
Brugada Syndrome: More than a Monogenic Channelopathy
 , , , , and
, , , , and
Abstract
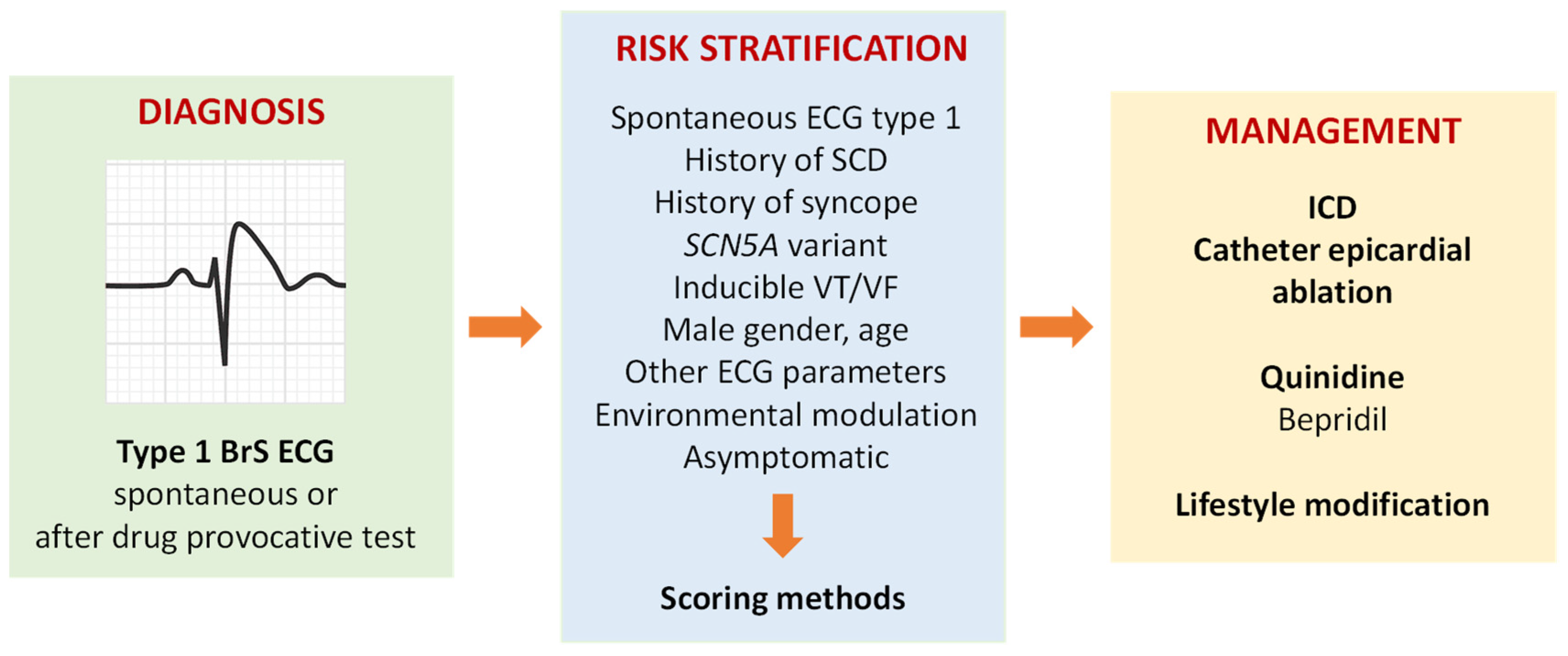
:1. Clinical Diagnosis and Risk Stratification
2. Management
3. Genetics and Molecular Mechanisms
3.1. Oligogenic Disease with Incomplete Penetrance and Expressivity
3.2. SCN5A Variants: Genotype–Phenotype Correlation
3.3. Variants in Genes Encoding for Other Ion Channels and Regulatory Proteins
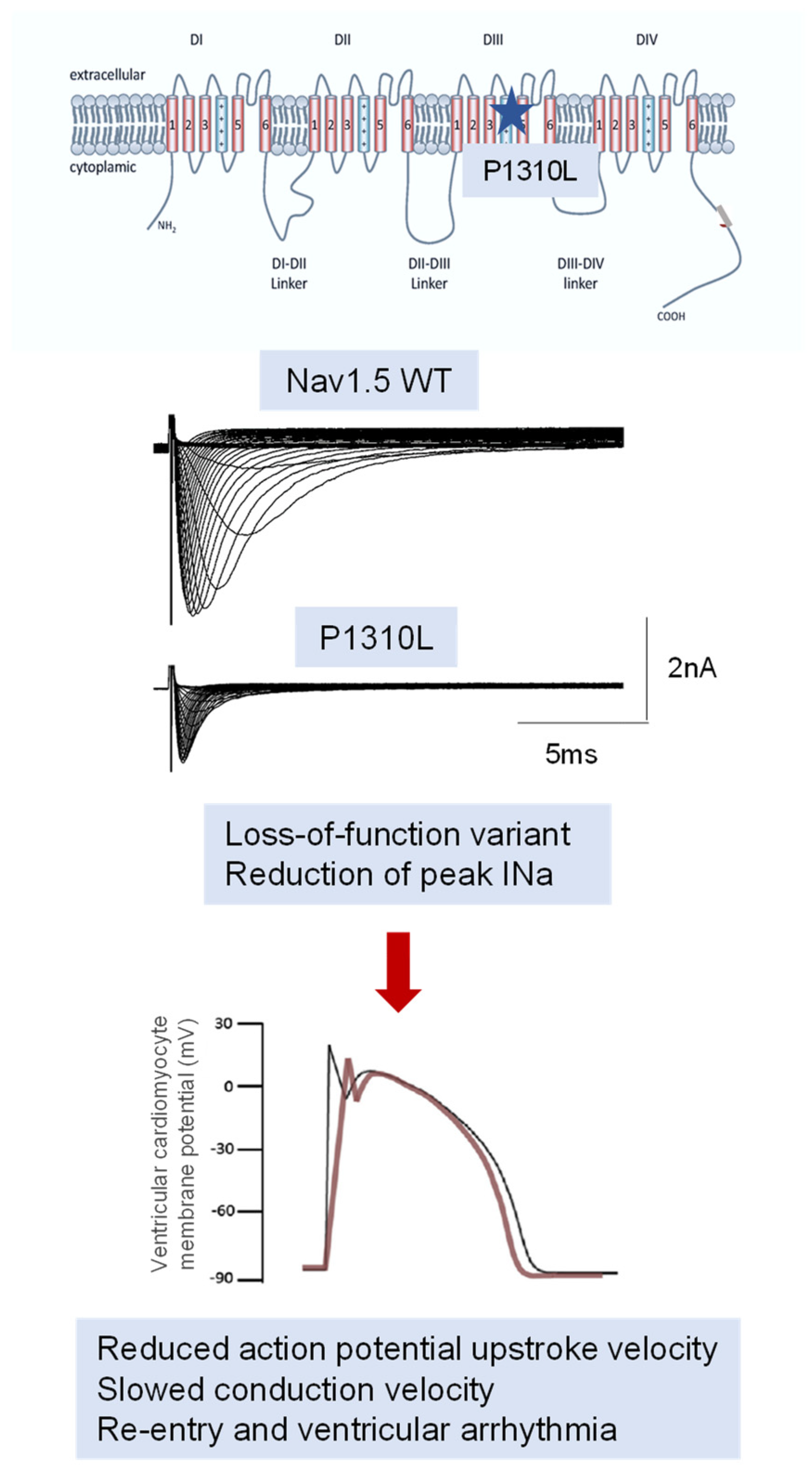
4. Pathophysiological Mechanisms
4.1. Preclinical Models
4.2. Pure Channelopathy or Concealed Cardiomyopathy: The Growing Role of SCN5A
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Elias, A.; Benito, B. Ion channel disorders and sudden cardiac death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Offerhaus, J.A.; Bezzina, C.R.; Wilde, A.A.M. Epidemiology of inherited arrhythmias. Nat. Rev. Cardiol. 2020, 17, 205–215. [Google Scholar] [CrossRef]
- Postema, P.G.; Wilde, A.A.M. Aging in Brugada Syndrome: What about risks? JACC Clin. Electrophysiol. 2017, 3, 68–70. [Google Scholar] [CrossRef]
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender differences in clinical manifestations of Brugada syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573. [Google Scholar] [CrossRef]
- Argenziano, M.; Tiscornia, G.; Moretta, R.; Casal, L.; Potilinski, C.; Amorena, C.; Gras, E.G. Arrhythmogenic effect of androgens on the rat heart. J. Physiol. Sci. 2017, 67, 217–225. [Google Scholar] [CrossRef]
- Mazzanti, A.; Underwood, K.; Nevelev, D.; Kofman, S.; Priori, S.G. The new kids on the block of arrhythmogenic disorders: Short QT syndrome and early repolarization. J. Cardiovasc. Electrophysiol. 2017, 28, 1226–1236. [Google Scholar] [CrossRef]
- Lieve, K.V.; Wilde, A.A. Inherited ion channel diseases: A brief review. Europace 2015, 17 (Suppl. 2), ii1–ii6. [Google Scholar] [CrossRef]
- Abdelsayed, M.; Peters, C.H.; Ruben, P.C. Differential thermosensitivity in mixed syndrome cardiac sodium channel mutants. J. Physiol. 2015, 593, 4201–4223. [Google Scholar] [CrossRef]
- Sarica, A.S.; Bor, S.; Orman, M.N.; Barajas-Martinez, H.; Juang, J.J.; Antzelevitch, C.; Hasdemir, C. Frequency of irritable bowel syndrome in patients with Brugada syndrome and drug-induced type 1 Brugada pattern. Am. J. Cardiol. 2021, 151, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Sieira, J.; Dendramis, G.; Brugada, P. Pathogenesis and management of Brugada syndrome. Nat. Rev. Cardiol. 2016, 13, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Patocskai, B. Brugada syndrome: Clinical, genetic, molecular, cellular, and ionic aspects. Curr. Probl. Cardiol. 2016, 41, 7–57. [Google Scholar] [CrossRef] [PubMed]
- Honarbakhsh, S.; Providencia, R.; Garcia-Hernandez, J.; Martin, C.A.; Hunter, R.J.; Lim, W.Y.; Kirkby, C.; Graham, A.J.; Sharifzadehgan, A.; Waldmann, V.; et al. A primary prevention clinical risk score model for patients with Brugada syndrome (BRUGADA-RISK). JACC Clin. Electrophysiol. 2021, 7, 210–222. [Google Scholar] [CrossRef]
- Honarbakhsh, S.; Providencia, R.; Lambiase, P.D. Risk stratification in Brugada syndrome: Current status and emerging approaches. Arrhythm. Electrophysiol. Rev. 2018, 7, 79. [Google Scholar] [CrossRef]
- Probst, V.; Goronflot, T.; Anys, S.; Tixier, R.; Briand, J.; Berthome, P.; Geoffroy, O.; Clementy, N.; Mansourati, J.; Jesel, L.; et al. Robustness and relevance of predictive score in sudden cardiac death for patients with Brugada syndrome. Eur. Heart J. 2021, 42, 1687–1695. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; Wilde, A.A.M. Channelopathies as causes of sudden cardiac death. Card. Electrophysiol. Clin. 2017, 9, 537–549. [Google Scholar] [CrossRef]
- Signore, F.; Simone, V.; Anaclerio, M.; Bozza, N.; Marulli, G.; De Palma, A. Successful uniportal thoracoscopic removal of a new generation implantable loop recorder accidentally migrated into the left pleural cavity and concomitant re-implantation: A case report. Int. J. Surg. Case Rep. 2023, 105, 108012. [Google Scholar] [CrossRef]
