
Compensatory Increase of Serum Hepassocin Protects Hyperthyroidism-Induced Hepatic Dysfunction
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Cell Culture
2.3. Lentiviral Vectors
2.4. Western Blot Analyses
2.5. Statistical Analysis
3. Results
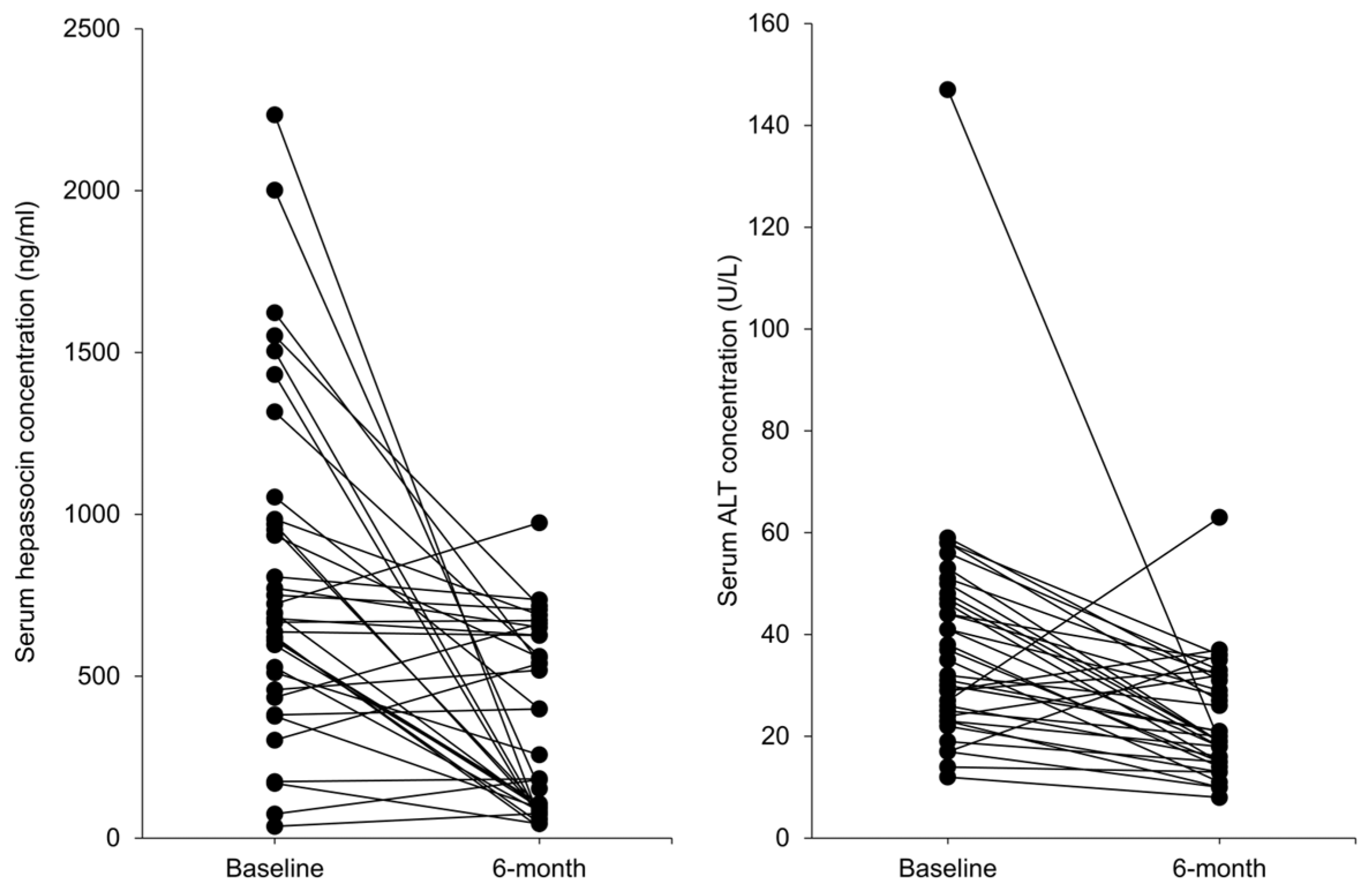
3.1. Serum Hepassocin Concentrations Were Significantly Decreased in Subjects with Graves’ Disease Received Standard Treatment
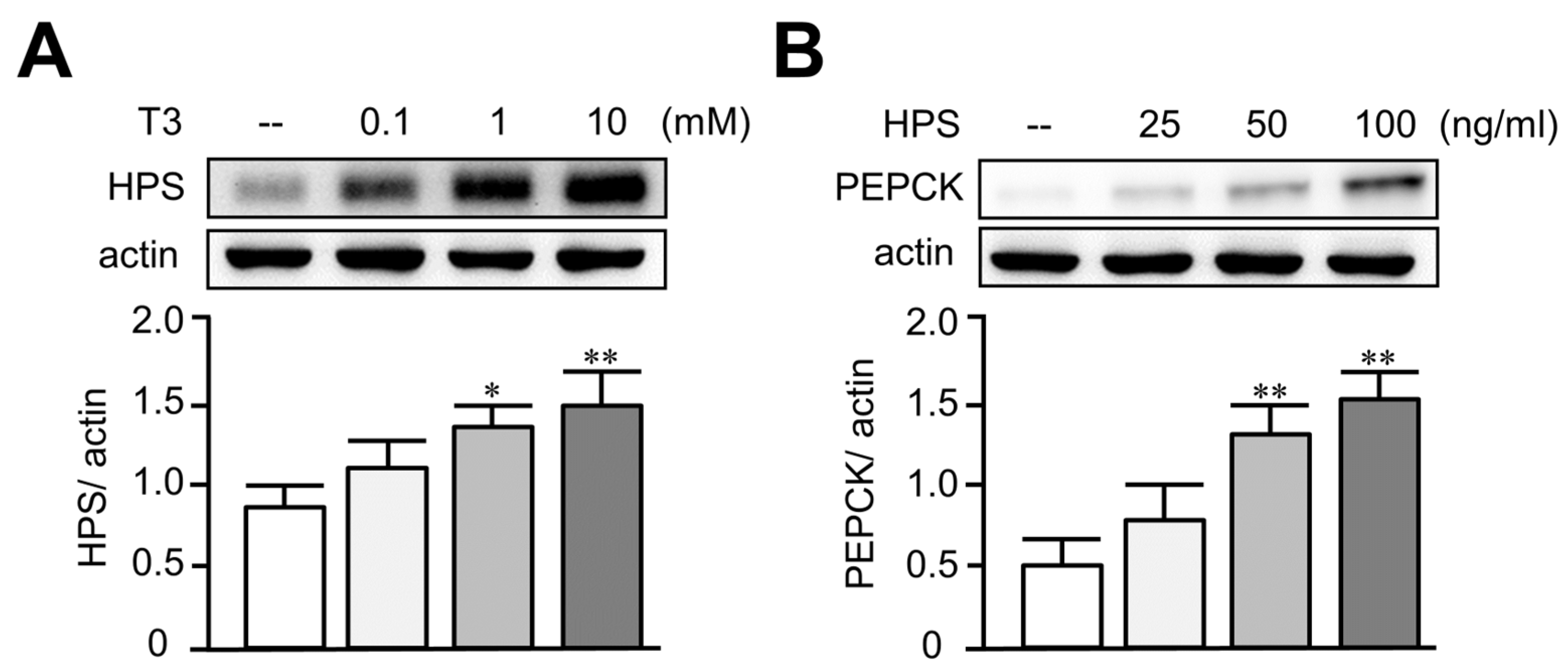
3.2. Treatment of T3 Dose-Dependently Increased Hepassocin and PEPCK Expressions in HepG2 Cells
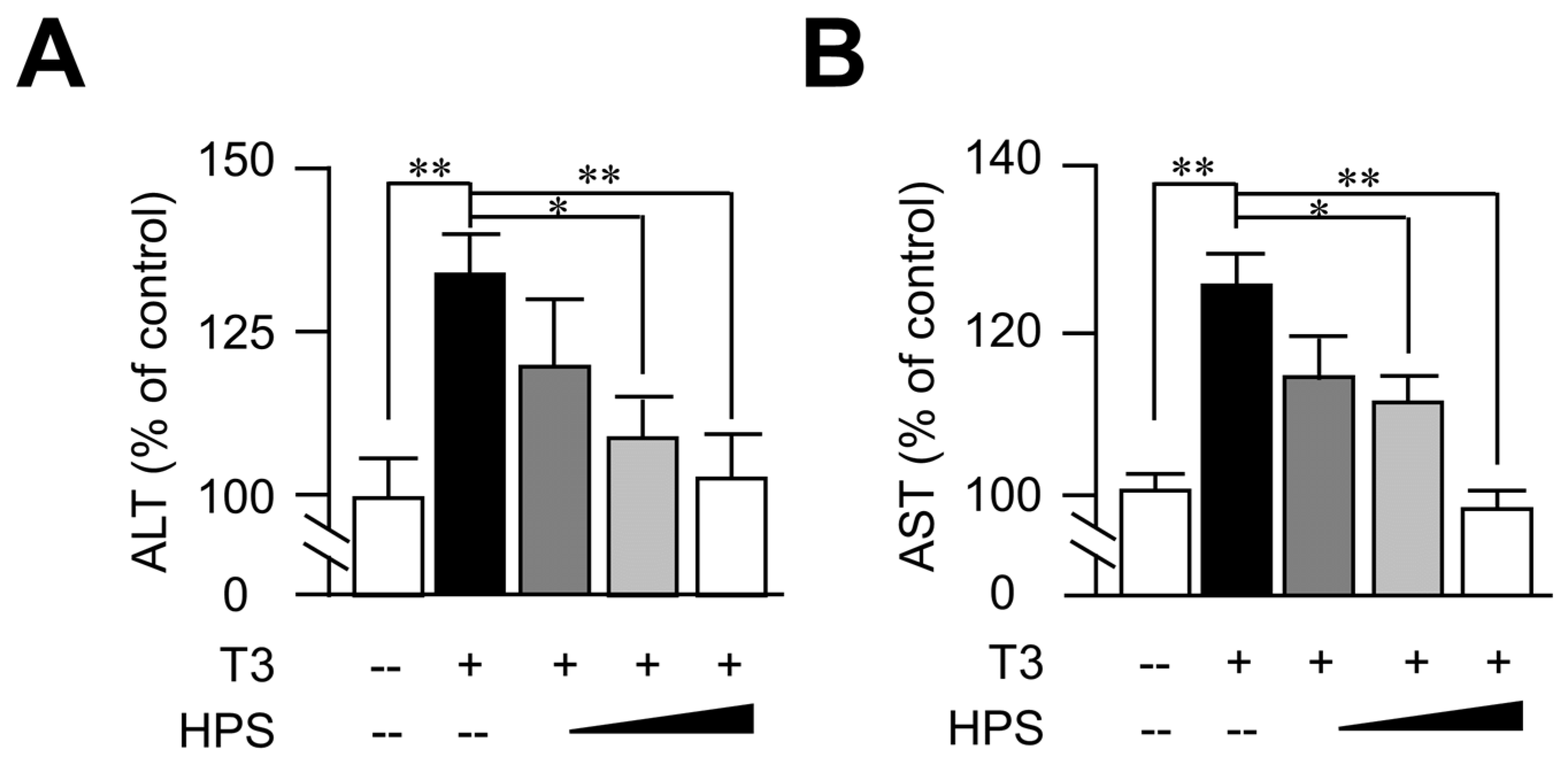
3.3. Hepassocin Reversed T3-Induced Hepatic Enzyme Elevation in a Dose-Dependent Manner
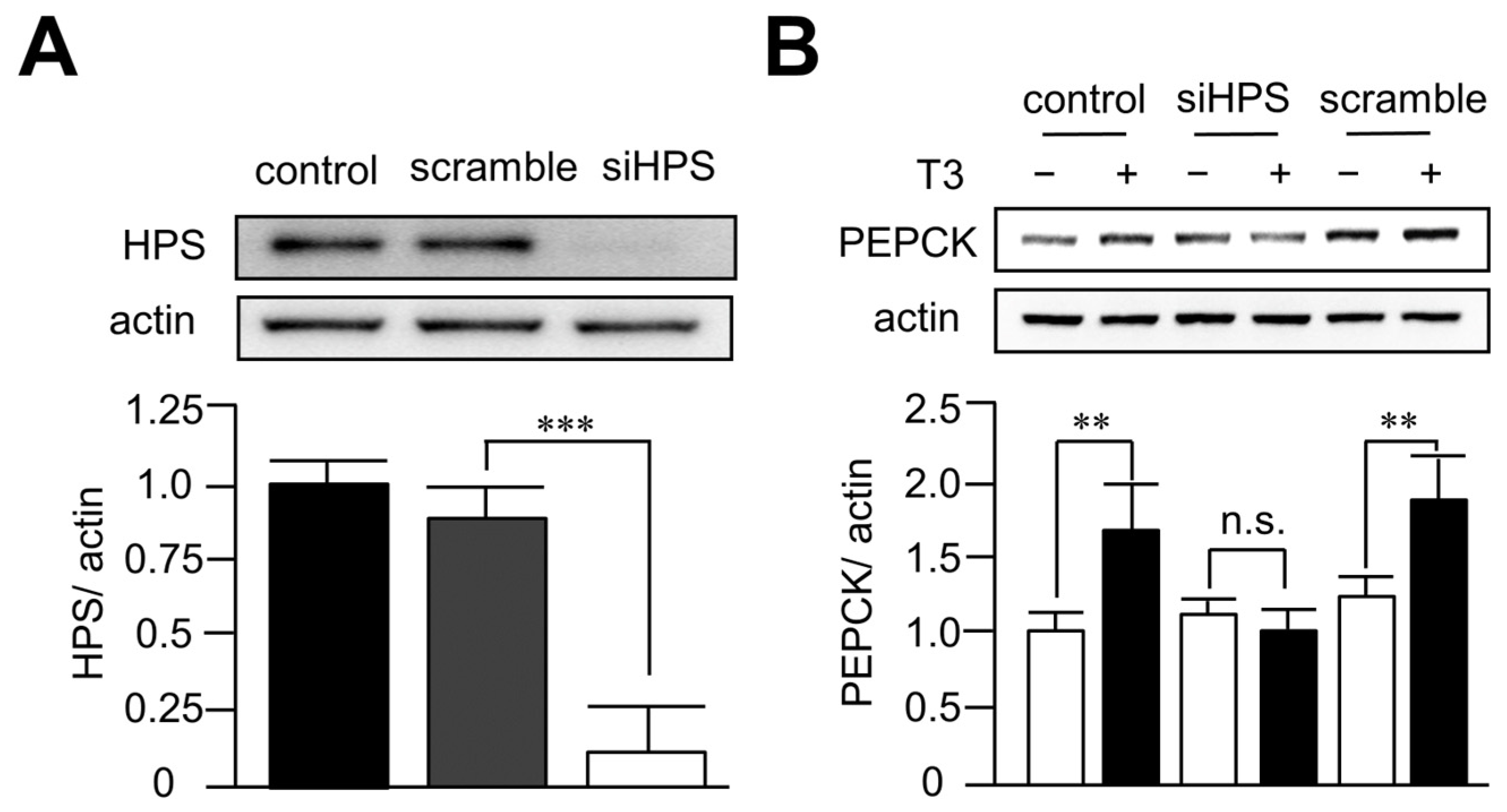
3.4. Knockdown of Hepassocin Diminished the Effects of T3 on PEPCK Expression
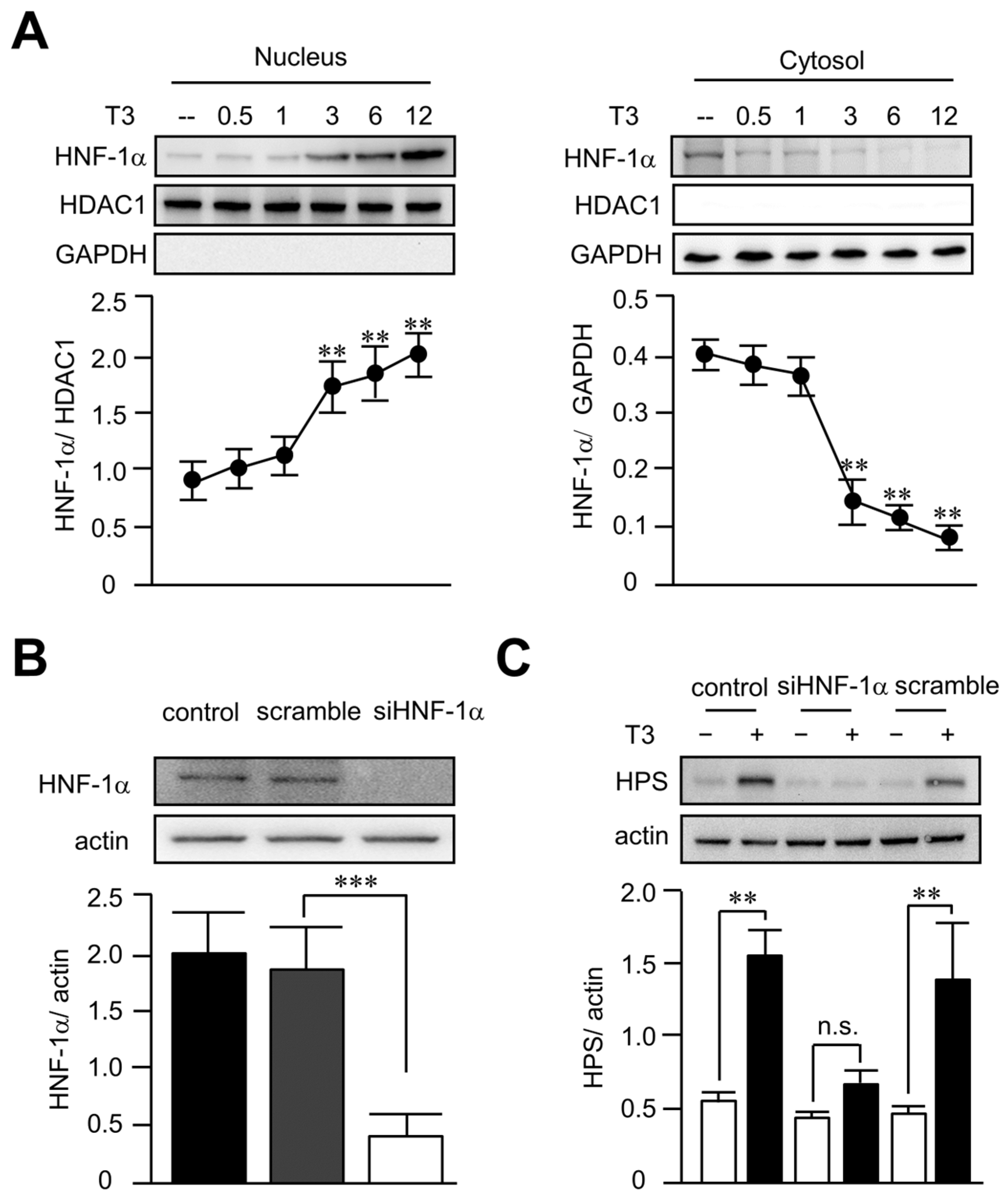
3.5. T3 Increased Hepassocin Expression through an HNF-1α-Dependent Pathway in HepG2 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Leo, S.; Lee, S.Y.; Braverman, L.E. Hyperthyroidism. Lancet 2016, 388, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scappaticcio, L.; Longo, M.; Maiorino, M.I.; Pernice, V.; Caruso, P.; Esposito, K.; Bellastella, G. Abnormal Liver Blood Tests in Patients with Hyperthyroidism: Systematic Review and Meta-Analysis. Thyroid 2021, 31, 884–894. [Google Scholar] [CrossRef]
- Khemichian, S.; Fong, T.L. Hepatic Dysfunction in Hyperthyroidism. Gastroenterol. Hepatol. 2011, 7, 337–339. [Google Scholar]
