
Genetic and Pharmacological Blockade of Sigma-1 Receptors Attenuates Inflammation-Associated Hypersensitivity during Acute Colitis in CD1 Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Colitis Induction
2.3. Drugs
2.4. Evaluation of Referred Mechanical Hypersensitivity: Von Frey Test
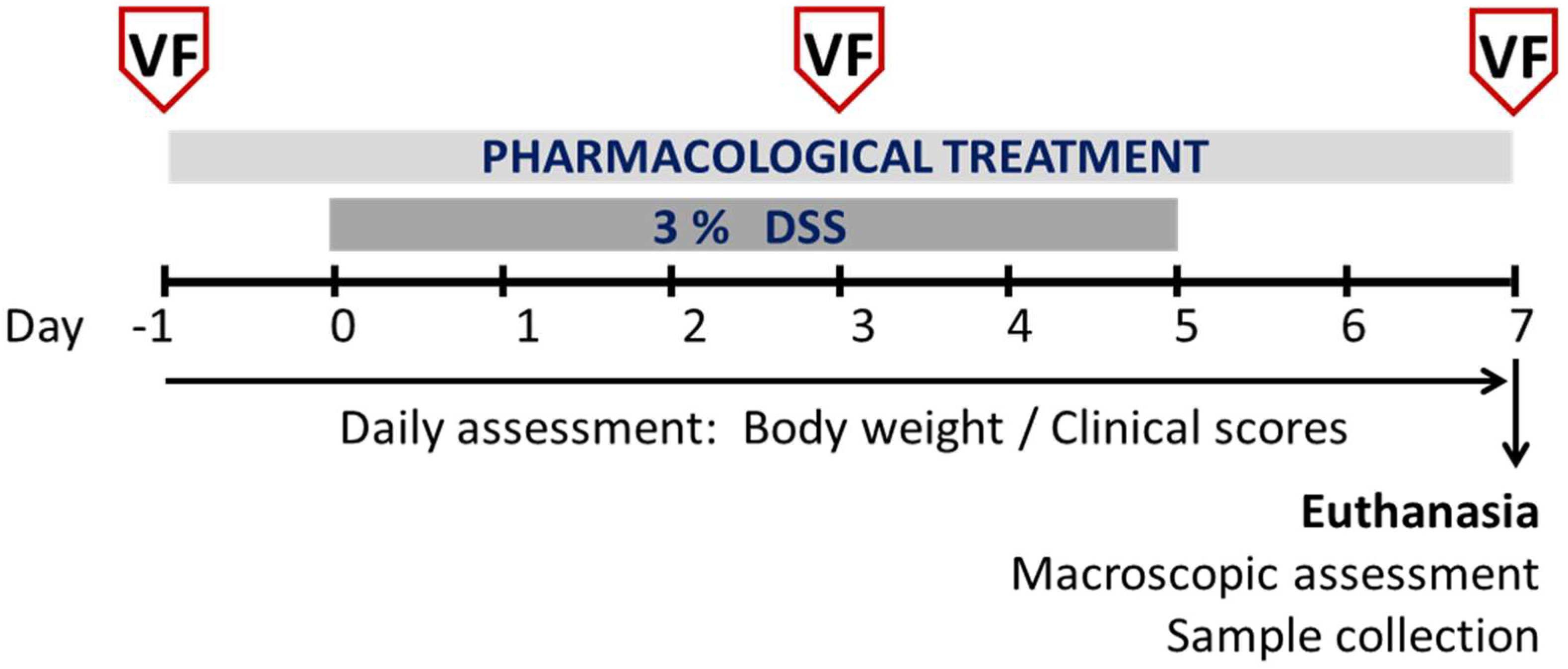
2.5. Experimental Protocols
2.6. Sample Collection
2.7. Clinical and Macroscopic Assessment of Inflammation
2.8. Histological Studies
2.9. Gene Expression: Quantitative Reverse Transcription-PCR
2.10. Protein Expression: Western Blot
2.11. Statistical Analysis
3. Results
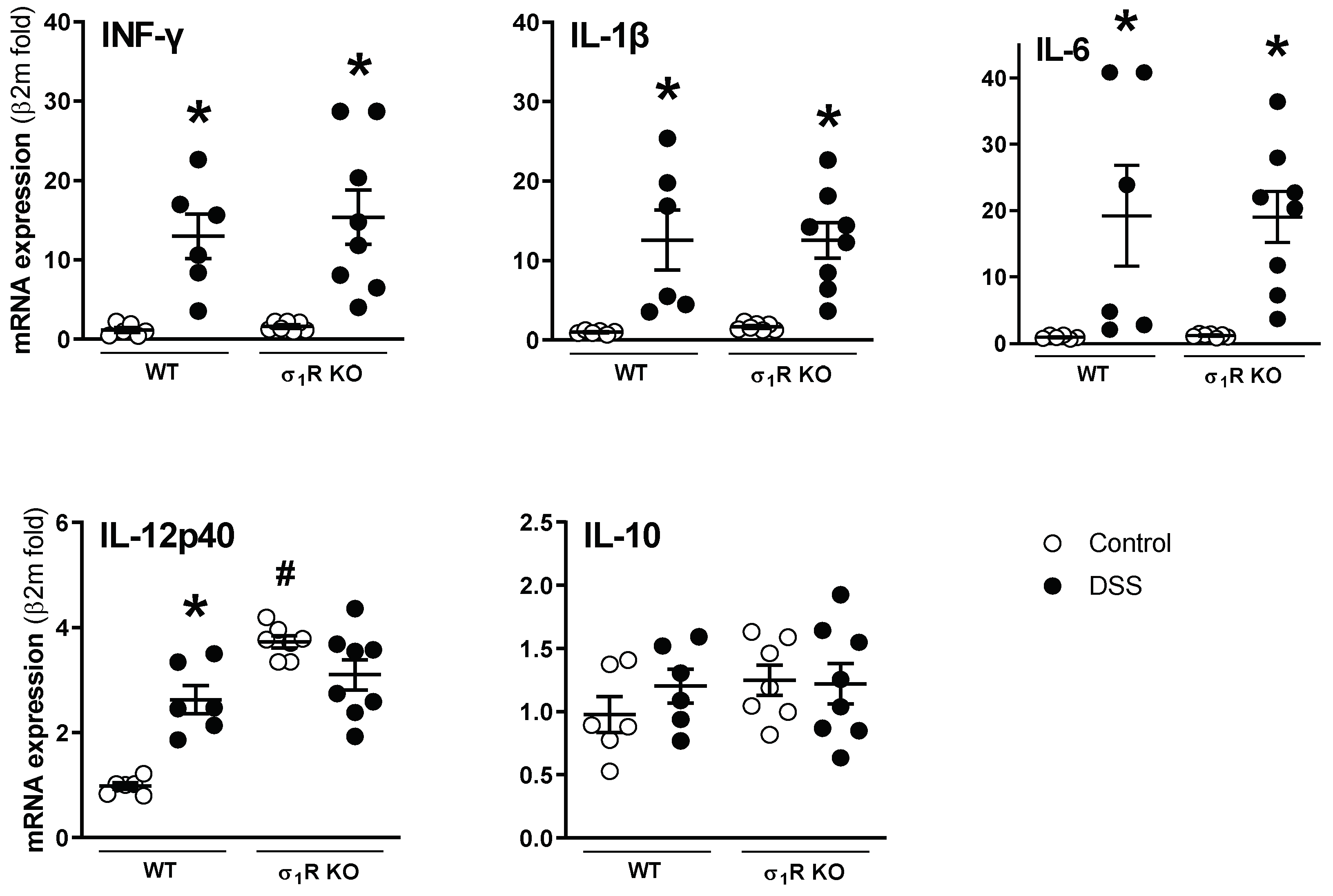
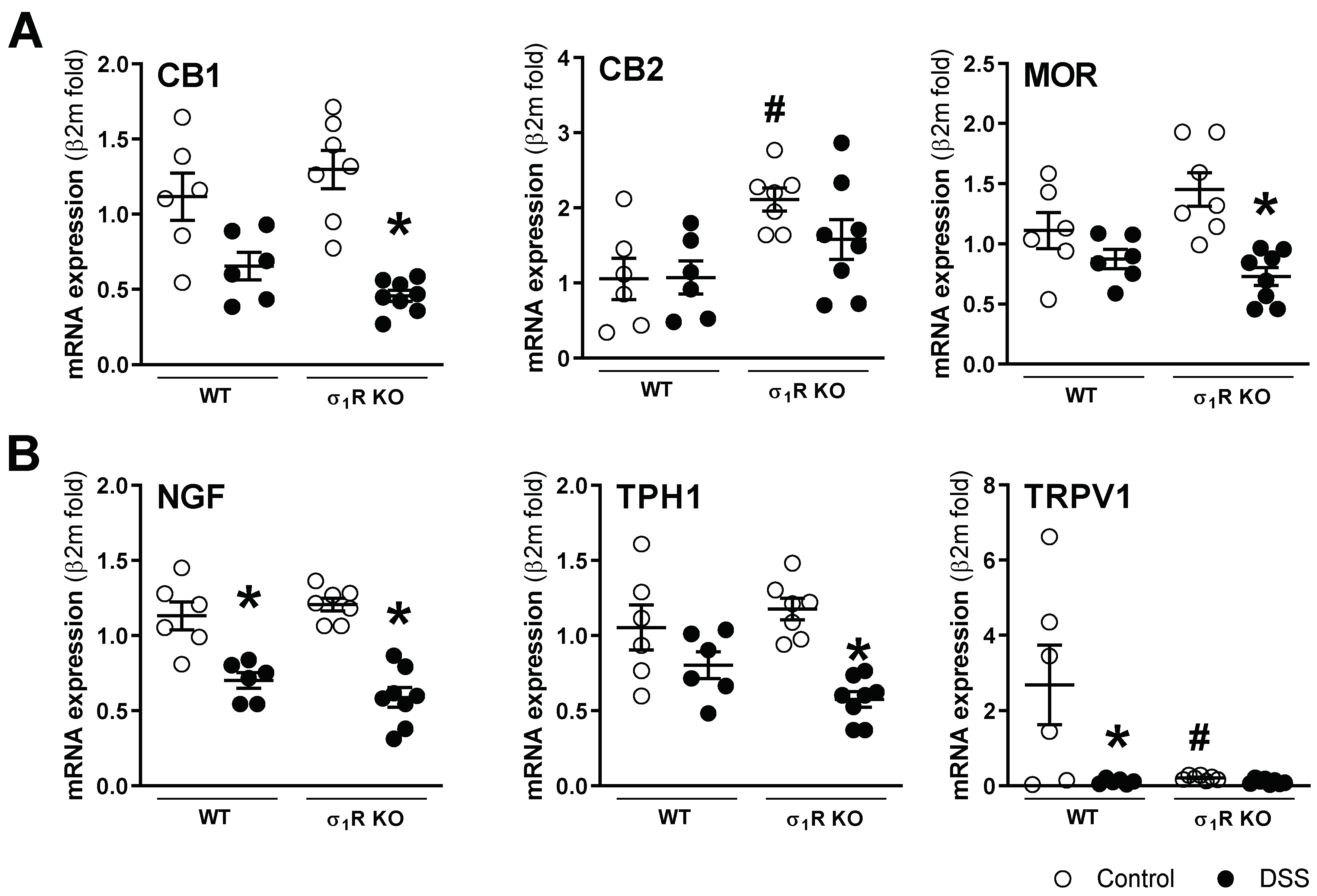
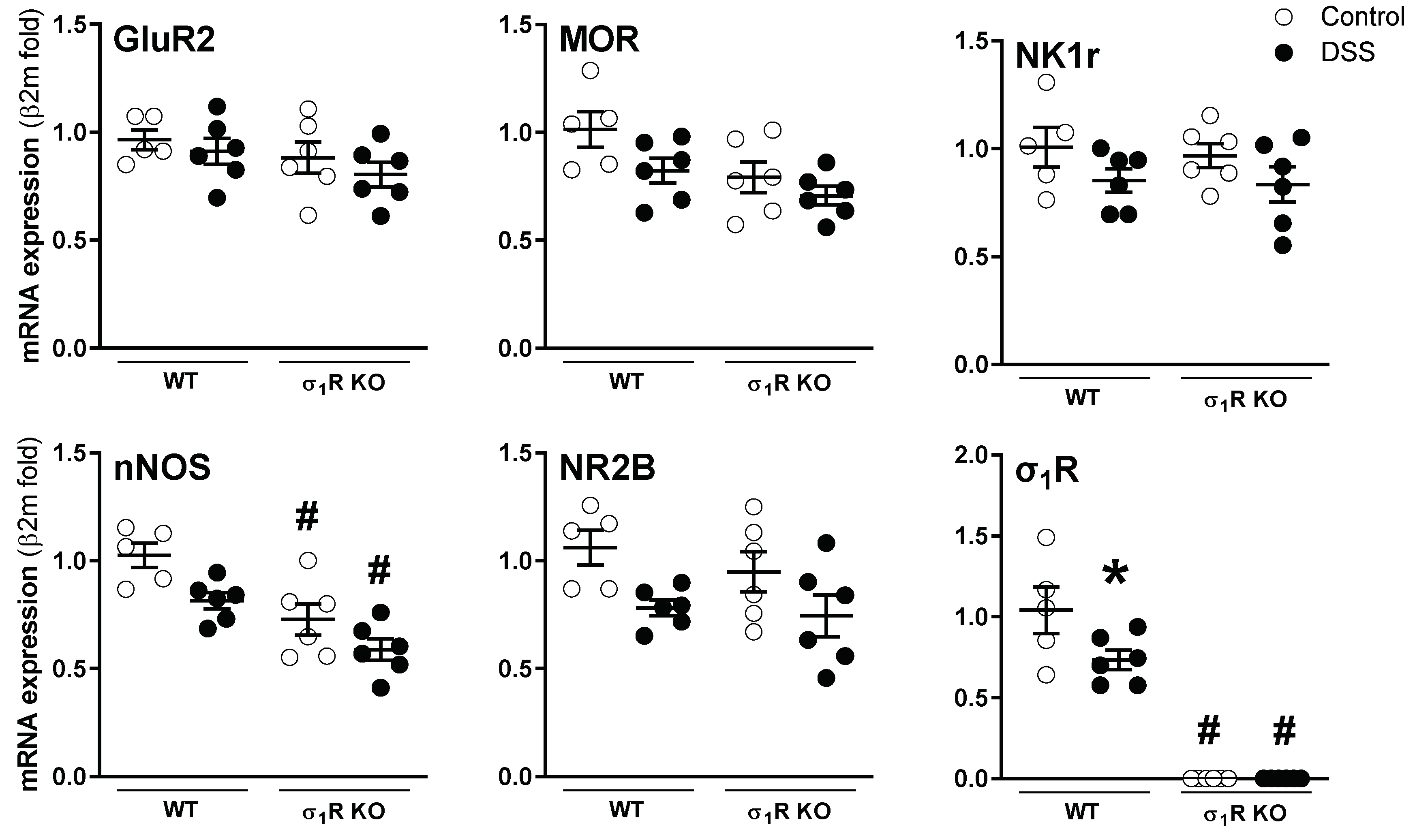
3.1. σ1R KO CD1 Mice Develop an Attenuated Acute Colitis
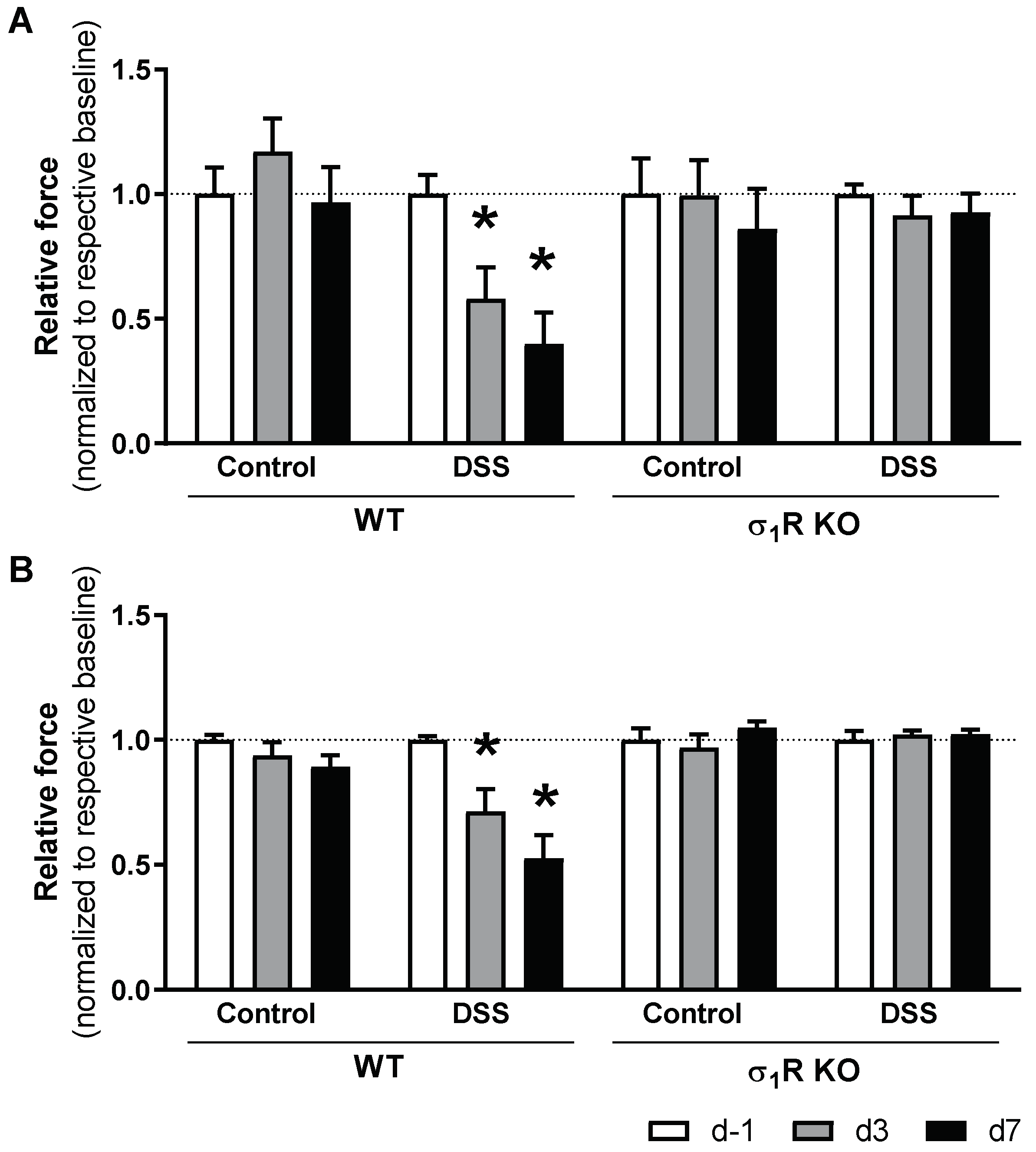
3.2. σ1R KO Mice Do Not Develop Acute Colitis-Associated Hypersensitivity
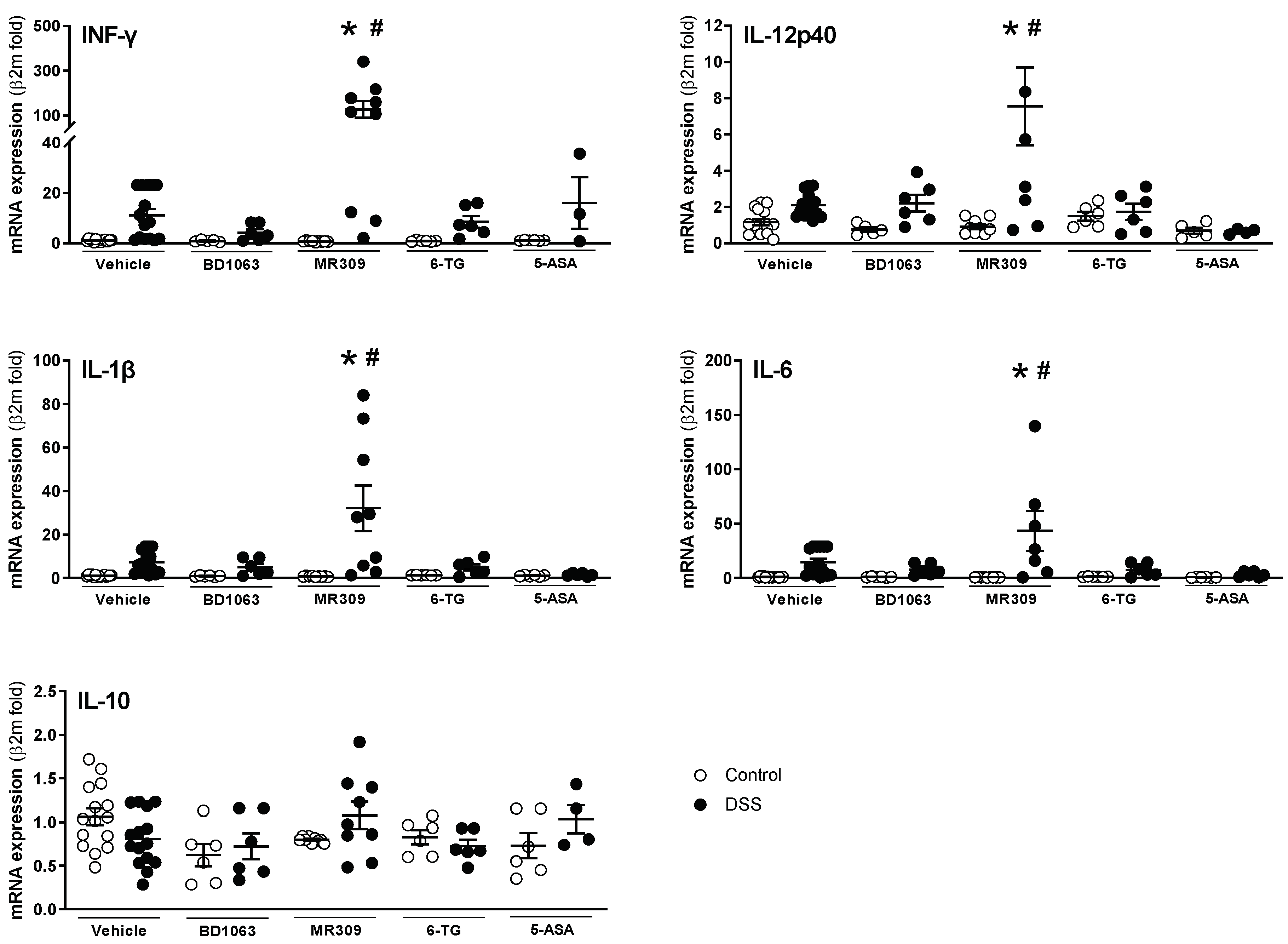
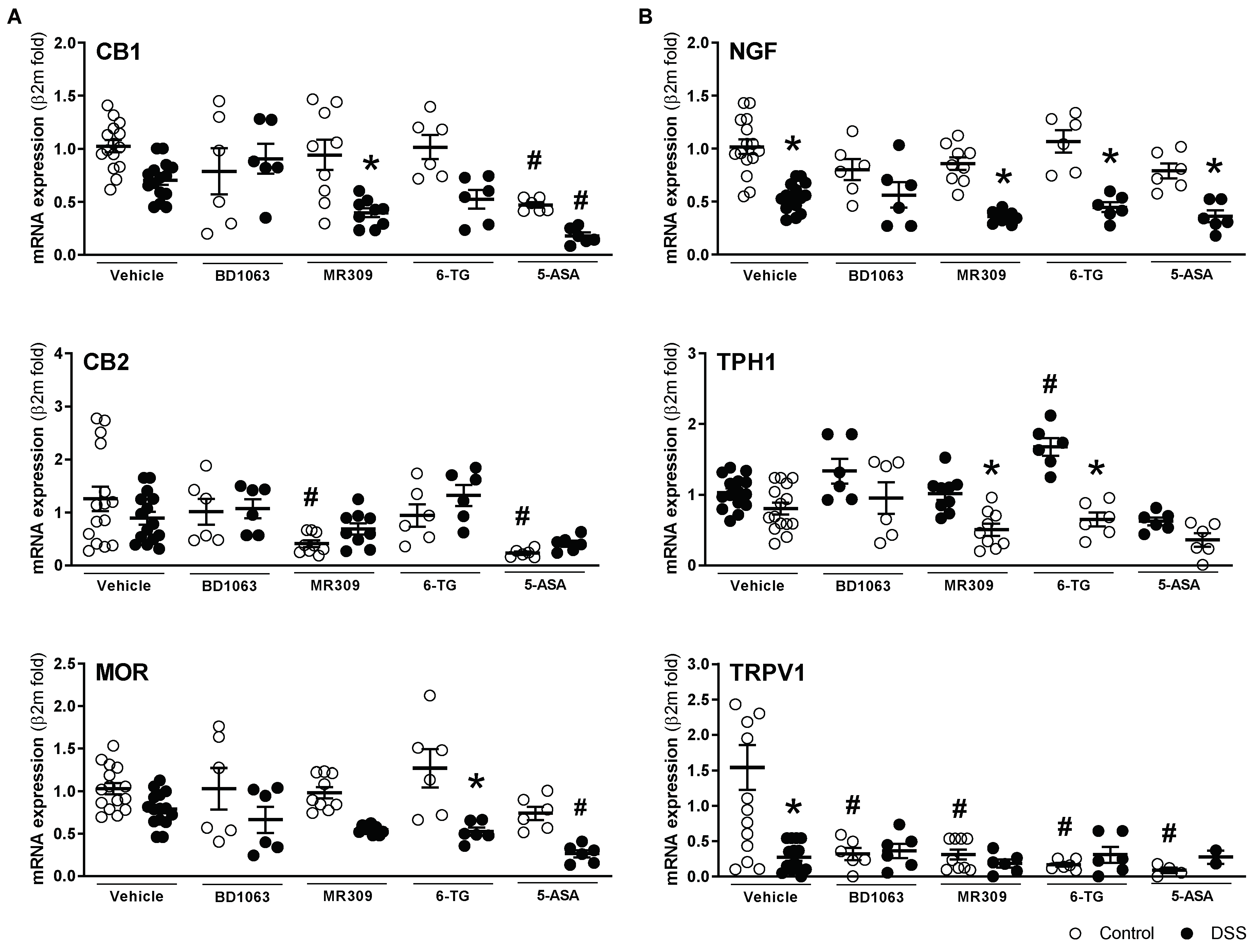
3.3. Antagonism of σ1Rs with BD1063 or E-52862 Attenuates DSS-Induced Colitis
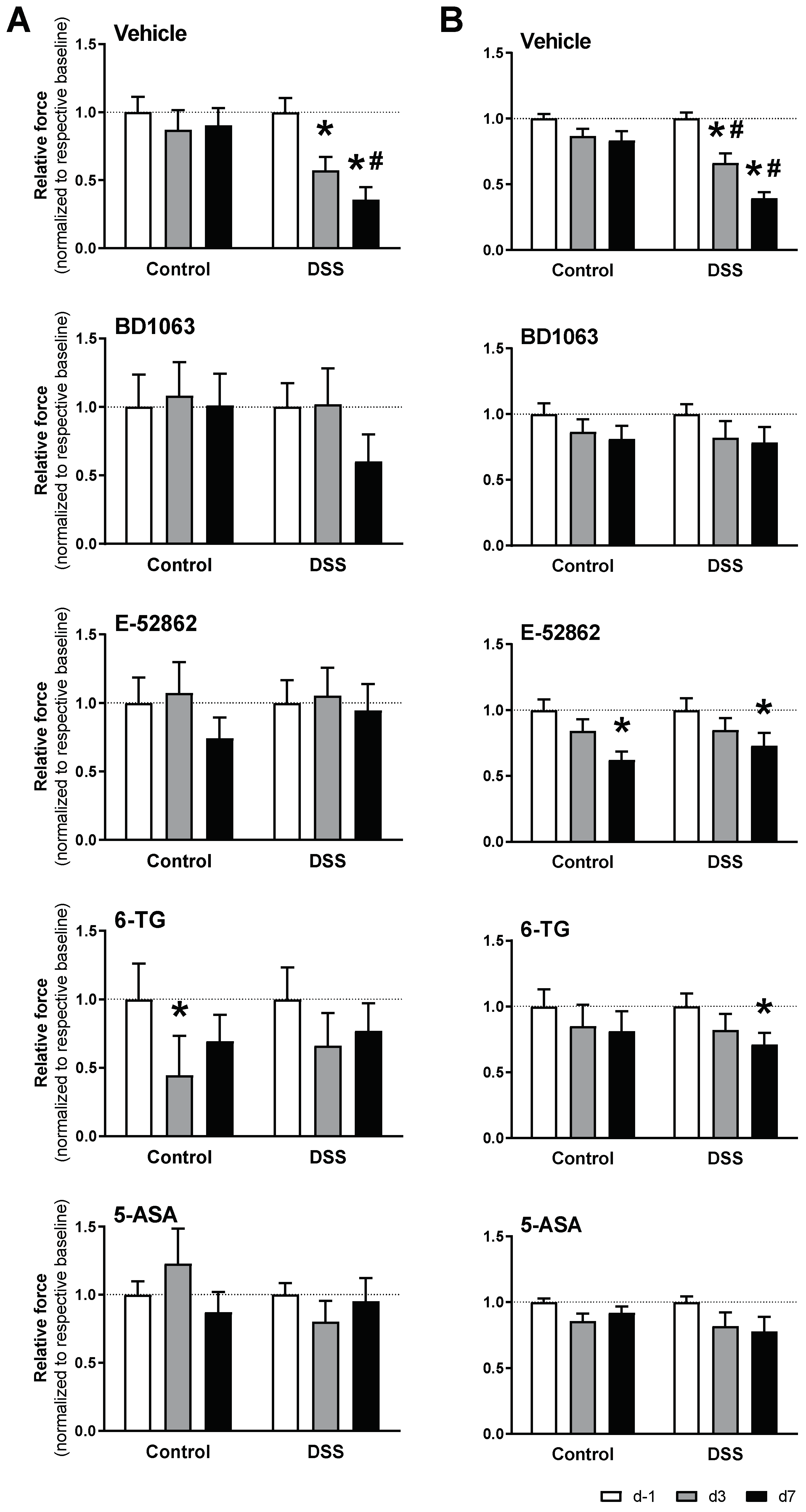
3.4. Antagonism of σ1Rs with BD1063 or E-52862 Attenuated Colitis-Associated Hypersensitivity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bosca-Watts, M.M.; Tosca, J.; Anton, R.; Mora, M.; Minguez, M.; Mora, F. Pathogenesis of Crohn’s Disease: Bug or No Bug. World J. Gastrointest. Pathophysiol. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Sadeghi, M.; Erickson, A.; Castro, J.; Deiteren, A.; Harrington, A.M.; Grundy, L.; Adams, D.J.; Brierley, S.M. Contribution of Membrane Receptor Signalling to Chronic Visceral Pain. Int. J. Biochem. Cell Biol. 2018, 98, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Drewes, A.M.; Olesen, A.E.; Farmer, A.D.; Szigethy, E.; Rebours, V.; Olesen, S.S. Gastrointestinal Pain. Nat. Rev. Dis. Prim. 2020, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, S.; Dickenson, A.H. Visceral Pain—The Ins and Outs, the Ups and Downs. Curr. Opin. Support. Palliat. Care 2012, 6, 17–26. [Google Scholar] [CrossRef]
- Knowles, C.H.; Aziz, Q. Basic and Clinical Aspects of Gastrointestinal Pain. Pain 2009, 141, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, G.F.; Bielefeldt, K. Physiology of Visceral Pain. Compr. Physiol. 2016, 6, 1609–1633. [Google Scholar] [CrossRef]
