas is influenced by patient age. J. Neurooncol. 2022, 158, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Bhutada, A.S.; Ladner, L.; Cuoco, J.A.; Entwistle, J.J.; Marvin, E.A.; Rogers, C.M. Prognostic Indicators for H3K27M-Mutant Diffuse Midline Glioma: A Population-Based Retrospective Surveillance, Epidemiology, and End Results Database Analysis. World Neurosurg. 2023, 178, e113–e121. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallee, V.P.; Hebert, J.; Sauvageau, G. H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Chia, N.; Wong, A.; Teo, K.; Tan, A.P.; Vellayappan, B.A.; Yeo, T.T.; Oh, S.Y.; Tan, C.L. H3K27M-mutant, hemispheric diffuse glioma in an adult patient with prolonged survival. Neurooncol. Adv. 2021, 3, vdab135. [Google Scholar] [CrossRef] [PubMed]
- Meyronet, D.; Esteban-Mader, M.; Bonnet, C.; Joly, M.O.; Uro-Coste, E.; Amiel-Benouaich, A.; Forest, F.; Rousselot-Denis, C.; Burel-Vandenbos, F.; Bourg, V.; et al. Characteristics of H3 K27M-mutant gliomas in adults. Neuro Oncol. 2017, 19, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Nobusawa, S.; Yamazaki, T.; Osawa, T.; Horiguchi, K.; Hashiba, Y.; Yaoita, H.; Matsumura, N.; Ikota, H.; Hirato, J.; et al. Histone H3 K27M mutations in adult cerebellar high-grade gliomas. Brain Tumor Pathol. 2017, 34, 113–119. [Google Scholar] [CrossRef] [PubMed]
- AlRayahi, J.; Alwalid, O.; Mubarak, W.; Maaz, A.U.R.; Mifsud, W. Pediatric Brain Tumors in the Molecular Era: Updates for the Radiologist. Semin. Roentgenol. 2023, 58, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Di Nunno, V.; Franceschi, E.; Gatto, L.; Tosoni, A.; Bartolini, S.; Brandes, A.A. How to treat histone 3 altered gliomas: Molecular landscape and therapeutic developments. Expert. Rev. Clin. Pharmacol. 2023, 16, 17–26. [Google Scholar] [CrossRef]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.; Perry, A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef]
- Bonner, E.R.; Dawood, A.; Gordish-Dressman, H.; Eze, A.; Bhattacharya, S.; Yadavilli, S.; Mueller, S.; Waszak, S.M.; Nazarian, J. Pan-cancer atlas of somatic core and linker histone mutations. NPJ Genom. Med. 2023, 8, 23. [Google Scholar] [CrossRef]
- Burkart, M.; Sanford, S.; Dinner, S.; Sharp, L.; Kinahan, K. Future health of AYA survivors. Pediatr. Blood Cancer 2019, 66, e27516. [Google Scholar] [CrossRef]
- Lasocki, A.; Abdalla, G.; Chow, G.; Thust, S.C. Imaging features associated with H3 K27-altered and H3 G34-mutant gliomas: A narrative systematic review. Cancer Imaging 2022, 22, 63. [Google Scholar] [CrossRef]
- Sareen, H.; Garrett, C.; Lynch, D.; Powter, B.; Brungs, D.; Cooper, A.; Po, J.; Koh, E.S.; Vessey, J.Y.; McKechnie, S.; et al. The Role of Liquid Biopsies in Detecting Molecular Tumor Biomarkers in Brain Cancer Patients. Cancers 2020, 12, 1831. [Google Scholar] [CrossRef]
- Wolter, M.; Felsberg, J.; Malzkorn, B.; Kaulich, K.; Reifenberger, G. Droplet digital PCR-based analyses for robust, rapid, and sensitive molecular diagnostics of gliomas. Acta Neuropathol. Commun. 2022, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.N.; Becker, T.; Bray, V.; Chua, W.; Ma, Y.; Xu, B.; Lynch, D.; de Souza, P.; Roberts, T. Plasma next generation sequencing and droplet digital PCR-based detection of epidermal growth factor receptor (EGFR) mutations in patients with advanced lung cancer treated with subsequent-line osimertinib. Thorac. Cancer 2019, 10, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ricarte, F.; Mayor, R.; Martinez-Saez, E.; Rubio-Perez, C.; Pineda, E.; Cordero, E.; Cicuendez, M.; Poca, M.A.; Lopez-Bigas, N.; Ramon, Y.C.S.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819. [Google Scholar] [CrossRef]