- Maratou, E.; Hadjidakis, D.J.; Peppa, M.; Alevizaki, M.; Tsegka, K.; Lambadiari, V.; Mitrou, P.; Boutati, E.; Kollias, A.; Economopoulos, T.; et al. Studies of insulin resistance in patients with clinical and subclinical hyperthyroidism. Eur. J. Endocrinol. 2010, 163, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadis, G.; Baker, B.; Marsh, H.; Mandarino, L.; Rizza, R.; Bergman, R.; Haymond, M.; Gerich, J. Effect of thyroid hormone excess on action, secretion, and metabolism of insulin in humans. Am. J. Physiol. 1985, 248, E593–E601. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.D.; Raptis, S.A. Thyroid hormone excess and glucose intolerance. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. S2), S225–S239. [Google Scholar] [CrossRef] [PubMed]
- Rochon, C.; Tauveron, I.; Dejax, C.; Benoit, P.; Capitan, P.; Fabricio, A.; Berry, C.; Champredon, C.; Thieblot, P.; Grizard, J. Response of glucose disposal to hyperinsulinaemia in human hypothyroidism and hyperthyroidism. Clin. Sci. 2003, 104, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Weranuj, R.; Praneet, W.; Monchaya, T.; Sutin, S. Hyperthyroidism induces glucose intolerance by lowering both insulin secretion and peripheral insulin sensitivity. J. Med. Assoc. Thai 2006, 89 (Suppl. S5), S133–S140. [Google Scholar]
