

Urinary Cysteinyl Leukotrienes as Biomarkers of Endothelial Activation, Inflammation and Oxidative Stress and Their Relationship with Organ Dysfunction in Human Septic Shock
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Clinical Data and Sample Collection
2.3. Routine Laboratory Procedures
2.4. Quantification of Urinary Cysteinyl Leukotrienes
2.5. Quantification of Urinary Isoprostanes
2.6. Measurement of Serum Endothelial Activation and Inflammatory Biomarkers
2.7. Quantification of Plasma and Urinary Angiotensinogen
2.8. Data and Statistical Analysis
3. Results
3.1. Demographic, Clinical, and Biochemical Characteristics
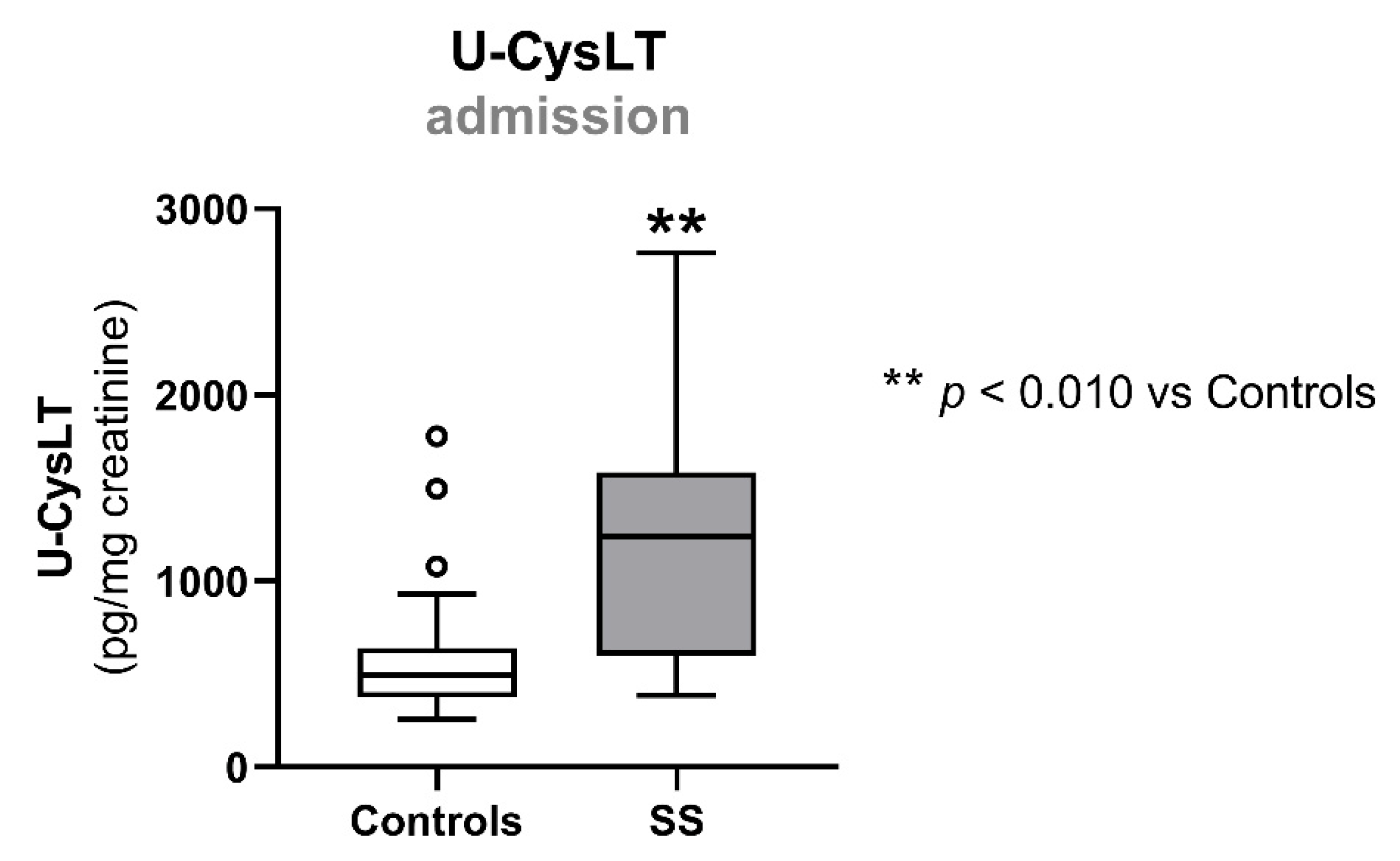
3.2. Urinary Cysteinyl Leukotrienes at Admission
3.3. Biomarkers of Endothelial Activation, Inflammation, Oxidative Stress, and RAAS at Admission
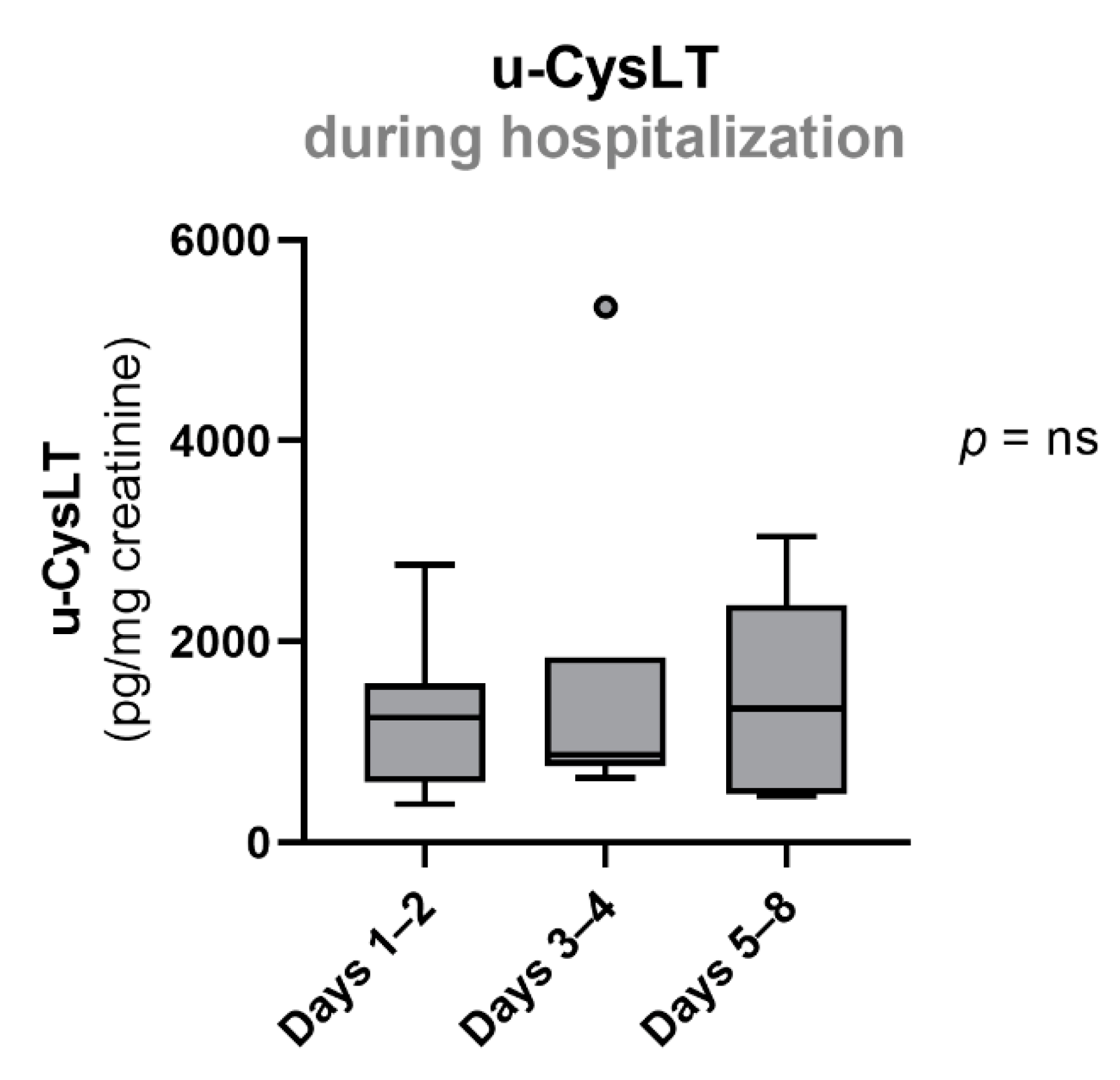
3.4. Evolution of Urinary Cysteinyl Leukotrienes during Hospitalization
3.5. Evolution of Endothelial Activation, Inflammation, Oxidative Stress, and RAAS during Hospitalization
3.6. Evolution of Clinical and Biochemical Parameters of Organ Dysfunction during Hospitalization in SS Patients
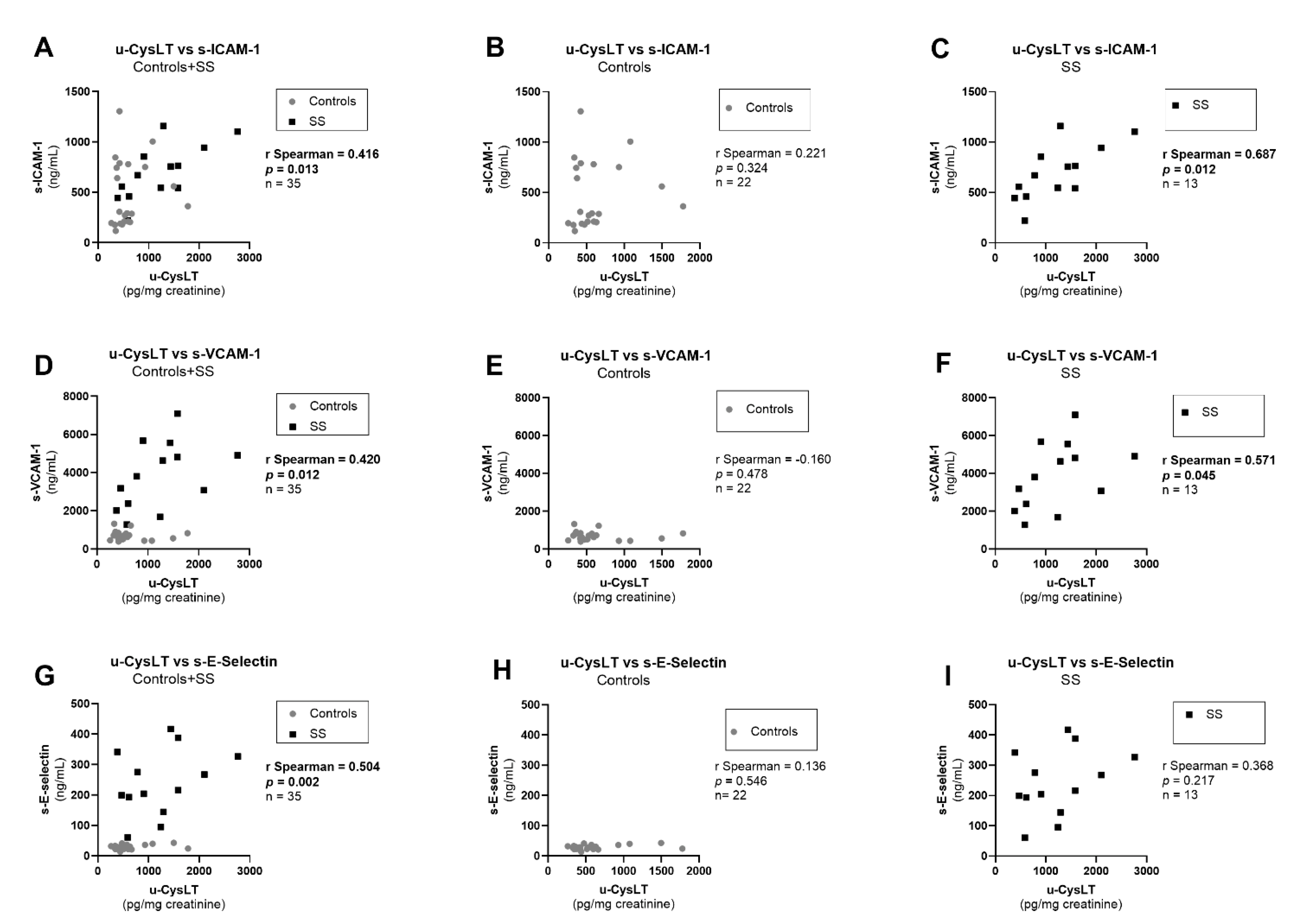
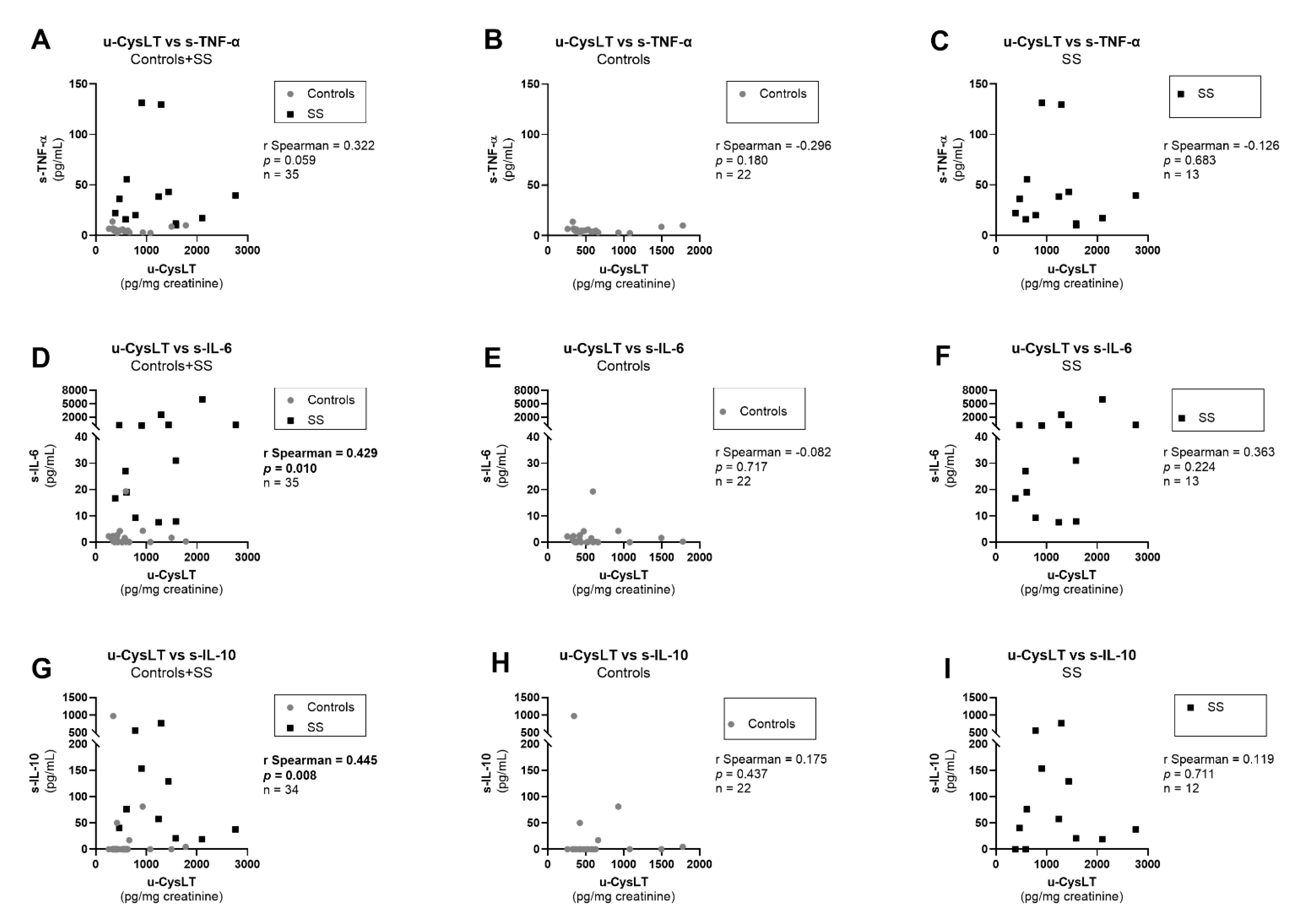
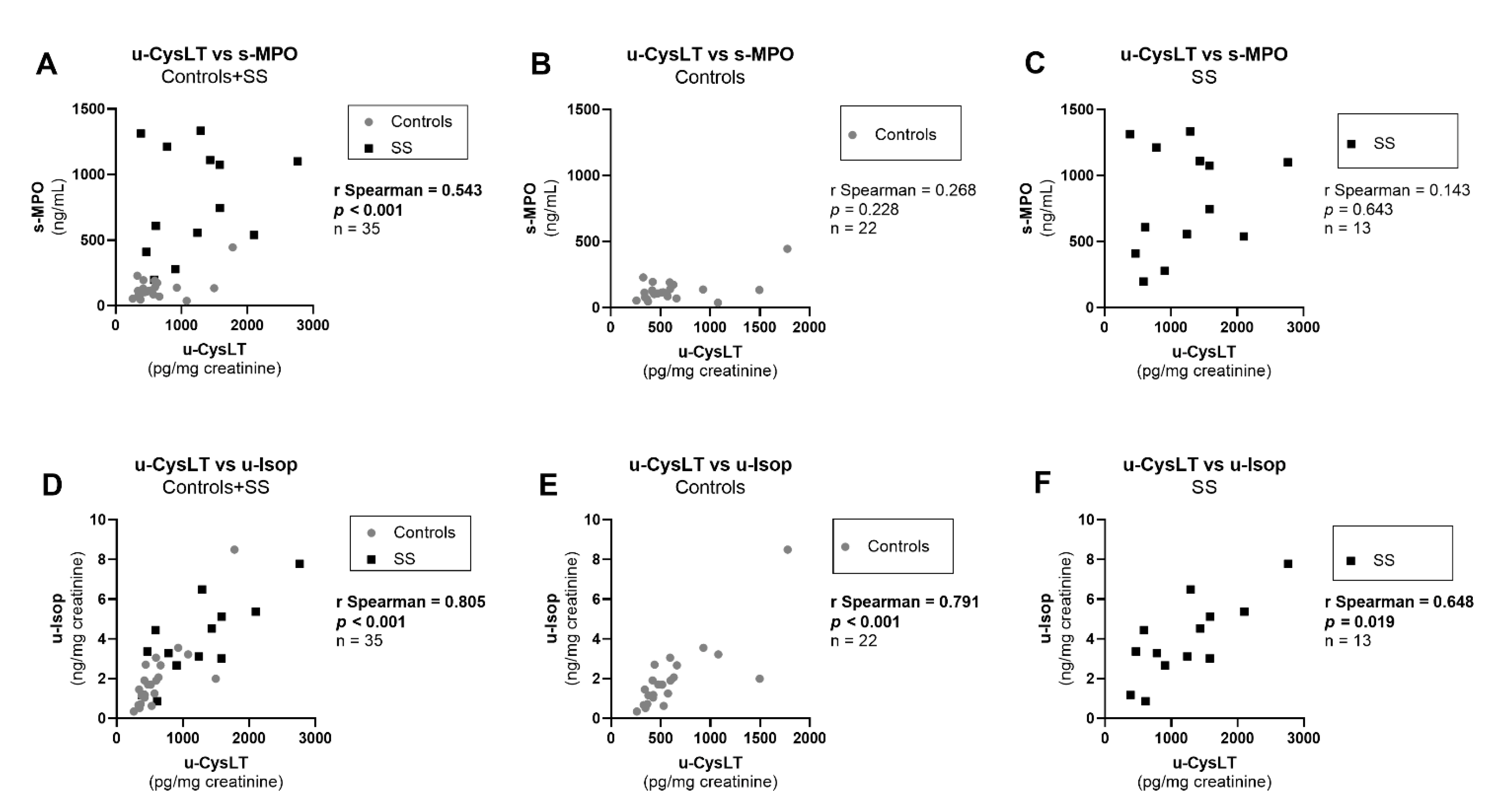
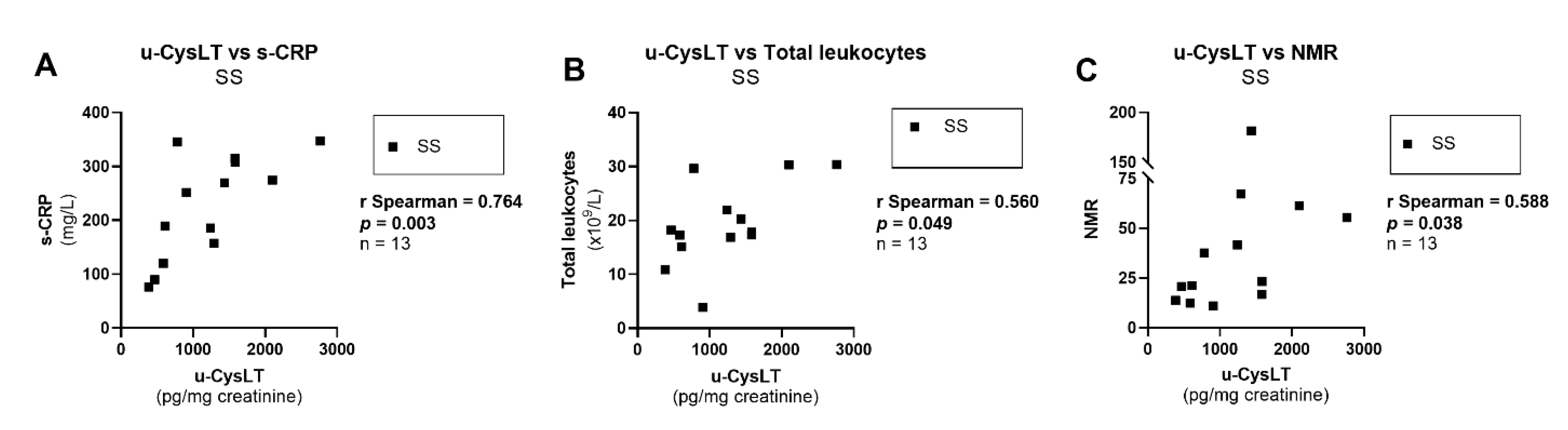
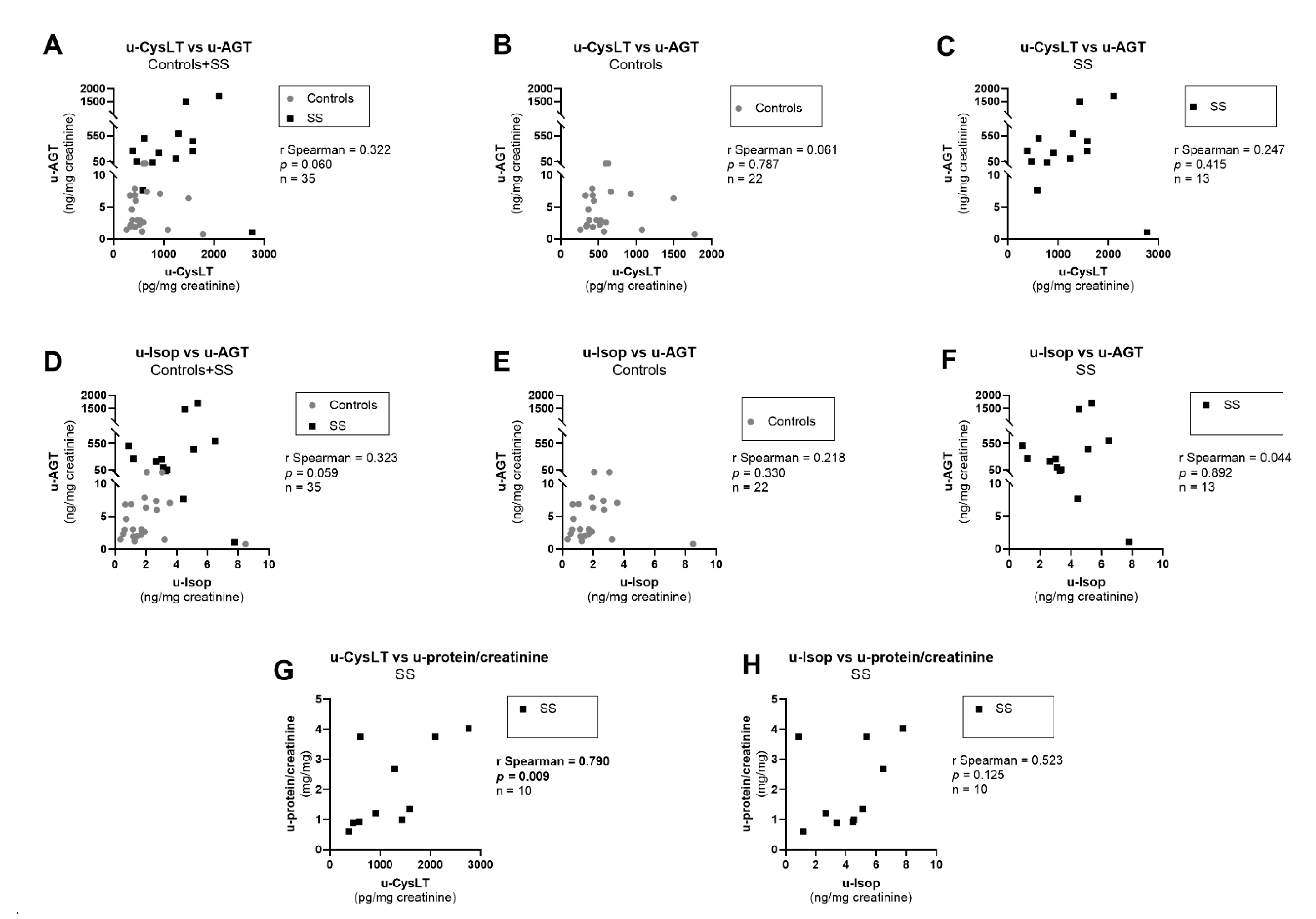
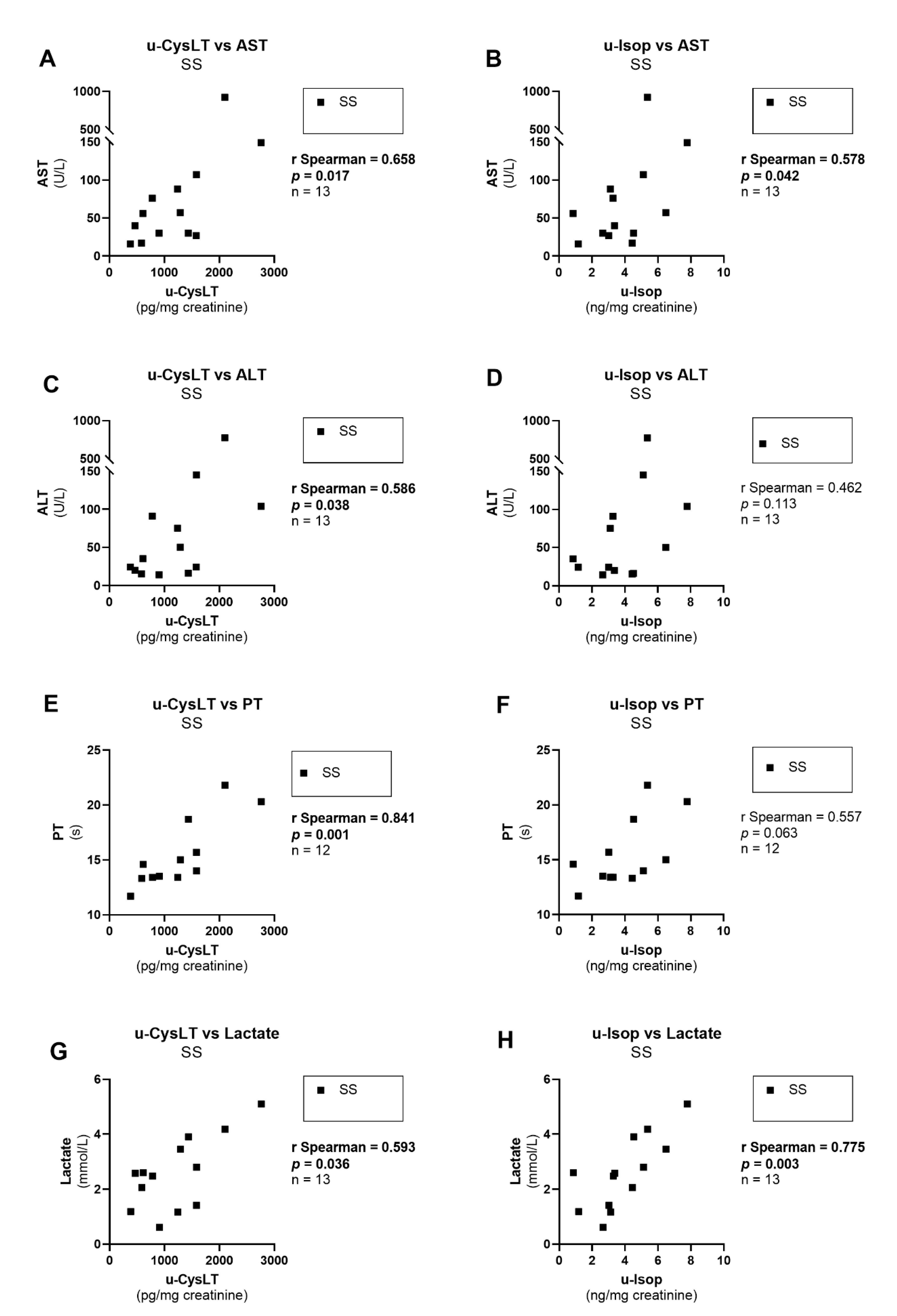
3.7. Correlation Analysis at Admission
3.8. Repeated-Measure Multivariate Analyses in SS Patients throughout Hospitalization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Leopold, S.J.; Cranendonk, D.R.; van der Poll, T. Host innate immune responses to sepsis. Virulence 2014, 5, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutschman, C.S.; Tracey, K.J. Sepsis: Current dogma and new perspectives. Immunity 2014, 40, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Lelubre, C.; Vincent, J.L. Mechanisms and treatment of organ failure in sepsis. Nat. Rev. Nephrol. 