- Ramalho, D.; Freitas, J. Drug-induced life-threatening arrhythmias and sudden cardiac death: A clinical perspective of long QT, short QT and Brugada syndromes. Rev. Port. Cardiol. (Engl. Ed.) 2018, 37, 435–446. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S. Clinical spectrum of SCN5A mutations: Long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 5569–5579. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef]
- Remme, C.A.; Wilde, A.A.; Bezzina, C.R. Cardiac sodium channel overlap syndromes: Different faces of SCN5A mutations. Trends Cardiovasc. Med. 2008, 18, 78–87. [Google Scholar] [CrossRef]
- Fonseca, D.J.; Vaz da Silva, M.J. Cardiac channelopathies: The role of sodium channel mutations. Rev. Port. Cardiol. (Engl. Ed.) 2018, 37, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Venkatesan, C.; Abdelhalim, H.; Zeeshan, S.; Arima, Y.; Linna-Kuosmanen, S.; Ahmed, Z. Genomic approaches to identify and investigate genes associated with atrial fibrillation and heart failure susceptibility. Hum. Genom. 2023, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Thompson, B.A.; Perrin, M.; James, P.; Zentner, D.; Kalman, J.M.; Vandenberg, J.I.; Fatkin, D. Arrhythmic phenotypes are a defining feature of dilated cardiomyopathy-associated SCN5A variants: A systematic review. Circ. Genom. Precis. Med. 2022, 15, e003432. [Google Scholar] [CrossRef]
- Kyndt, F.; Probst, V.; Potet, F.; Demolombe, S.; Chevallier, J.C.; Baro, I.; Moisan, J.P.; Boisseau, P.; Schott, J.J.; Escande, D.; et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 2001, 104, 3081–3086. [Google Scholar] [CrossRef]
- Probst, V.; Wilde, A.A.; Barc, J.; Sacher, F.; Babuty, D.; Mabo, P.; Mansourati, J.; Le Scouarnec, S.; Kyndt, F.; Le Caignec, C.; et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet. 2009, 2, 552–557. [Google Scholar] [CrossRef]
- Balla, C.; Conte, E.; Selvatici, R.; Marsano, R.M.; Gerbino, A.; Farnè, M.; Blunck, R.; Vitali, F.; Armaroli, A.; Brieda, A.; et al. Functional characterization of two novel mutations in SCN5A associated with Brugada syndrome identified in Italian patients. Int. J. Mol. Sci. 2021, 22, 6513. [Google Scholar] [CrossRef]
- Balla, C.; Mele, D.; Vitali, F.; Andreoli, C.; Tonet, E.; Sanchini, M.; Ferlini, A.; Rapezzi, C.; Gualandi, F.; Bertini, M. Novel SCN5A variant shows multiple phenotypic expression in the same family. Circ. Genom. Precis. Med. 2021, 14, e003481. [Google Scholar] [CrossRef]
- Béziau, D.M.; Barc, J.; O’Hara, T.; Le Gloan, L.; Amarouch, M.Y.; Solnon, A.; Pavin, D.; Lecointe, S.; Bouillet, P.; Gourraud, J.B.; et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res. Cardiol. 2014, 109, 446. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada syndrome: Oligogenic or mendelian disease? Int. J. Mol. Sci. 2020, 21, 1687. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present status of Brugada syndrome: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Barc, J.; Tadros, R.; Glinge, C.; Chiang, D.Y.; Jouni, M.; Simonet, F.; Jurgens, S.J.; Baudic, M.; Nicastro, M.; Potet, F.; et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat. Genet. 2022, 54, 232–239. [Google Scholar] [CrossRef]
- Martínez-Campelo, L.; Cruz, R.; Blanco-Verea, A.; Moscoso, I.; Ramos-Luis, E.; Lage, R.; Álvarez-Barredo, M.; Sabater-Molina, M.; Peñafiel-Verdú, P.; Jiménez-Jáimez, J.; et al. Searching for genetic modulators of the phenotypic heterogeneity in Brugada syndrome. PLoS ONE 2022, 17, e0263469. [Google Scholar] [CrossRef]
- Jimenez-Vazquez, E.N.; Arad, M.; Macías, Á.; Vera-Pedrosa, M.L.; Cruz, F.M.; Gutierrez, L.K.; Cuttita, A.J.; Monteiro da Rocha, A.; Herron, T.J.; Ponce-Balbuena, D.; et al. SNTA1 gene rescues ion channel function and is antiarrhythmic in cardiomyocytes derived from induced pluripotent stem cells from muscular dystrophy patients. eLife 2022, 11, e76576. [Google Scholar] [CrossRef]
- Makara, M.A.; Curran, J.; Little, S.C.; Musa, H.; Polina, I.; Smith, S.A.; Wright, P.J.; Unudurthi, S.D.; Snyder, J.; Bennett, V.; et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 2014, 115, 929–938. [Google Scholar] [CrossRef]
- Wang, L.; Meng, X.; Yuchi, Z.; Zhao, Z.; Xu, D.; Fedida, D.; Wang, Z.; Huang, C. De novo mutation in the SCN5A gene associated with Brugada syndrome. Cell. Physiol. Biochem. 2015, 36, 2250–2262. [Google Scholar] [CrossRef]