- Biondi, B.; Kahaly, G.J.; Robertson, R.P. Thyroid Dysfunction and Diabetes Mellitus: Two Closely Associated Disorders. Endocr. Rev. 2019, 40, 789–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, G.W.; Holman, G.D. The glucose transporter family: Structure, function and tissue-specific expression. Biochem. J. 1995, 295 Pt 2, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Chandrasekaran, K. Hyperglycemia induced changes in liver: In vivo and in vitro studies. Curr. Diabetes Rev. 2009, 5, 67–78. [Google Scholar] [CrossRef]
- Dey, A.; Swaminathan, K. Hyperglycemia-induced mitochondrial alterations in liver. Life Sci. 2010, 87, 197–214. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Hu, Y.; Xu, X.H.; Mao, X.M. Hepatic dysfunction related to thyrotropin receptor antibody in patients with Graves’ disease. Exp. Clin. Endocrinol. Diabetes 2014, 122, 368–372. [Google Scholar] [CrossRef] [PubMed]
- de Campos Mazo, D.F.; de Vasconcelos, G.B.; Pereira, M.A.; de Mello, E.S.; Bacchella, T.; Carrilho, F.J.; Cancado, E.L. Clinical spectrum and therapeutic approach to hepatocellular injury in patients with hyperthyroidism. Clin. Exp. Gastroenterol. 2013, 6, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansourian, A.R. Liver functional behavior during thyrotoxicosis: A review. J. Biol. Sci. 2013, 13, 665–678. [Google Scholar] [CrossRef]