2018, 14, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Liu, V.X.; Iwashyna, T.J.; Brunkhorst, F.M.; Rea, T.D.; Scherag, A.; Rubenfeld, G.; Kahn, J.M.; Shankar-Hari, M.; Singer, M.; et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.A.; Rush, B.; Boyd, J. Pathophysiology of Septic Shock. Crit. Care Clin. 2018, 34, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Shankar-Hari, M.; Phillips, G.S.; Levy, M.L.; Seymour, C.W.; Liu, V.X.; Deutschman, C.S.; Angus, D.C.; Rubenfeld, G.D.; Singer, M. Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Pierrakos, C.; Velissaris, D.; Bisdorff, M.; Marshall, J.C.; Vincent, J.L. Biomarkers of sepsis: Time for a reappraisal. Crit. Care 2020, 24, 287. [Google Scholar] [CrossRef]
- Hamade, B.; Huang, D.T. Procalcitonin: Where Are We Now? Crit. Care Clin. 2020, 36, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; McIntyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 47, 1181–1247. [Google Scholar] [CrossRef]
- Fleischmann-Struzek, C.; Mellhammar, L.; Rose, N.; Cassini, A.; Rudd, K.E.; Schlattmann, P.; Allegranzi, B.; Reinhart, K. Incidence and mortality of hospital- and ICU-treated sepsis: Results from an updated and expanded systematic review and meta-analysis. Intensive Care Med. 2020, 46, 1552–1562. [Google Scholar] [CrossRef]
- Rhee, C.; Dantes, R.; Epstein, L.; Murphy, D.J.; Seymour, C.W.; Iwashyna, T.J.; Kadri, S.S.; Angus, D.C.; Danner, R.L.; Fiore, A.E.; et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs. Claims Data, 2009–2014. JAMA 2017, 318, 1241–1249. [Google Scholar] [CrossRef]
- Pro, C.I.; Yealy, D.M.; Kellum, J.A.; Huang, D.T.; Barnato, A.E.; Weissfeld, L.A.; Pike, F.; Terndrup, T.; Wang, H.E.; Hou, P.C.; et al. A randomized trial of protocol-based care for early septic shock. N. Engl. J. Med. 2014, 370, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Investigators, A.; Group, A.C.T.; Peake, S.L.; Delaney, A.; Bailey, M.; Bellomo, R.; Cameron, P.A.; Cooper, D.J.; Higgins, A.M.; Holdgate, A.; et al. Goal-directed resuscitation for patients with early septic shock. N. Engl. J. Med. 2014, 371, 1496–1506. [Google Scholar] [CrossRef] [Green Version]