- Gando, I.; Morganstein, J.; Jana, K.; McDonald, T.V.; Tang, Y.; Coetzee, W.A. Infant sudden death: Mutations responsible for impaired Nav1.5 channel trafficking and function. Pacing Clin. Electrophysiol. 2017, 40, 703–712. [Google Scholar] [CrossRef]
- Chen, J.; Li, H.; Guo, S.; Yang, Z.; Sun, S.; Zeng, J.; Gou, H.; Chen, Y.; Wang, F.; Lin, Y.; et al. Whole exome sequencing in Brugada and long QT syndromes revealed novel rare and potential pathogenic mutations related to the dysfunction of the cardiac sodium channel. Orphanet J. Rare Dis. 2022, 17, 394. [Google Scholar] [CrossRef] [PubMed]
- Simons, E.; Nijak, A.; Loeys, B.; Alaerts, M. Generation of two induced pluripotent stem cell (iPSC) lines (BBANTWi006-A, BBANTWi007-A) from Brugada syndrome patients carrying an SCN5A mutation. Stem Cell Res. 2022, 60, 102719. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, K.; Ohno, S.; Ozawa, J.; Hayano, M.; Hattori, T.; Kobori, A.; Yahata, M.; Aburadani, I.; Watanabe, S.; Matsumoto, Y.; et al. Copy number variations of SCN5A in Brugada syndrome. Heart Rhythm 2018, 15, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Nishii, N.; Ogawa, M.; Morita, H.; Nakamura, K.; Banba, K.; Miura, D.; Kumagai, N.; Matsunaga, A.; Kawamura, H.; Urakawa, S.; et al. SCN5A mutation is associated with early and frequent recurrence of ventricular fibrillation in patients with Brugada syndrome. Circ. J. 2010, 74, 2572–2578. [Google Scholar] [CrossRef]
- Rudic, B.; Schimpf, R.; Veltmann, C.; Doesch, C.; Tülümen, E.; Schoenberg, S.O.; Borggrefe, M.; Papavassiliu, T. Brugada syndrome: Clinical presentation and genotype-correlation with magnetic resonance imaging parameters. Europace 2016, 18, 1411–1419. [Google Scholar] [CrossRef]
- Yamagata, K.; Horie, M.; Aiba, T.; Ogawa, S.; Aizawa, Y.; Ohe, T.; Yamagishi, M.; Makita, N.; Sakurada, H.; Tanaka, T.; et al. Genotype-phenotype correlation of SCN5A mutation for the clinical and electrocardiographic characteristics of probands with Brugada syndrome: A Japanese multicenter registry. Circulation 2017, 135, 2255–2270. [Google Scholar] [CrossRef]
- Sommariva, E.; Pappone, C.; Martinelli Boneschi, F.; Di Resta, C.; Carbone, R.M.; Salvi, E.; Vergara, P.; Sala, S.; Cusi, D.; Ferrari, M.; et al. Genetics can contribute to the prognosis of Brugada syndrome: A pilot model for risk stratification. Eur. J. Hum. Genet. 2013, 21, 911–917. [Google Scholar] [CrossRef]
- Meregalli, P.G.; Tan, H.L.; Probst, V.; Koopmann, T.T.; Tanck, M.W.; Bhuiyan, Z.A.; Sacher, F.; Kyndt, F.; Schott, J.J.; Albuisson, J.; et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009, 6, 341–348. [Google Scholar] [CrossRef]
- Pfahnl, A.E.; Viswanathan, P.C.; Weiss, R.; Shang, L.L.; Sanyal, S.; Shusterman, V.; Kornblit, C.; London, B.; Dudley, S.C., Jr. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm 2007, 4, 46–53. [Google Scholar] [CrossRef]
- Itoh, H.; Shimizu, M.; Takata, S.; Mabuchi, H.; Imoto, K. A novel missense mutation in the SCN5A gene associated with Brugada syndrome bidirectionally affecting blocking actions of antiarrhythmic drugs. J. Cardiovasc. Electrophysiol. 2005, 16, 486–493. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Muhammad, A.; Li, B.; Wada, Y.; Hall, L.; Solus, J.F.; Short, L.; Roden, D.M.; Glazer, A.M. Dominant negative effects of SCN5A missense variants. Genet. Med. 2022, 24, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Veltmann, C.; Eckardt, L.; Meregalli, P.G.; Gaita, F.; Tan, H.L.; Babuty, D.; Sacher, F.; Giustetto, C.; Schulze-Bahr, E.; et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada syndrome registry. Circulation 2010, 121, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef] [PubMed]
- Gualandi, F.; Zaraket, F.; Malagù, M.; Parmeggiani, G.; Trabanelli, C.; Fini, S.; Dang, X.; Wei, X.; Fang, M.; Bertini, M.; et al. Mutation load of multiple ion channel gene mutations in Brugada syndrome. Cardiology 2017, 137, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sallé, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.; Rosso, R.; Chorin, E.; Havakuk, O.; Antzelevitch, C.; Viskin, S. Risk stratification in Brugada syndrome: Clinical characteristics, electrocardiographic parameters, and auxiliary testing. Heart Rhythm 2016, 13, 299–310. [Google Scholar] [CrossRef]