- Ruiz-Cantero, M.C.; González-Cano, R.; Tejada, M.Á.; Santos-Caballero, M.; Perazzoli, G.; Nieto, F.R.; Cobos, E.J. Sigma-1 Receptor: A Drug Target for the Modulation of Neuroimmune and Neuroglial Interactions during Chronic Pain. Pharmacol. Res. 2021, 163, 105339. [Google Scholar] [CrossRef]
- Almansa, C.; Vela, J.M. Selective Sigma-1 Receptor Antagonists for the Treatment of Pain. Futur. Med. Chem. 2014, 6, 675–695. [Google Scholar] [CrossRef]
- Gris, G.; Cobos, E.J.; Zamanillo, D.; Portillo-Salido, E. Sigma-1 Receptor and Inflammatory Pain. Inflamm. Res. 2015, 64, 377–381. [Google Scholar] [CrossRef]
- Puente, B.; Nadal, X.; Portillo-Salido, E.; Sánchez-Arroyos, R.; Ovalle, S.; Palacios, G.; Muro, A.; Romero, L.; Entrena, J.M.; Baeyens, J.M.; et al. Sigma-1 Receptors Regulate Activity-Induced Spinal Sensitization and Neuropathic Pain after Peripheral Nerve Injury. Pain 2009, 145, 294–303. [Google Scholar] [CrossRef]
- Gris, G.; Merlos, M.; Vela, J.M.; Zamanillo, D.; Portillo-Salido, E. S1RA, a Selective Sigma-1 Receptor Antagonist, Inhibits Inflammatory Pain in the Carrageenan and Complete Freund’s Adjuvant Models in Mice. Behav. Pharmacol. 2014, 25, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Cendán, C.M.; Pujalte, J.M.; Portillo-Salido, E.; Montoliu, L.; Baeyens, J.M. Formalin-Induced Pain Is Reduced in Σ1 Receptor Knockout Mice. Eur. J. Pharmacol. 2005, 511, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Torres, A.; Fernández-Pastor, B.; Carceller, A.; Vela, J.M.; Merlos, M.; Zamanillo, D. Effects of the Selective Sigma-1 Receptor Antagonist S1RA on Formalin-Induced Pain Behavior and Neurotransmitter Release in the Spinal Cord in Rats. J. Neurochem. 2014, 129, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Montilla-García, Á.; Tejada, M.; Carmen Ruiz-Cantero, M.; Bravo-Caparrós, I.; Yeste, S.; Zamanillo, D.; Cobos, E.J. Modulation by Sigma-1 Receptor of Morphine Analgesia and Tolerance: Nociceptive Pain, Tactile Allodynia and Grip Strength Deficits during Joint Inflammation. Front. Pharmacol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tejada, M.A.; Montilla-García, A.; Sánchez-Fernández, C.; Entrena, J.M.; Perazzoli, G.; Baeyens, J.M.; Cobos, E.J. Sigma-1 Receptor Inhibition Reverses Acute Inflammatory Hyperalgesia in Mice: Role of Peripheral Sigma-1 Receptors. Psychopharmacology 2014, 231, 3855–3869. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.; Guadagnoli, N.; McCurdy, C.R. Advances with the Discovery and Development of Novel Sigma 1 Receptor Antagonists for the Management of Pain. Expert Opin. Drug Discov. 2023, 18, 693–705. [Google Scholar] [CrossRef] [PubMed]
- González-Cano, R.; Merlos, M.; Baeyens, J.M.; Cendan, C.M. Σ1 Receptors Are Involved in the Visceral Pain Induced by Intracolonic Administration of Capsaicin in Mice. Anesthesiology 2013, 118, 691–700. [Google Scholar] [CrossRef] [PubMed]
- González-Cano, R.; Artacho-Cordón, A.; Romero, L.; Tejada, M.A.; Nieto, F.R.; Merlos, M.; Cañizares, F.J.; Cendán, C.M.; Fernández-Segura, E.; Baeyens, J.M. Urinary Bladder Sigma-1 Receptors: A New Target for Cystitis Treatment. Pharmacol. Res. 2020, 155, 104724. [Google Scholar] [CrossRef]
- López-Estévez, S.; Gris, G.; De, B.; Carceller, A.; Martínez, V. Intestinal Inflammation-Associated Hypersensitivity Is Attenuated in a DSS Model of Colitis in Sigma-1 Knockout C57BL/6 Mice. Biomed. Pharmacother. 2021, 143, 112126. [Google Scholar] [CrossRef]
- Langa, F.; Codony, X.; Tovar, V.; Lavado, A.; Giménez, E.; Cozar, P.; Cantero, M.; Dordal, A.; Hernández, E.; Pérez, R.; et al. Generation and Phenotypic Analysis of Sigma Receptor Type I (Sigma 1) Knockout Mice. Eur. J. Neurosci. 2003, 18, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.T. Speed Congenics: Applications for Transgenic and Knock-out Mouse Strains. Neuropeptides 2002, 36, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Melgar, S.; Karlsson, A.; Michaëlsson, E. Acute Colitis Induced by Dextran Sulfate Sodium Progresses to Chronicity in C57BL/6 but Not in BALB/c Mice: Correlation between Symptoms and Inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef]
- Melgar, S.; Engström, K.; Jägervall, A.; Martinez, V. Psychological Stress Reactivates Dextran Sulfate Sodium-Induced Chronic Colitis in Mice. Stress 2008, 11, 348–362. [Google Scholar] [CrossRef]