- Mouncey, P.R.; Osborn, T.M.; Power, G.S.; Harrison, D.A.; Sadique, M.Z.; Grieve, R.D.; Jahan, R.; Harvey, S.E.; Bell, D.; Bion, J.F.; et al. Trial of early, goal-directed resuscitation for septic shock. N. Engl. J. Med. 2015, 372, 1301–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.; Vincent, J.L.; Adhikari, N.K.; Machado, F.R.; Angus, D.C.; Calandra, T.; Jaton, K.; Giulieri, S.; Delaloye, J.; Opal, S.; et al. Sepsis: A roadmap for future research. Lancet Infect. Dis. 2015, 15, 581–614. [Google Scholar] [CrossRef]
- Font, M.D.; Thyagarajan, B.; Khanna, A.K. Sepsis and Septic Shock-Basics of diagnosis, pathophysiology and clinical decision making. Med. Clin. North. Am. 2020, 104, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Bone, R.C.; Fisher, C.J., Jr.; Clemmer, T.P.; Slotman, G.J.; Metz, C.A.; Balk, R.A. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N. Engl. J. Med. 1987, 317, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Wunderink, R.; Silverman, H.; Perl, T.M.; Nasraway, S.; Levy, H.; Bone, R.; Wenzel, R.P.; Balk, R.; Allred, R.; et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA 1995, 273, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.J., Jr.; Slotman, G.J.; Opal, S.M.; Pribble, J.P.; Bone, R.C.; Emmanuel, G.; Ng, D.; Bloedow, D.C.; Catalano, M.A. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: A randomized, open-label, placebo-controlled multicenter trial. Crit. Care Med. 1994, 22, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Wheeler, A.P.; Russell, J.A.; Schein, R.; Summer, W.R.; Steinberg, K.P.; Fulkerson, W.J.; Wright, P.E.; Christman, B.W.; Dupont, W.D.; et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N. Engl. J. Med. 1997, 336, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Nedeva, C. Inflammation and Cell Death of the Innate and Adaptive Immune System during Sepsis. Biomolecules 2021, 11, 1011. [Google Scholar] [CrossRef] [PubMed]