- Clatot, J.; Neyroud, N.; Cox, R.; Souil, C.; Huang, J.; Guicheney, P.; Antzelevitch, C. Inter-regulation of Kv4.3 and voltage-gated sodium channels underlies predisposition to cardiac and neuronal channelopathies. Int. J. Mol. Sci. 2020, 21, 5057. [Google Scholar] [CrossRef]
- Ozhathil, L.C.; Rougier, J.S.; Arullampalam, P.; Essers, M.C.; Ross-Kaschitza, D.; Abriel, H. Deletion of Trpm4 alters the function of the Nav1.5 channel in murine cardiac myocytes. Int. J. Mol. Sci. 2021, 22, 3401. [Google Scholar] [CrossRef]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Díez-Guerra, F.J.; Jiménez-Vázquez, E.N.; Ramirez, R.J.; Monteiro da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and NaV1.5 channels traffic together to the sarcolemma to control excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Marchal, G.A.; Wolswinkel, R.; Jansen, J.A.; van der Made, I.; Beekman, L.; Ruiz-Villalba, A.; Baartscheer, A.; Rajamani, S.; Belardinelli, L.; et al. Functional modulation of atrio-ventricular conduction by enhanced late sodium current and calcium-dependent mechanisms in Scn5a1798insD/+ mice. Europace 2020, 22, 1579–1589. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G. Is it time to include ion channel diseases among cardiomyopathies? J. Electrocardiol. 2005, 38, 81–87. [Google Scholar] [CrossRef]
- Ishikawa, T.; Takahashi, N.; Ohno, S.; Sakurada, H.; Nakamura, K.; On, Y.K.; Park, J.E.; Makiyama, T.; Horie, M.; Arimura, T.; et al. Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ. J. 2013, 77, 959–967. [Google Scholar] [CrossRef]
- Angsutararux, P.; Zhu, W.; Voelker, T.L.; Silva, J.R. Molecular pathology of sodium channel beta-subunit variants. Front. Pharmacol. 2021, 12, 761275. [Google Scholar] [CrossRef] [PubMed]
- Salvage, S.C.; Jeevaratnam, K.; Huang, C.L.; Jackson, A.P. Cardiac sodium channel complexes and arrhythmia: Structural and functional roles of the β1 and β3 subunits. J. Physiol. 2023, 601, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Micaglio, E.; Vicedomini, G.; Locati, E.T.; Ciconte, G.; Giannelli, L.; Giordano, F.; Crisà, S.; Vecchi, M.; Borrelli, V.; et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace 2019, 21, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, X.M.; Barajas-Martinez, H.; Jiang, H.; Antzelevitch, C.; Hu, D. Common variants in SCN10A gene associated with Brugada syndrome. Hum. Mol. Genet. 2021, 31, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef]
- van den Boogaard, M.; Smemo, S.; Burnicka-Turek, O.; Arnolds, D.E.; van de Werken, H.J.; Klous, P.; McKean, D.; Muehlschlegel, J.D.; Moosmann, J.; Toka, O.; et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Investig. 2014, 124, 1844–1852. [Google Scholar] [CrossRef]
- Novelli, V.; Memmi, M.; Malovini, A.; Mazzanti, A.; Liu, N.; Yanfei, R.; Bongianino, R.; Denegri, M.; Monteforte, N.; Bloise, R.; et al. Role of CACNA1C in Brugada syndrome: Prevalence and phenotype of probands referred for genetic testing. Heart Rhythm 2022, 19, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Adler, A.; Green, S.; Al-Zoughool, F.; Doroshenko, P.; Orr, N.; Uppal, S.; Healey, J.S.; Birnie, D.; Sanatani, S.; et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ. Cardiovasc. Genet. 2014, 7, 782–789. [Google Scholar] [CrossRef]
- Nakajima, T.; Wu, J.; Kaneko, Y.; Ashihara, T.; Ohno, S.; Irie, T.; Ding, W.G.; Matsuura, H.; Kurabayashi, M.; Horie, M. KCNE3 T4A as the genetic basis of Brugada-pattern electrocardiogram. Circ. J. 2012, 76, 2763–2772. [Google Scholar] [CrossRef] [PubMed]
- Portero, V.; Le Scouarnec, S.; Es-Salah-Lamoureux, Z.; Burel, S.; Gourraud, J.B.; Bonnaud, S.; Lindenbaum, P.; Simonet, F.; Violleau, J.; Baron, E.; et al. Dysfunction of the voltage-gated K+ channel β2 subunit in a familial case of Brugada syndrome. J. Am. Heart Assoc. 2016, 5, e003122. [Google Scholar] [CrossRef]
- Abbott, G.W. KCNE4 and KCNE5: K+ channel regulation and cardiac arrhythmogenesis. Gene 2016, 593, 249–260. [Google Scholar] [CrossRef]
- Biel, S.; Aquila, M.; Hertel, B.; Berthold, A.; Neumann, T.; DiFrancesco, D.; Moroni, A.; Thiel, G.; Kauferstein, S. Mutation in S6 domain of HCN4 channel in patient with suspected Brugada syndrome modifies channel function. Pflug. Arch. 2016, 468, 1663–1671. [Google Scholar] [CrossRef]