- Yorke, E. Hyperthyroidism and Liver Dysfunction: A Review of a Common Comorbidity. Clin. Med. Insights Endocrinol. Diabetes 2022, 15, 11795514221074672. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.T.; Yu, M.; Li, C.Y.; Zhan, Y.Q.; Xu, W.X.; Li, Y.H.; Li, W.; Wang, Z.D.; Ge, C.H.; Yang, X.M. Specific expression and regulation of hepassocin in the liver and down-regulation of the correlation of HNF1alpha with decreased levels of hepassocin in human hepatocellular carcinoma. J. Biol. Chem. 2009, 284, 13335–13347. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Uchida, S.; Yoshimura, H.; Aoki, M.; Toyoda, Y.; Sakai, Y.; Morimoto, S.; Fukamachi, H.; Shiokawa, K.; Hanada, K. Isolation and characterization of a novel liver-specific gene, hepassocin, upregulated during liver regeneration. Biochim. Biophys. Acta 2000, 1492, 31–44. [Google Scholar] [CrossRef]
- Hara, H.; Yoshimura, H.; Uchida, S.; Toyoda, Y.; Aoki, M.; Sakai, Y.; Morimoto, S.; Shiokawa, K. Molecular cloning and functional expression analysis of a cDNA for human hepassocin, a liver-specific protein with hepatocyte mitogenic activity. Biochim. Biophys. Acta 2001, 1520, 45–53. [Google Scholar] [CrossRef]
- Li, C.Y.; Cao, C.Z.; Xu, W.X.; Cao, M.M.; Yang, F.; Dong, L.; Yu, M.; Zhan, Y.Q.; Gao, Y.B.; Li, W.; et al. Recombinant human hepassocin stimulates proliferation of hepatocytes in vivo and improves survival in rats with fulminant hepatic failure. Gut 2010, 59, 817–826. [Google Scholar] [CrossRef]