- Matsumoto, R.R.; McCracken, K.A.; Friedman, M.J.; Pouw, B.; De Costa, B.R.; Bowen, W.D. Conformationally Restricted Analogs of BD1008 and an Antisense Oligodeoxynucleotide Targeting Σ1 Receptors Produce Anti-Cocaine Effects in Mice. Eur. J. Pharmacol. 2001, 419, 163–174. [Google Scholar] [CrossRef]
- Díaz, J.L.; Cuberes, R.; Berrocal, J.; Contijoch, M.; Christmann, U.; Fernández, A.; Port, A.; Holenz, J.; Buschmann, H.; Laggner, C.; et al. Synthesis and Biological Evaluation of the 1-Arylpyrazole Class of σ(1) Receptor Antagonists: Identification of 4-{2-[5-Methyl-1-(Naphthalen-2-Yl)-1H-Pyrazol-3-Yloxy]Ethyl}morpholine (S1RA, E-52862). J Med. Chem. 2012, 55, 8211–8224. [Google Scholar] [CrossRef] [PubMed]
- Derijks, L.J.J.; Gilissen, L.P.L.; Hooymans, P.M.; Hommes, D.W. Review Article: Thiopurines in Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2006, 24, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Hauso, Ø.; Martinsen, T.C.; Waldum, H. 5-Aminosalicylic Acid, a Specific Drug for Ulcerative Colitis. Scand. J. Gastroenterol. 2015, 50, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Laird, J.M.A.; Martinez-Caro, L.; Garcia-Nicas, E.; Cervero, F. A New Model of Visceral Pain and Referred Hyperalgesia in the Mouse. Pain 2001, 92, 335–342. [Google Scholar] [CrossRef]
- Chaplan, S.; Bach, F.; Pogrel, J.; Chung, J.; Yaksh, T. Quantitative Assessment of Tactile Allodynia in the Rat Paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Bonin, R.P.; Bories, C.; De Koninck, Y. A Simplified Up-down Method (SUDO) for Measuring Mechanical Nociception in Rodents Using von Frey Filaments. Mol. Pain 2014, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, H.; Wang, C.; Hu, Y.; Wang, S.; Shen, L. Aquaporin 4 Deficiency Alleviates Experimental Colitis in Mice. FASEB J. 2019, 33, 8935–8944. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, C.; Pagano, E.; Lacroix, S.; Venneri, T.; Cristiano, C.; Calignano, A.; Parisi, O.A.; Izzo, A.A.; Di Marzo, V.; Borrelli, F. Fish Oil, Cannabidiol and the Gut Microbiota: An Investigation in a Murine Model of Colitis. Front. Pharmacol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.H.; Rapp, L.; Lindström, E. Effect of DSS-Induced Colitis on Visceral Sensitivity to Colorectal Distension in Mice. Neurogastroenterol. Motil. 