- Auricchio, A.; Demarchi, A.; Özkartal, T.; Campanale, D.; Caputo, M.L.; di Valentino, M.; Menafoglio, A.; Regoli, F.; Facchini, M.; Del Bufalo, A.; et al. Role of genetic testing in young patients with idiopathic atrioventricular conduction disease. Europace 2023, 25, 643–650. [Google Scholar] [CrossRef]
- Wang, C.; Hennessey, J.A.; Kirkton, R.D.; Wang, C.; Graham, V.; Puranam, R.S.; Rosenberg, P.B.; Bursac, N.; Pitt, G.S. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 2011, 109, 775–782. [Google Scholar] [CrossRef]
- Hennessey, J.A.; Marcou, C.A.; Wang, C.; Wei, E.Q.; Wang, C.; Tester, D.J.; Torchio, M.; Dagradi, F.; Crotti, L.; Schwartz, P.J.; et al. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013, 10, 1886–1894. [Google Scholar] [CrossRef]
- London, B.; Michalec, M.; Mehdi, H.; Zhu, X.; Kerchner, L.; Sanyal, S.; Viswanathan, P.C.; Pfahnl, A.E.; Shang, L.L.; Madhusudanan, M.; et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007, 116, 2260–2268. [Google Scholar] [CrossRef]
- Ishikawa, T.; Sato, A.; Marcou, C.A.; Tester, D.J.; Ackerman, M.J.; Crotti, L.; Schwartz, P.J.; On, Y.K.; Park, J.E.; Nakamura, K.; et al. A novel disease gene for Brugada syndrome: Sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. MOG1: A new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef]
- Xiong, H.; Bai, X.; Quan, Z.; Yu, D.; Zhang, H.; Zhang, C.; Liang, L.; Yao, Y.; Yang, Q.; Wang, Z.; et al. Mechanistic insights into the interaction of cardiac sodium channel Na(v)1.5 with MOG1 and a new molecular mechanism for Brugada syndrome. Heart Rhythm 2022, 19, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Juang, J.J.; Binda, A.; Lee, S.J.; Hwang, J.J.; Chen, W.J.; Liu, Y.B.; Lin, L.Y.; Yu, C.C.; Ho, L.T.; Huang, H.C.; et al. GSTM3 variant is a novel genetic modifier in Brugada syndrome, a disease with risk of sudden cardiac death. EBioMedicine 2020, 57, 102843. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Alarcón, M.; Cámara-Checa, A.; Dago, M.; Crespo-García, T.; Nieto-Marín, P.; Marín, M.; Merino, J.L.; Toquero, J.; Salguero-Bodes, R.; Tamargo, J.; et al. Zfhx3 transcription factor represses the expression of SCN5A gene and decreases sodium current density (INa). Int. J. Mol. Sci. 2021, 22, 13031. [Google Scholar] [CrossRef]
- Bersell, K.R.; Yang, T.; Mosley, J.D.; Glazer, A.M.; Hale, A.T.; Kryshtal, D.O.; Kim, K.; Steimle, J.D.; Brown, J.D.; Salem, J.E.; et al. Transcriptional dysregulation underlies both monogenic arrhythmia syndrome and common modifiers of cardiac repolarization. Circulation 2023, 147, 824–840. [Google Scholar] [CrossRef]
- Boczek, N.J.; Ye, D.; Johnson, E.K.; Wang, W.; Crotti, L.; Tester, D.J.; Dagradi, F.; Mizusawa, Y.; Torchio, M.; Alders, M.; et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ. Res. 2014, 115, 460–469. [Google Scholar] [CrossRef]
- Veerman, C.C.; Podliesna, S.; Tadros, R.; Lodder, E.M.; Mengarelli, I.; de Jonge, B.; Beekman, L.; Barc, J.; Wilders, R.; Wilde, A.A.M.; et al. The Brugada syndrome susceptibility gene HEY2 modulates cardiac transmural ion channel patterning and electrical heterogeneity. Circ. Res. 2017, 121, e21. [Google Scholar] [CrossRef]
- Iop, L.; Iliceto, S.; Civieri, G.; Tona, F. Inherited and acquired rhythm disturbances in sick sinus syndrome, Brugada syndrome, and atrial fibrillation: Lessons from preclinical modeling. Cells 2021, 10, 3175. [Google Scholar] [CrossRef]
- Mele, A.; Fonzino, A.; Rana, F.; Camerino, G.M.; De Bellis, M.; Conte, E.; Giustino, A.; Conte Camerino, D.; Desaphy, J.F. In vivo longitudinal study of rodent skeletal muscle atrophy using ultrasonography. Sci. Rep. 2016, 6, 20061. [Google Scholar] [CrossRef] [PubMed]
- Papadatos, G.A.; Wallerstein, P.M.; Head, C.E.; Ratcliff, R.; Brady, P.A.; Benndorf, K.; Saumarez, R.C.; Trezise, A.E.; Huang, C.L.; Vandenberg, J.I.; et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. USA 2002, 99, 6210–6215. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Salerno, S.; Connolly, A.; Bishop, M.; Charpentier, F.; Stølen, T.; Smith, G.L. Normal interventricular differences in tissue architecture underlies right ventricular susceptibility to conduction abnormalities in a mouse model of Brugada syndrome. Cardiovasc. Res. 2018, 114, 724–736. [Google Scholar] [CrossRef] [PubMed]