- Gao, M.; Zhan, Y.Q.; Yu, M.; Ge, C.H.; Li, C.Y.; Zhang, J.H.; Wang, X.H.; Ge, Z.Q.; Yang, X.M. Hepassocin activates the EGFR/ERK cascade and induces proliferation of L02 cells through the Src-dependent pathway. Cell Signal. 2014, 26, 2161–2166. [Google Scholar] [CrossRef]
- Wu, H.T.; Chen, S.C.; Fan, K.C.; Kuo, C.H.; Lin, S.Y.; Wang, S.H.; Chang, C.J.; Li, H.Y. Targeting fibrinogen-like protein 1 is a novel therapeutic strategy to combat obesity. FASEB J. 2020, 34, 2958–2967. [Google Scholar] [CrossRef]
- Wu, H.T.; Lu, F.H.; Ou, H.Y.; Su, Y.C.; Hung, H.C.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. The role of hepassocin in the development of non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 1065–1072. [Google Scholar] [CrossRef]
- Kang, L.; Li, H.Y.; Ou, H.Y.; Wu, P.; Wang, S.H.; Chang, C.J.; Lin, S.Y.; Wu, C.L.; Wu, H.T. Role of placental fibrinogen-like protein 1 in gestational diabetes. Transl. Res. 2020, 218, 73–80. [Google Scholar] [CrossRef]
- Ou, H.Y.; Wu, H.T.; Lin, C.H.; Du, Y.F.; Hu, C.Y.; Hung, H.C.; Wu, P.; Li, H.Y.; Wang, S.H.; Chang, C.J. The Hepatic Protection Effects of Hepassocin in Hyperglycemic Crisis. J. Clin. Endocrinol. Metab. 2017, 102, 2407–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.T.; Ou, H.Y.; Hung, H.C.; Su, Y.C.; Lu, F.H.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. A novel hepatokine, HFREP1, plays a crucial role in the development of insulin resistance and type 2 diabetes. Diabetologia 2016, 59, 1732–1742. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.S.; Burch, H.B.; Cooper, D.S.; Greenlee, M.C.; Laurberg, P.; Maia, A.L.; Rivkees, S.A.; Samuels, M.; Sosa, J.A.; Stan, M.N.; et al. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid 2016, 26, 1343–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gürlek, A.; Cobankara, V.; Bayraktar, M. Liver Tests in Hyperthyroidism: Effect of Antithyroid Therapy. J. Clin. Gastroenterol. 1997, 24, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, S.; Bayraktaroglu, T.; Demircan, N.; Sert, M.; Açikgoz, S.; Tekin, I.O.; Ustundag, Y. Effect of hyperthyroidism and propylthiouracil treatment on liver biochemical tests. Int. J. Clin. Pract. 2005, 59, 1304–1308. [Google Scholar] [CrossRef]
- Kumar, A.; Sinha, R.A.; Tiwari, M.; Singh, R.; Koji, T.; Manhas, N.; Rastogi, L.; Pal, L.; Shrivastava, A.; Sahu, R.P.; et al. Hyperthyroidism induces apoptosis in rat liver through activation of death receptor-mediated pathways. J. Hepatol. 2007, 46, 888–898. [Google Scholar] [CrossRef]
- Piantanida, E.; Ippolito, S.; Gallo, D.; Masiello, E.; Premoli, P.; Cusini, C.; Rosetti, S.; Sabatino, J.; Segato, S.; Trimarchi, F.; et al. The interplay between thyroid and liver: Implications for clinical practice. J. Endocrinol. Investig. 2020, 43, 885–899. [Google Scholar] [CrossRef]