2006, 18, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zamanillo, D.; Romero, L.; Merlos, M.; Vela, J.M. Sigma 1 Receptor: A New Therapeutic Target for Pain. Eur. J. Pharmacol. 2013, 716, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.; Merlos, M.; Vela, J.M. Antinociception by Sigma-1 Receptor Antagonists. Central and Peripheral Effects. Adv. Pharmacol. 2016, 75, 181–215. [Google Scholar] [CrossRef]
- Parenti, C.; Marrazzo, A.; Aricò, G.; Cantarella, G.; Prezzavento, O.; Ronsisvalle, S.; Scoto, G.M.; Ronsisvalle, G. Effects of a Selective Sigma 1 Antagonist Compound on Inflammatory Pain. Inflammation 2014, 37, 261–266. [Google Scholar] [CrossRef]
- Parenti, C.; Marrazzo, A.; Aricò, G.; Parenti, R.; Pasquinucci, L.; Ronsisvalle, S.; Ronsisvalle, G.; Scoto, G.M. The Antagonistic Effect of the Sigma 1 Receptor Ligand (+)-MR200 on Persistent Pain Induced by Inflammation. Inflamm. Res. 2014, 63, 231–237. [Google Scholar] [CrossRef]
- Szabo, A.; Kovacs, A.; Frecska, E.; Rajnavolgyi, E. Psychedelic N,N-Dimethyltryptamine and 5-Methoxy-N,N-Dimethyltryptamine Modulate Innate and Adaptive Inflammatory Responses through the Sigma-1 Receptor of Human Monocyte-Derived Dendritic Cells. PLoS ONE 2014, 9, e106533. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, J.A.; Hen, R. The Broken Mouse: The Role of Development, Plasticity and Environment in the Interpretation of Phenotypic Changes in Knockout Mice. Curr. Opin. Neurobiol. 2000, 10, 146–152. [Google Scholar] [CrossRef]
- Costa, R.; Motta, E.M.; Manjavachi, M.N.; Cola, M.; Calixto, J.B. Activation of the Alpha-7 Nicotinic Acetylcholine Receptor (A7 NAchR) Reverses Referred Mechanical Hyperalgesia Induced by Colonic Inflammation in Mice. Neuropharmacology 2012, 63, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B. Introduction to Sigma Receptors: Their Role in Disease and as Therapeutic Targets. In Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology; Smith, S.B., Su, T.-P., Eds.; Springer: Cham, Switzerland, 2017; Volume 964, ISBN 978-3-319-50172-7. [Google Scholar]
- Racz, I.; Nadal, X.; Alferink, J.; Baños, J.E.; Rehnelt, J.; Martín, M.; Pintado, B.; Gutierrez-Adan, A.; Sanguino, E.; Manzanares, J.; et al. Crucial Role of CB2 Cannabinoid Receptor in the Regulation of Central Immune Responses during Neuropathic Pain. J. Neurosci. 2008, 28, 12125–12135. [Google Scholar] [CrossRef]