- van Veen, T.A.; van Rijen, H.V.; van Kempen, M.J.; Miquerol, L.; Opthof, T.; Gros, D.; Vos, M.A.; Jongsma, H.J.; de Bakker, J.M. Discontinuous conduction in mouse bundle branches is caused by bundle-branch architecture. Circulation 2005, 112, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Derangeon, M.; Montnach, J.; Cerpa, C.O.; Jagu, B.; Patin, J.; Toumaniantz, G.; Girardeau, A.; Huang, C.L.H.; College, W.H.; Grace, A.A.; et al. Transforming growth factor β receptor inhibition prevents ventricular fibrosis in a mouse model of progressive cardiac conduction disease. Cardiovasc. Res. 2017, 113, 464–474. [Google Scholar] [CrossRef]
- Patin, J.; Castro, C.; Steenman, M.; Hivonnait, A.; Carcouët, A.; Tessier, A.; Lebreton, J.; Bihouée, A.; Donnart, A.; Le Marec, H.; et al. Gap-134, a Connexin43 activator, prevents age-related development of ventricular fibrosis in Scn5a+/− mice. Pharmacol. Res. 2020, 159, 104922. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, L.; Cui, C.; Qin, H.; Chen, H.; Chen, S.; Lin, Y.; Cheng, H.; Jiang, X.; Chen, M. Pathogenesis and drug response of iPSC-derived cardiomyocytes from two Brugada syndrome patients with different Nav1.5-subunit mutations. J. Biomed. Res. 2021, 35, 395–407. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Albers, S.; Cyganek, L.; Zhao, Z.; Lan, H.; Li, X.; Xu, Q.; Kleinsorge, M.; Huang, M.; Liao, Z.; et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace 2019, 21, 1410–1421. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Muller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada syndrome with an SCN1B variants in human-induced pluripotent stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Wada, Y.; Hall, L.D.; Mitchell, D.W.; Glazer, A.M.; Roden, D.M. Functional Assays Reclassify Suspected Splice-Altering Variants of Uncertain Significance in Mendelian Channelopathies. Circ. Genom. Precis. Med. 2022, 15, e003782. [Google Scholar] [CrossRef]
- Martini, B.; Martini, N.; De Mattia, L.; Buja, G. Delayed depolarization and histologic abnormalities underlie the Brugada syndrome. Pacing Clin. Electrophysiol. 2023, 46, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Catalano, O.; Antonaci, S.; Moro, G.; Mussida, M.; Frascaroli, M.; Baldi, M.; Cobelli, F.; Baiardi, P.; Nastoli, J.; Bloise, R.; et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Heart J. 2009, 30, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, F.; Bertini, M.; Balla, C.; Pestelli, G.; Luisi, A.; Smarrazzo, V.; Farnè, M.; Ferlini, A.; Gualandi, F.; Mele, D. Type 1 Brugada pattern is associated with echocardiography-detected delayed right ventricular outflow tract contraction. J. Am. Coll. Cardiol. 2021, 77, 2865–2867. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef]
- Frustaci, A.; Priori, S.G.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef]
- Pappone, C.; Micaglio, E.; Locati, E.T.; Monasky, M.M. The omics of channelopathies and cardiomyopathies: What we know and how they are useful. Eur. Heart J. Suppl. 2020, 22, L105–L109. [Google Scholar] [CrossRef]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef]
- Farnè, M.; Balla, C.; Margutti, A.; Selvatici, R.; De Raffele, M.; Di Domenico, A.; Imbrici, P.; De Maria, E.; Biffi, M.; Bertini, M.; et al. Mutations in MYBPC3 and MYH7 in association with Brugada type 1 ECG pattern: Overlap between Brugada syndrome and hypertrophic cardiomyopathy? Cardiogenetics 2021, 11, 139–147. [Google Scholar] [CrossRef]
- De Maria, E.; Borghi, A.; Tonelli, L.; Selvatici, R.; Cappelli, S.; Gualandi, F. Brugada ECG pattern in hypertrophic cardiomyopathy: Brugada phenocopy or overlapping syndrome? J. Electrocardiol. 2021, 69, 132–135. [Google Scholar] [CrossRef]
- Armaroli, A.; Balla, C.; Trabanelli, C.; Selvatici, R.; Brieda, A.; Sette, E.; Bertini, M.; Mele, D.; Biffi, M.; Campo, G.C.; et al. Lamin A/C missense mutation R216C pinpoints overlapping features between Brugada syndrome and laminopathies. Circ. Genom. Precis. Med. 2020, 13, e002751. [Google Scholar] [CrossRef]