- Dolmatova, E.V.; Wang, K.; Mandavilli, R.; Griendling, K.K. The effects of sepsis on endothelium and clinical implications. Cardiovasc. Res. 2021, 117, 60–73. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Sprague, R.S.; Stephenson, A.H.; Dahms, T.E.; Lonigro, A.J. Proposed role for leukotrienes in the pathophysiology of multiple systems organ failure. Crit. Care Clin. 1989, 5, 315–329. [Google Scholar] [CrossRef]
- Denzlinger, C.; Rapp, S.; Hagmann, W.; Keppler, D. Leukotrienes as mediators in tissue trauma. Science 1985, 230, 330–332. [Google Scholar] [CrossRef]
- Lefer, A.M. Significance of lipid mediators in shock states. Circ. Shock 1989, 27, 3–12. [Google Scholar]
- Goetzl, E.J.; An, S.; Smith, W.L. Specificity of expression and effects of eicosanoid mediators in normal physiology and human diseases. FASEB J. 1995, 9, 1051–1058. [Google Scholar] [CrossRef]
- Lewis, R.A.; Austen, K.F. The biologically active leukotrienes. Biosynthesis, metabolism, receptors, functions, and pharmacology. J. Clin. Investig. 1984, 73, 889–897. [Google Scholar] [CrossRef]
- Clark, J.D.; Lin, L.L.; Kriz, R.W.; Ramesha, C.S.; Sultzman, L.A.; Lin, A.Y.; Milona, N.; Knopf, J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell 1991, 65, 1043–1051. [Google Scholar] [CrossRef]
- Reid, G.K.; Kargman, S.; Vickers, P.J.; Mancini, J.A.; Léveillé, C.; Ethier, D.; Miller, D.K.; Gillard, J.W.; Dixon, R.A.; Evans, J.F. Correlation between expression of 5-lipoxygenase-activating protein, 5-lipoxygenase, and cellular leukotriene synthesis. J. Biol. Chem. 1990, 265, 19818–19823. [Google Scholar] [CrossRef]
- Evans, J.F.; Dupuis, P.; Ford-Hutchinson, A.W. Purification and characterisation of leukotriene A4 hydrolase from rat neutrophils. Biochim. Biophys. Acta 1985, 840, 43–50. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Ali, A.; Vaillancourt, J.P.; Calaycay, J.R.; Mumford, R.A.; Zamboni, R.J.; Ford-Hutchinson, A.W. Purification to homogeneity and the N-terminal sequence of human leukotriene C4 synthase: A homodimeric glutathione S-transferase composed of 18-kDa subunits. Proc. Natl. Acad. Sci. USA 1993, 90, 2015–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.E.; Allison, R.D.; Meister, A. Interconversion of leukotrienes catalyzed by purified gamma-glutamyl transpeptidase: Concomitant formation of leukotriene D4 and gamma-glutamyl amino acids. Proc. Natl. Acad. Sci. USA 1982, 79, 1088–1091. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Lewis, R.A.; Corey, E.J.; Austen, K.F. Conversion of leukotriene D4 to leukotriene E4 by a dipeptidase released from the specific granule of human polymorphonuclear leucocytes. Immunology 1983, 48, 27–35. [Google Scholar]
- Hammarström, S.; Bernström, K.; Orning, L.; Dahlén, S.E.; Hedqvist, P. Rapid in vivo metabolism of leukotriene C3 in the monkey Macaca irus. Biochem. Biophys. Res. Commun. 1981, 101, 1109–1115. [Google Scholar] [CrossRef]
- Mayatepek, E.; Hoffmann, G.F. Leukotrienes: Biosynthesis, Metabolism, and Pathophysiologic Significance. Pediatr. Res. 1995, 37, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Maclouf, J.; Antoine, C.; De Caterina, R.; Sicari, R.; Murphy, R.C.; Patrignani, P.; Loizzo, S.; Patrono, C. Entry rate and metabolism of leukotriene C4 into vascular compartment in healthy subjects. Am. J. Physiol. 1992, 263, H244–H249. [Google Scholar] [CrossRef]
- Colazzo, F.; Gelosa, P.; Tremoli, E.; Sironi, L.; Castiglioni, L. Role of the Cysteinyl Leukotrienes in the Pathogenesis and Progression of Cardiovascular Diseases. Mediat. Inflamm. 2017, 2017, 2432958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, A.; Austen, K.F.; Kanaoka, Y. Targeted gene disruption reveals the role of cysteinyl leukotriene 1 receptor in the enhanced vascular permeability of mice undergoing acute inflammatory responses. J. Biol. Chem. 2002, 277, 20820–20824. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Boyce, J.A. Cysteinyl Leukotrienes and Their Receptors; Emerging Concepts. Allergy Asthma Immunol. Res. 2014, 6, 288. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, A.; Balestrieri, B.; Austen, K.F.; Kanaoka, Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc. Natl. Acad. Sci. USA 2009, 106, 11685–11690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaoka, Y.; Maekawa, A.; Austen, K.F. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J. Biol. Chem. 2013, 288, 10967–10972. [Google Scholar] [CrossRef] [Green Version]