- Holzer, P. TRPV1 and the Gut: From a Tasty Receptor for a Painful Vanilloid to a Key Player in Hyperalgesia. Eur. J. Pharmacol. 2004, 500, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Phillis, B.D.; Martin, C.M.; Kang, D.; Larsson, H.; Lindström, E.A.; Martinez, V.; Blackshaw, L.A. Role of TRPV1 in High-Threshold Rat Colonic Splanchnic Afferents Is Revealed by Inflammation. Neurosci. Lett. 2009, 459, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Rentería, M.; Juárez-Contreras, R.; González-Ramírez, R.; Islas, L.D.; Sierra-Ramírez, F.; Llorente, I.; Simon, S.A.; Hiriart, M.; Rosenbaum, T.; Morales-Lázaro, S.L. TRPV1 Channels and the Progesterone Receptor Sig-1R Interact to Regulate Pain. Proc. Natl. Acad. Sci. USA 2018, 115, E1657–E1666. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Montero, E.; Sánchez-Blázquez, P.; Onetti, Y.; Merlos, M.; Garzón, J. Ligands Exert Biased Activity to Regulate Sigma 1 Receptor Interactions with Cationic TRPA1, TRPV1, and TRPM8 Channels. Front. Pharmacol. 2019, 10, 634. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Cantero, M.C.; Cortés-Montero, E.; Jain, A.; Montilla-García, Á.; Bravo-Caparrós, I.; Shim, J.; Sánchez-Blázquez, P.; Woolf, C.J.; Baeyens, J.M.; Cobos, E.J. The Sigma-1 Receptor Curtails Endogenous Opioid Analgesia during Sensitization of TRPV1 Nociceptors. Br. J. Pharmacol. 2023, 180, 1148–1167. [Google Scholar] [CrossRef]
- Lapointe, T.K.; Basso, L.; Iftinca, M.C.; Flynn, R.; Chapman, K.; Dietrich, G.; Vergnolle, N.; Altier, C. TRPV1 Sensitization Mediates Postinflammatory Visceral Pain Following Acute Colitis. Am. J. Physiol.—Gastrointest. Liver Physiol. 2015, 309, G87–G99. [Google Scholar] [CrossRef]
- Csekő, K.; Beckers, B.; Keszthelyi, D.; Helyes, Z. Role of TRPV1 and TRPA1 Ion Channels in Inflammatory Bowel Diseases: Potential Therapeutic Targets? Pharmaceuticals 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, M.; Vergara, P.; Martínez, V. Stress and Antibiotics Alter Luminal and Wall-Adhered Microbiota and Enhance the Local Expression of Visceral Sensory-Related Systems in Mice. Neurogastroenterol. Motil. 2013, 25, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, M.; Vergara, P.; Martínez, V. Environment-Related Adaptive Changes of Gut Commensal Microbiota Do Not Alter Colonic Toll-Like Receptors but Modulate the Local Expression of Sensory-Related Systems in Rats. Microb. Ecol. 2013, 66, 232–243. [Google Scholar] [CrossRef]