- Li, D.; Bonner, E.R.; Wierzbicki, K.; Panditharatna, E.; Huang, T.; Lulla, R.; Mueller, S.; Koschmann, C.; Nazarian, J.; Saratsis, A.M. Standardization of the liquid biopsy for pediatric diffuse midline glioma using ddPCR. Sci. Rep. 2021, 11, 5098. [Google Scholar] [CrossRef] [PubMed]
- Panditharatna, E.; Kilburn, L.B.; Aboian, M.S.; Kambhampati, M.; Gordish-Dressman, H.; Magge, S.N.; Gupta, N.; Myseros, J.S.; Hwang, E.I.; Kline, C.; et al. Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy. Clin. Cancer Res. 2018, 24, 5850–5859. [Google Scholar] [CrossRef]
- Garcia-Romero, N.; Carrion-Navarro, J.; Areal-Hidalgo, P.; Ortiz de Mendivil, A.; Asensi-Puig, A.; Madurga, R.; Nunez-Torres, R.; Gonzalez-Neira, A.; Belda-Iniesta, C.; Gonzalez-Rumayor, V.; et al. BRAF V600E Detection in Liquid Biopsies from Pediatric Central Nervous System Tumors. Cancers 2019, 12, 66. [Google Scholar] [CrossRef]
- Izquierdo, E.; Proszek, P.; Pericoli, G.; Temelso, S.; Clarke, M.; Carvalho, D.M.; Mackay, A.; Marshall, L.V.; Carceller, F.; Hargrave, D.; et al. Droplet digital PCR-based detection of circulating tumor DNA from pediatric high grade and diffuse midline glioma patients. Neurooncol. Adv. 2021, 3, vdab013. [Google Scholar] [CrossRef]
- Friedman, J.S.; Hertz, C.A.J.; Karajannis, M.A.; Miller, A.M. Tapping into the genome: The role of CSF ctDNA liquid biopsy in glioma. Neurooncol. Adv. 2022, 4, ii33–ii40. [Google Scholar] [CrossRef]
- Fontanilles, M.; Marguet, F.; Beaussire, L.; Magne, N.; Pepin, L.F.; Alexandru, C.; Tennevet, I.; Hanzen, C.; Langlois, O.; Jardin, F.; et al. Cell-free DNA and circulating TERT promoter mutation for disease monitoring in newly-diagnosed glioblastoma. Acta Neuropathol. Commun. 2020, 8, 179. [Google Scholar] [CrossRef]
- Juratli, T.A.; Stasik, S.; Zolal, A.; Schuster, C.; Richter, S.; Daubner, D.; Juratli, M.A.; Thowe, R.; Hennig, S.; Makina, M.; et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clin. Cancer Res. 2018, 24, 5282–5291. [Google Scholar] [CrossRef]
- Muralidharan, K.; Yekula, A.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Wang, L.; Lau, S.; Zhang, H.; Lee, H.; Bettegowda, C.; et al. TERT Promoter Mutation Analysis for Blood-Based Diagnosis and Monitoring of Gliomas. Clin. Cancer Res. 2021, 27, 169–178. [Google Scholar] [CrossRef]
- Boisselier, B.; Gallego Perez-Larraya, J.; Rossetto, M.; Labussiere, M.; Ciccarino, P.; Marie, Y.; Delattre, J.Y.; Sanson, M. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology 2012, 79, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Kahana-Edwin, S.; Cain, L.E.; Karpelowsky, J. Roadmap to Liquid Biopsy Biobanking from Pediatric Cancers-Challenges and Opportunities. Biopreserv. Biobank. 2021, 19, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A.; Brastianos, P.K.; Wakimoto, H.; Zolal, A.; Filbin, M.G.; Cahill, D.P.; Santagata, S.; Juratli, T.A. A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol. 2023, 146, 515–525. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Zhang, M.; Piao, Y.; Chen, L.; Liang, H.; Wei, Y.; Hu, Z.; Zhao, L.; Teng, L.; et al. H3 K27M-mutant diffuse midline gliomas in different anatomical locations. Hum. Pathol. 2018, 78, 89–96. [Google Scholar] [CrossRef]
- Lopez, G.; Oberheim Bush, N.A.; Berger, M.S.; Perry, A.; Solomon, D.A. Diffuse non-midline glioma with H3F3A K27M mutation: A prognostic and treatment dilemma. Acta Neuropathol. Commun. 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Onishi, S.; Ohba, S.; Kuraoka, K.; Kurashige, T.; Sugiyama, K.; Yamasaki, F. Molecular and clinical characterization of H3 K27M-mutant "non-midline" glioblastoma: A case report and literature review. Neurocirugia 2022, 33, 356–360. [Google Scholar] [CrossRef]