- Jeevaratnam, K.; Guzadhur, L.; Goh, Y.M.; Grace, A.A.; Huang, C.L. Sodium channel haploinsufficiency and structural change in ventricular arrhythmogenesis. Acta Physiol. 2016, 216, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Isbister, J.C.; Gray, B.; Offen, S.; Yeates, L.; Naoum, C.; Medi, C.; Raju, H.; Semsarian, C.; Puranik, R.; Sy, R.W. Longitudinal assessment of structural phenotype in Brugada syndrome using cardiac magnetic resonance imaging. Heart Rhythm O2 2022, 4, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Scala, R.; Maqoud, F.; Zizzo, N.; Mele, A.; Camerino, G.M.; Zito, F.A.; Ranieri, G.; McClenaghan, C.; Harter, T.M.; Nichols, C.G.; et al. Pathophysiological consequences of KATP channel overactivity and pharmacological response to glibenclamide in skeletal muscle of murine model of Cantù syndrome. Front. Pharmacol. 2020, 11, 604885. [Google Scholar] [CrossRef] [PubMed]
- Maqoud, F.; Cetrone, M.; Mele, A.; Tricarico, D. Molecular structure and function of big calcium-activated potassium channels in skeletal muscle: Pharmacological perspectives. Physiol. Genom. 2017, 6, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Marsman, E.M.J.; Postema, P.G.; Remme, C.A. Brugada syndrome: Update and future perspectives. Heart 2022, 108, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Andorin, A.; Behr, E.R.; Denjoy, I.; Crotti, L.; Dagradi, F.; Jesel, L.; Sacher, F.; Petit, B.; Mabo, P.; Maltret, A.; et al. Impact of clinical and genetic findings on the management of young patients with Brugada syndrome. Heart Rhythm 2016, 13, 1274–1282. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Class of Genes/Proteins | Gene | Protein | Functional Defect | BrS and Other Related Diseases | References |
|---|---|---|---|---|---|
| Sodium channels and accessory subunits | SCN5A | Sodium channel alpha subunit Nav1.5 | Loss-of-function variants reduce Nav1.5 expression and alter gating properties or kinetics, causing reduced INa | BrS 1, Sudden Infant Death Syndrome and LQTS 3 | [2] |
| SCN10A | Sodium channel alpha subunit Nav1.8 | Involvement in BrS controversial due to low expression in the heart; may modulate SCN5A gene expression level? | BrS, Familial Episodic Pain Syndrome 2 and Sodium Channelopathy-Related Small Fiber Neuropathy | [65,66,70] | |
| SCN1B | Sodium channel beta 1 subunit | Loss-of-function variants cause reduced INa | BrS 5, Familial AF 13, LQTS, SCD and DEE 52 | [64] | |
| SCN3B | Sodium channel beta 3 subunit | Loss-of-function variants cause reduced INa | BrS 7, Familial AF, LQTS and SCD | [64] | |
| Potassium channels and accessory subunits | KCND3 | Voltage-Gated Potassium Channel Kv4.3 | Gain-of-function variants increase Ito | BrS 9 and Spinocerebellar Ataxia 19 and 22 | [57] |
| KCNE3 | Voltage-Gated Potassium Channel Regulatory Subunit MiRP2 | Gain-of-function variants increase Ito mediated by Kv4.3 | BrS 6 and Hypokalemic Periodic Paralysis Type 1 | [73] | |
| KCNE5 | Cardiac Voltage-Gated Potassium Channel Regulatory Beta Subunit 5 | Gain-of-function variants increase Ito mediated by Kv4.3 | BrS and Amme Complex | [75] | |
| KCNAB2 | Voltage-Gated Potassium Channel Regulatory Beta Subunit 2 | Gain-of-function variants increase Ito mediated by Kv4.3 | BrS, Chromosome 1P36 Deletion Syndrome and Partial Trisomy Distal 4Q | [74] | |
| KCND2 | Voltage-Gated Potassium Channel Kv4.2 | Gain-of-function variants increase Ito mediated by Kv4.2 | BrS, LQTS and Early Myoclonic Encephalopathy | [72] | |
| KCNJ8 | Inwardly Rectifying Potassium Channel Kir6.1 | Gain-of-function variants increase the IK-ATP | BrS, Cantu Syndrome and Infant SD | [67] | |
| ABCC9 | ATP Binding Cassette Subfamily C Member 9 SUR2 | Gain-of-function variants increase the IK-ATP mediated by Kir6.1 | BrS, Cantu Syndrome and Familial AF 12 | [67] | |
| KCNH2 | Voltage-Gated Potassium Channel Kv11.1 (HERG) | Gain-of-function variants increase IKr | BrS, LQTS 2 and SQTS 1 | [41] | |
| Calcium channels and accessory subunits | CACNA1C | Voltage-Gated Calcium Channel Subunit Alpha Cav1.2 | Loss-of-function variants reduce ICaL | BrS 3, Timothy Syndrome and LQTS 8 | [71] |
| CACNB2 | Voltage-Gated Calcium Channel Beta 2 Subunit | Loss-of-function variants reduce ICaL | BrS 4 and Lambert-Eaton Myasthenic Syndrome | [71] | |