- Liu, Z.; Ukomadu, C. Fibrinogen-like protein 1, a hepatocyte derived protein is an acute phase reactant. Biochem. Biophys. Res. Commun. 2008, 365, 729–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreines, K.; Jett, M.; Knowles, H.C. Observations in hyperthyroidism of abnormal glucose tolerance and other traits related to diabetes mellitus. Diabetes 1965, 14, 740–744. [Google Scholar] [CrossRef]
- Maxon, H.R.; Kreines, K.W.; Goldsmith, R.E.; Knowles, H.C. Long-Term Observations of Glucose Tolerance in Thyrotoxic Patients. Arch. Intern. Med. 1975, 135, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.H.; Ou, H.Y.; Wu, H.T.; Hung, H.C.; Wu, J.S.; Yang, Y.C.; Chang, C.J. Serum hepassocin concentrations in diabetic patients with or without nonalcoholic fatty liver disease. Diabetes Manag. 2014, 4, 255–261. [Google Scholar] [CrossRef]
- Perna, S.; Peroni, G.; Miccono, A.; Riva, A.; Morazzoni, P.; Allegrini, P.; Preda, S.; Baldiraghi, V.; Guido, D.; Rondanelli, M. Multidimensional Effects of Soy Isoflavone by Food or Supplements in Menopause Women: A Systematic Review and Bibliometric Analysis. Nat. Prod. Commun. 2016, 11, 1733–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farebrother, J.; Zimmermann, M.B.; Andersson, M. Excess iodine intake: Sources, assessment, and effects on thyroid function. Ann. N. Y. Acad. Sci. 2019, 1446, 44–65. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | |
|---|---|
| n | 36 |
| Age (y) | 37.9 ± 11.5 |
| Sex (male/female) | 9/27 (25/75) |
| Body weight (kg) | 55.98 (9.46) |
| BMI (kg/m2) | 21.72 (2.79) |
| Systolic blood pressure (mmHg) | 124.2 (13.3) |