- Yuile, A.; Khasraw, M.; Low, J.T.; Walsh, K.M.; Lipp, E.; Sy, J.; Satgunaseelan, L.; Kastelan, M.A.; De Silva, M.; Lee, A.; et al. Patterns of care in adult histone mutant gliomas: Results of an international survey. Neurooncol. Pract. 2022, 9, 520–525. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Y.; Wang, L.; Gao, Y.; Xu, J. The clinicopathological features and prognosis of multifocal high-grade gliomas in adults with H3F3A mutation. Neurosciences 2023, 28, 42–47. [Google Scholar] [CrossRef]
- Schulte, J.D.; Buerki, R.A.; Lapointe, S.; Molinaro, A.M.; Zhang, Y.; Villanueva-Meyer, J.E.; Perry, A.; Phillips, J.J.; Tihan, T.; Bollen, A.W.; et al. Clinical, radiologic, and genetic characteristics of histone H3 K27M-mutant diffuse midline gliomas in adults. Neurooncol. Adv. 2020, 2, vdaa142. [Google Scholar] [CrossRef]
- Chen, C.C.L.; Deshmukh, S.; Jessa, S.; Hadjadj, D.; Lisi, V.; Andrade, A.F.; Faury, D.; Jawhar, W.; Dali, R.; Suzuki, H.; et al. Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 2020, 183, 1617–1633.e1622. [Google Scholar] [CrossRef] [PubMed]
- Sledzinska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef] [PubMed]
- Powter, B.; Jeffreys, S.A.; Sareen, H.; Cooper, A.; Brungs, D.; Po, J.; Roberts, T.; Koh, E.S.; Scott, K.F.; Sajinovic, M.; et al. Human TERT promoter mutations as a prognostic biomarker in glioma. J. Cancer Res. Clin. Oncol. 2021, 147, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Ozair, A.; Bhat, V.; Alisch, R.S.; Khosla, A.A.; Kotecha, R.R.; Odia, Y.; McDermott, M.W.; Ahluwalia, M.S. DNA Methylation and Histone Modification in Low-Grade Gliomas: Current Understanding and Potential Clinical Targets. Cancers 2023, 15, 1342. [Google Scholar] [CrossRef]
- Hegi, M.E.; Genbrugge, E.; Gorlia, T.; Stupp, R.; Gilbert, M.R.; Chinot, O.L.; Nabors, L.B.; Jones, G.; Van Criekinge, W.; Straub, J.; et al. MGMT Promoter Methylation Cutoff with Safety Margin for Selecting Glioblastoma Patients into Trials Omitting Temozolomide: A Pooled Analysis of Four Clinical Trials. Clin. Cancer Res. 2019, 25, 1809–1816. [Google Scholar] [CrossRef]
- Majchrzak-Celinska, A.; Dybska, E.; Barciszewska, A.M. DNA methylation analysis with methylation-sensitive high-resolution melting (MS-HRM) reveals gene panel for glioma characteristics. CNS Neurosci. Ther. 2020, 26, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Weiser, A.; Sanchez Bergman, A.; Machaalani, C.; Bennett, J.; Roth, P.; Reimann, R.R.; Nazarian, J.; Guerreiro Stucklin, A.S. Bridging the age gap: A review of molecularly informed treatments for glioma in adolescents and young adults. Front. Oncol. 2023, 13, 1254645. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wu, Q.; Hao, Z.; Chen, L. Identification of novel prognostic targets in glioblastoma using bioinformatics analysis. Biomed. Eng. Online 2022, 21, 26. [Google Scholar] [CrossRef]
- Vuong, H.G.; Nguyen, T.Q.; Ngo, T.N.M.; Nguyen, H.C.; Fung, K.M.; Dunn, I.F. The interaction between TERT promoter mutation and MGMT promoter methylation on overall survival of glioma patients: A meta-analysis. BMC Cancer 2020, 20, 897. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primers and Probes | |||
|---|---|---|---|
| H3.1-K27M | H3.3-K27M | H3.3-G34R | |
| Forward primer | 5′-AACAGACAGCTCGGAAATC-3′ | 5′-AAATCGACCGGTGGTAAAGC-3′ | |