- Morlion, B.J.; Torwesten, E.; Kuhn, K.S.; Puchstein, C.; Fürst, P. Cysteinyl-leukotriene generation as a biomarker for survival in the critically ill. Crit. Care Med. 2000, 28, 3655–3658. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Maekawa, A.; Penrose, J.F.; Austen, K.F.; Lam, B.K. Attenuated zymosan-induced peritoneal vascular permeability and IgE-dependent passive cutaneous anaphylaxis in mice lacking leukotriene C4 synthase. J. Biol. Chem. 2001, 276, 22608–22613. [Google Scholar] [CrossRef] [Green Version]
- Beller, T.C.; Maekawa, A.; Friend, D.S.; Austen, K.F.; Kanaoka, Y. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J. Biol. Chem. 2004, 279, 46129–46134. [Google Scholar] [CrossRef] [Green Version]
- Hui, Y.; Cheng, Y.; Smalera, I.; Jian, W.; Goldhahn, L.; Fitzgerald, G.A.; Funk, C.D. Directed vascular expression of human cysteinyl leukotriene 2 receptor modulates endothelial permeability and systemic blood pressure. Circulation 2004, 110, 3360–3366. [Google Scholar] [CrossRef] [Green Version]
- Mellor, E.A.; Austen, K.F.; Boyce, J.A. Cysteinyl Leukotrienes and Uridine Diphosphate Induce Cytokine Generation by Human Mast Cells Through an Interleukin 4–regulated Pathway that Is Inhibited by Leukotriene Receptor Antagonists. J. Exp. Med. 2002, 195, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Keppler, D.; Hagmann, W.; Rapp, S. Role of leukotrienes in endotoxin action in vivo. Rev. Infect. Dis 1987, 9 (Suppl. S5), S580–S584. [Google Scholar] [CrossRef]
- Benjamim, C.F.; Canetti, C.; Cunha, F.Q.; Kunkel, S.L.; Peters-Golden, M. Opposing and hierarchical roles of leukotrienes in local innate immune versus vascular responses in a model of sepsis. J. Immunol. 2005, 174, 1616–1620. [Google Scholar] [CrossRef]
- Khodir, A.E.; Ghoneim, H.A.; Rahim, M.A.; Suddek, G.M. Montelukast reduces sepsis-induced lung and renal injury in rats. Can. J. Physiol. Pharmacol. 2014, 92, 839–847. [Google Scholar] [CrossRef]