- Aguilera, M.; Cerdà-Cuéllar, M.; Martínez, V. Antibiotic-Induced Dysbiosis Alters Host-Bacterial Interactions and Leads to Colonic Sensory and Motor Changes in Mice. Gut Microbes 2015, 6, 10–23. [Google Scholar] [CrossRef]
- Choi, S.-R.; Han, H.-J.; Beitz, A.J.; Lee, J.-H. NNOS-PSD95 Interactions Activate the PKC-ε Isoform Leading to Increased GluN1 Phosphorylation and the Development of Neuropathic Mechanical Allodynia in Mice. Neurosci. Lett. 2019, 703, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-R.; Kwon, S.-G.; Choi, H.-S.; Han, H.-J.; Beitz, A.J.; Lee, J.-H. Neuronal NOS Activates Spinal NADPH Oxidase 2 Contributing to Central Sigma-1 Receptor-Induced Pain Hypersensitivity in Mice. Biol. Pharm. Bull. 2016, 39, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.; Díaz, A.F.; Gallardo, N.; Leánez, S.; Balboni, G.; Pol, O. Treatment with the Delta Opioid Agonist UFP-512 Alleviates Chronic Inflammatory and Neuropathic Pain: Mechanisms Implicated. Front. Pharmacol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Son, J.S.; Kwon, Y.B. Sigma-1 Receptor Antagonist BD1047 Reduces Allodynia and Spinal ERK Phosphorylation Following Chronic Compression of Dorsal Root Ganglion in Rats. Korean J. Physiol. Pharmacol. 2010, 14, 359–364. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, J.; Sun, C. MicroRNA-211-5p Enhances Analgesic Effect of Dexmedetomidine on Inflammatory Visceral Pain in Rats by Suppressing ERK Signaling. J. Mol. Neurosci. 2019, 68, 19–28. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Colitis Induction | Treatment | n | |
|---|---|---|---|---|
| Study 1 | Wild-type | No (Tap water) | - | 6 |
| Yes (3% DSS) | - | 6 | ||
| σ1R knockout | No (Tap water) | - | 8 | |
| Yes (3% DSS) | - | 8 | ||
| Study 2 | Wild-type | No (Tap water) | Vehicle (5 mL/kg, po, BID) | 15 |
| BD1063 (20 mg/Kg, po, BID) | 6 | |||
| E-52862 (20 mg/Kg, po, BID) | 9 | |||
| 6-TG (2 mg/Kg, po, SID) | 6 | |||
| 5-ASA (50 mg/Kg, po, BID) | 6 | |||