| CACNA2D1 | Voltage-Gated Calcium Channel Auxiliary Subunit Alpha2delta 1 | Loss-of-function variants reduce ICaL | BrS, Familial SQTS and DEE 110 | [71] | |
| Other ion channels | TRPM4 | Transient Receptor Potential Cation Channel Subfamily M Member 4 contributes to depolarization that gives rise to the AP in the SAN | Both gain-of-function and loss-of-function variants cause BrS with unclear mechanisms | BrS, Progressive Familial Heart Block Type Ib and Erythrokeratodermia Variabilis Et Progressiva 6 | [54,58,77] |
| HCN4 | Hyperpolarization Activated Cyclic Nucleotide Gated Potassium Channel 4, | Loss-of-function variant reduces If in the SAN | BrS 8 and SSS 2 | [76] | |
| Non-ion channel proteins that affect Nav1.5 traffick and INa | GPD1L | Glycerol-3-Phosphate Dehydrogenase 1 Like | Variants cause trafficking defects of Nav1.5 and reduction in INa | BrS 2 | [80] |
| RANGRF | RAN Guanine Nucleotide Release Factor (MOG1) (chaperone that binds to Nav1.5 and facilitates Nav1.5 trafficking to the cell surface) | Variants cause trafficking defects of Nav1.5 and likely reduce INa | BrS and SSS | [82,83] | |
| SLMAP | Sarcolemma Associated Protein (Golgi) | Variants cause trafficking defects of Nav1.5 and reduction in INa | BrS and lung cancer | [81] | |
| PKP2 | Plakophilin 2 | Variants reduce the number of Nav1.5 channels at the intercalated disc and likely reduce INa | BrS, Familial Arrhythmogenic Right Ventricular Dysplasia 9 and ARVC | [84] | |
| GPD1L | Glycerol-3-Phosphate Dehydrogenase 1 Like | Variants cause trafficking defects of Nav1.5 and reduction in INa | BrS 2 | [80] | |
| FGF12B | Fibroblast Growth Factor FGF-12b (potent regulator of Nav1.5 traffic and function) | Variants reduce INa but not ICaL | BrS, DEE 47 and Non-Specific Early-Onset Epileptic Encephalopathy | [78,79] | |
| MAPRE2 | Microtubule-Associated Protein RP/EB Family Member 2 | Variants cause microtubule-related trafficking effects on Nav1.5 expression | BrS, Skin Creases, Congenital Symmetric Circumferential 2 and Multiple Benign Circumferential Skin Creases On Limbs | [35] | |
| GSTM3 | Glutathione S-Transferase Mu 3 | Copy number deletions cause reduction in INa and higher rates of syncope and SCD | BrS, Larynx Cancer and Pharynx Cancer | [85] | |
| Transcription factors that regulate SCN5A transcription and INa | ZFHX3 | zinc finger homeobox 3 | Variants downregulate SCN5A transcription and Nav1.5 expression and can modify BrS phenotype | Genetic modifier in BrS, Prostate Cancer and Small Cell Cancer Of The Lung | [86] |
| TBX5 | T-box transcription factor 5 | Variants downregulate SCN5A transcription, decrease cardiac peak INa and enhance “late” INa | BrS, Holt-Oram Syndrome and Patent Foramen Ovale | [87] | |
| Non-ion channel proteins that affect Ito | SEMA3A | semaphorin-3A binds to Kv4.3 and reduces peak current densities without perturbing cell surface expression | Loss-of-function variants increase Ito mediated by Kv4.3 | BrS, Hypogonadotropic Hypogonadism 16 with or without Anosmia | [88] |
| HEY2 | Hes Related Family BHLH Transcription Factor With YRPW Motif 2 affects cardiac ion channel gene expression in mice and humans | SNP increasing HEY2 transcript increases KCNIP2 expression and Ito | BrS, Aortic Aneurysm, Familial Thoracic 1 and Tricuspid Atresia | [68,89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liantonio, A.; Bertini, M.; Mele, A.; Balla, C.; Dinoi, G.; Selvatici, R.; Mele, M.; De Luca, A.; Gualandi, F.; Imbrici, P. Brugada Syndrome: More than a Monogenic Channelopathy. Biomedicines 2023, 11, 2297. https://doi.org/10.3390/biomedicines11082297
Liantonio A, Bertini M, Mele A, Balla C, Dinoi G, Selvatici R, Mele M, De Luca A, Gualandi F, Imbrici P. Brugada Syndrome: More than a Monogenic Channelopathy. Biomedicines. 2023; 11(8):2297. https://doi.org/10.3390/biomedicines11082297
Chicago/Turabian StyleLiantonio, Antonella, Matteo Bertini, Antonietta Mele, Cristina Balla, Giorgia Dinoi, Rita Selvatici, Marco Mele, Annamaria De Luca, Francesca Gualandi, and Paola Imbrici. 2023. "Brugada Syndrome: More than a Monogenic Channelopathy" Biomedicines 11, no. 8: 2297. https://doi.org/10.3390/biomedicines11082297