- Mohamadin, A.M.; Elberry, A.A.; Elkablawy, M.A.; Gawad, H.S.; Al-Abbasi, F.A. Montelukast, a leukotriene receptor antagonist abrogates lipopolysaccharide-induced toxicity and oxidative stress in rat liver. Pathophysiology 2011, 18, 235–242. [Google Scholar] [CrossRef]
- Coskun, A.K.; Yigiter, M.; Oral, A.; Odabasoglu, F.; Halici, Z.; Mentes, O.; Cadirci, E.; Atalay, F.; Suleyman, H. The effects of montelukast on antioxidant enzymes and proinflammatory cytokines on the heart, liver, lungs, and kidneys in a rat model of cecal ligation and puncture-induced sepsis. Sci. World J. 2011, 11, 1341–1356. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Simera, I.; Moher, D.; Hoey, J.; Schulz, K.F.; Altman, D.G. A catalogue of reporting guidelines for health research. Eur. J. Clin. Investig. 2010, 40, 35–53. [Google Scholar] [CrossRef]
- Shinohara, M.; Kibi, M.; Riley, I.R.; Chiang, N.; Dalli, J.; Kraft, B.D.; Piantadosi, C.A.; Choi, A.M.; Serhan, C.N. Cell-cell interactions and bronchoconstrictor eicosanoid reduction with inhaled carbon monoxide and resolvin D1. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L746–L757. [Google Scholar] [CrossRef]
- Zeltsman, D.; Quinn, J.V.; Mores, C.; Slotman, G.J. Pathophysiologic plasma levels of leukotriene C4 in relation to the hemodynamic dysfunction and mediator release of graded bacteremia. Shock 1997, 7, 282–287. [Google Scholar] [CrossRef]
- Tran, H.S.; Quinn, J.V.; Puc, M.M.; Woolley, D.S.; Puglisi, R.N.; Slotman, G.J. The cardiovascular hemodynamics and leukotriene kinetics during prostacyclin and anti-prostacyclin antibody infusions in septic shock. Shock 2000, 13, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, S.M.; Ofenstein, J.P.; Sarnaik, A.P. Ibuprofen attenuates cardiopulmonary dysfunction by modifying vascular tone in endotoxemia. Prostaglandins Leukot. Essent. Fat. Acids 1999, 60, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Buchholz, U.; Kojima, H.; Keppler, A.; Hafkemeyer, P.; Fukui, H.; Tsujii, T.; Keppler, D. Cysteinyl