| Reverse primer | 5′-TAACGGTGAGGCTTTTTCA-3′ | 5′-AATACCTGTAACGATGAGGT-3′ | |
| Wild-type probe | 5′-HEX-CTCGCAAGAGCGCGCCG-BQ1-3′ | 5′-HEX-CACTCTTGCGAGCGGCTT-BQ1-3′ | 5′-HEX-TCTACTGGAGGGGTGAAGAA-BQ1-3′ |
| Mutant probe | 5′-FAM-CTCGCATGAGCGCGCCG-BQ1-3′ | 5′-FAM-CACTCATGCGAGCGGCTT-BQ1-3′ | 5′-FAM-TCTACTGGAAGGGTGAAGAA-BQ1-3′ |
| gBlock Sequence (NIH Nucleotide GenBank ID) | |||
| H3.1-K27M | AF531275.1 Range 391–537; base 493 changed (A to T) | ||
| H3.3-K27M | NG065173.1 Range 6638–6809; base 6729 changed (A to T) | ||
| H3.3-G34R | NG065173.1 Range 6638–6809; base 6749 changed (G to A) | ||
| Cell Line | Mutation Detected * |
|---|---|
| SU-DIPG21 | H3.1 H3B K27M |
| HSJD-GBM2 | H3.3 H3F3A G34R |
| SU-DIPG24 | H3.3 H3F3A K27M |
| HSJD-DIPG007 | H3.3 H3F3A K27M |
| P00208 | H3.3 H3F3A K27M |
| RA055 | H3.3 H3F3A K27M |
| SU-DIPG17 | H3.3 H3F3A K27M |
| SU-DIPG6 | H3.3 H3F3A K27M |
| RA021 | wild-type |
| VUMC-DIPG10 | wild-type |
| P000106 | wild-type |
| P001003 | wild-type |
| P001105 | wild-type |
| P001302 | wild-type |
| P001802 | wild-type |
| RA034 | wild-type |
| Characteristics | Patients, n (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Age group | 18–39 years | 40–65 years | 66–85 years | Total | ||||
| No. of patients | 8 | 41 | 40 | 89 | ||||
| Mean age in years at diagnosis [Range] | 30 [18–36] | 55 [43–65] | 73 [66–85] | 61 [18–85] | ||||
| Sex | ||||||||
| Male | 8 | (100) | 26 | (63.4) | 25 | (63.5) | 59 | (66.3) |
| Female | 0 | (0) | 15 | (36.6) | 15 | (37.5) | 30 | (33.7) |
| Grade | ||||||||
| WHO grade 4 | 7 | (87.5) | 40 | (97.6) | 40 | (100) | 87 | (97.8) |
| WHO grade 2 or 3 | 1 | (12.5) | 1 | (2.4) | 0 | (0) | 2 | (2.2) |
| Surgical Resection | ||||||||
| GTR | 3 | (37.5) | 16 | (39) | 12 | (30) | 31 | (34.8) |
| STR | 5 | (62.5) | 23 | (56.1) | 22 | (55) | 50 | (56.2) |
| Biopsy | 0 | (0) | 2 | (4.9) | 6 | (15) | 8 | (9) |
| Radiation | ||||||||
| Yes | 8 | (100) | 35 | (85.4) | 24 | (60) | 67 | (75.3) |
| No | (0) | 6 | (14.6) | 16 | (40) | 22 | (24.7) | |
| Concurrent TMZ | ||||||||
| Yes | 8 | (100) | 36 | (87.8) | 21 | (52.5) | 65 | (73) |
| No | (0) | 5 | (12.2) | 19 | (47.5) | 24 | (27) | |
| Tumour Location | ||||||||
| Frontal | 4 | (50) | 19 | (46.3) | 11 | (27.5) | 34 | (38.2) |
| Parietal | 2 | (25) | 5 | (12.2) | 6 | (15) | 13 | (14.6) |
| Temporal | 1 | (12.5) | 10 | (24.4) | 15 | (37.5) | 26 | (29.2) |
| Other | 1 | (12.5) | 7 | (17.1) | 8 | (20) | 16 | (18) |
| Mean overall survival in days [Range] | 1067 [429–2100] | 602 [23–2879] | 300 [27–1430] | 493 [23–2879] | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grebstad Tune, B.; Sareen, H.; Powter, B.; Kahana-Edwin, S.; Cooper, A.; Koh, E.-S.; Lee, C.S.; Po, J.W.; McCowage, G.; Dexter, M.; et al. From Pediatric to Adult Brain Cancer: Exploring Histone H3 Mutations in Australian Brain Cancer Patients. Biomedicines 2023, 11, 2907. https://doi.org/10.3390/biomedicines11112907
Grebstad Tune B, Sareen H, Powter B, Kahana-Edwin S, Cooper A, Koh E-S, Lee CS, Po JW, McCowage G, Dexter M, et al. From Pediatric to Adult Brain Cancer: Exploring Histone H3 Mutations in Australian Brain Cancer Patients. Biomedicines. 2023; 11(11):2907. https://doi.org/10.3390/biomedicines11112907
Chicago/Turabian StyleGrebstad Tune, Benedicte, Heena Sareen, Branka Powter, Smadar Kahana-Edwin, Adam Cooper, Eng-Siew Koh, Cheok S. Lee, Joseph W. Po, Geoff McCowage, Mark Dexter, and et al. 2023. "From Pediatric to Adult Brain Cancer: Exploring Histone H3 Mutations in Australian Brain Cancer Patients" Biomedicines 11, no. 11: 2907. https://doi.org/10.3390/biomedicines11112907