| Yes (3% DSS) | Vehicle (5 mL/kg, po, BID) | 15 | ||
| BD1063 (20 mg/Kg, po, BID) | 6 | |||
| E-52862 (20 mg/Kg, po, BID) | 9 | |||
| 6-TG (2 mg/Kg, po, SID) | 6 | |||
| 5-ASA (50 mg/Kg, po, BID) | 6 |
| Type | Reactivity | Host | Dilution | Source |
|---|---|---|---|---|
| Primary | β-tubulin | Goat polyclonal | 1:1000 | Santa Cruz Biotech. (#sc-9935) |
| Primary | CaMKII | Mouse monoclonal | 1:2000 | Invitrogen (#MA1-048) |
| Primary | pCaMKII | Mouse monoclonal | 1:1000 | Invitrogen (#MA1-047) |
| Primary | GFAP | Mouse monoclonal | 1:10,000 | Cell Signaling (#3670) |
| Primary | GAPDH | Mouse monoclonal | 1:80,000 | Sigma-Aldrich (#G8795) |
| Primary | GAPDH | Rabbit polyclonal | 1:20,000 | Sigma-Aldrich (#G9545) |
| Primary | tERK | Rabbit polyclonal | 1:30,000 | Sigma-Aldrich (#M5670) |
| Primary | pERK | Mouse monoclonal | 1:1000 | Sigma-Aldrich (#M8159) |
| Primary | p38 | Rabbit polyclonal | 1:1000 | Invitrogen (#AHO1202) |
| Primary | pp38 | Rabbit monoclonal | 1:1000 | Invitrogen (#MA5-15177) |
| Secondary | Anti-Mouse IgG | Goat polyclonal | 1:2000 | Sigma-Aldrich (#A5278) |
| Secondary | Anti-Rabbit IgG | Goat polyclonal | 1:4000 | Sigma-Aldrich (#A9169) |
| Secondary | Anti-Goat IgG | Donkey polyclonal | 1:2000 | Abcam (#ab97110) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Estévez, S.; Aguilera, M.; Gris, G.; de la Puente, B.; Carceller, A.; Martínez, V. Genetic and Pharmacological Blockade of Sigma-1 Receptors Attenuates Inflammation-Associated Hypersensitivity during Acute Colitis in CD1 Mice. Biomedicines 2023, 11, 2758. https://doi.org/10.3390/biomedicines11102758
López-Estévez S, Aguilera M, Gris G, de la Puente B, Carceller A, Martínez V. Genetic and Pharmacological Blockade of Sigma-1 Receptors Attenuates Inflammation-Associated Hypersensitivity during Acute Colitis in CD1 Mice. Biomedicines. 2023; 11(10):2758. https://doi.org/10.3390/biomedicines11102758
Chicago/Turabian StyleLópez-Estévez, Sergio, Mònica Aguilera, Georgia Gris, Beatriz de la Puente, Alicia Carceller, and Vicente Martínez. 2023. "Genetic and Pharmacological Blockade of Sigma-1 Receptors Attenuates Inflammation-Associated Hypersensitivity during Acute Colitis in CD1 Mice" Biomedicines 11, no. 10: 2758. https://doi.org/10.3390/biomedicines11102758