| Diastolic blood pressure (mmHg) | 71.8 (11.2) |
| Fasting plasma glucose (mg/dL) | 108.0 (19.6) |
| Creatinine (mg/dL) | 0.42 (0.18) |
| eGFR (mL/min/1.73 m2) | 89.2 (5.0) |
| ALT (U/L) | 38.4 (23.0) |
| AST (U/L) | 28.9 (15.9) |
| TSH * (μU/mL) | <0.03 (<0.03 ~ <0.03) |
| Total T3 (ng/dl) | 371.24 (158.57) |
| Free T4 (ng/dL, mean) | 3.45 (0.91) |
| TRAb (U/L) | 12.11 (12.34) |
| Hepassocin (ng/mL) | 799.99 (513.71) |
| Total cholesterol (mg/dL) | 173.1 (33.0) |
| Triglyceride (mg/dL) | 93.0 (32.5) |
| HDL-c (mg/dL) | 63.9 (13.5) |
| LDL-c (mg/dL) | 104.8 (30.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-C.; Lin, C.-H.; Chou, H.-W.; Wang, C.-T.; Liang, Y.-C.; Wu, H.-T.; Ou, H.-Y. Compensatory Increase of Serum Hepassocin Protects Hyperthyroidism-Induced Hepatic Dysfunction. Biomedicines 2023, 11, 1936. https://doi.org/10.3390/biomedicines11071936
Wang C-C, Lin C-H, Chou H-W, Wang C-T, Liang Y-C, Wu H-T, Ou H-Y. Compensatory Increase of Serum Hepassocin Protects Hyperthyroidism-Induced Hepatic Dysfunction. Biomedicines. 2023; 11(7):1936. https://doi.org/10.3390/biomedicines11071936
Chicago/Turabian StyleWang, Chih-Chen, Ching-Han Lin, Hsuan-Wen Chou, Chung-Teng Wang, Yu-Cheng Liang, Hung-Tsung Wu, and Horng-Yih Ou. 2023. "Compensatory Increase of Serum Hepassocin Protects Hyperthyroidism-Induced Hepatic Dysfunction" Biomedicines 11, no. 7: 1936. https://doi.org/10.3390/biomedicines11071936