leukotrienes in the urine of patients with liver diseases. Hepatology 1994, 20, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Fauler, J.; Tsikas, D.; Holch, M.; Seekamp, A.; Nerlich, M.L.; Sturm, J.; Frölich, J.C. Enhanced urinary excretion of leukotriene E4 by patients with multiple trauma with or without adult respiratory distress syndrome. Clin. Sci. 1991, 80, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Hellman, J. Oxidative stress and endothelial dysfunction in sepsis and acute inflammation. Antioxid Redox Signal. 2021, 35, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Amalakuhan, B.; Habib, S.A.; Mangat, M.; Reyes, L.F.; Rodriguez, A.H.; Hinojosa, C.A.; Soni, N.J.; Gilley, R.P.; Bustamante, C.A.; Anzueto, A.; et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine 2016, 88, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Cummings, C.J.; Sessler, C.N.; Beall, L.D.; Fisher, B.J.; Best, A.M.; Fowler, A.A., 3rd. Soluble E-selectin levels in sepsis and critical illness. Correlation with infection and hemodynamic dysfunction. Am. J. Respir. Crit. Care Med. 1997, 156, 431–437. [Google Scholar] [CrossRef]
- Duah, E.; Adapala, R.K.; Al-Azzam, N.; Kondeti, V.; Gombedza, F.; Thodeti, C.K.; Paruchuri, S. Cysteinyl leukotrienes regulate endothelial cell inflammatory and proliferative signals through CysLT2 and CysLT1 receptors. Sci. Rep. 2013, 3, 3274. [Google Scholar] [CrossRef] [Green Version]
- Carson, W.E.; Yu, H.; Dierksheide, J.; Pfeffer, K.; Bouchard, P.; Clark, R.; Durbin, J.; Baldwin, A.S.; Peschon, J.; Johnson, P.R.; et al. A fatal cytokine-induced systemic inflammatory response reveals a critical role for NK cells. J. Immunol. 1999, 162, 4943–4951. [Google Scholar]
- Dinarello, C.A. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 1997, 112, 321s–329s. [Google Scholar] [CrossRef] [Green Version]
- Hauptmann, B.; Van Damme, J.; Dayer, J.M. Modulation of IL-1 inflammatory and immunomodulatory properties by IL-6. Eur. Cytokine Netw. 1991, 2, 39–46. [Google Scholar]
- Van der Poll, T.; Büller, H.R.; Ten Cate, H.; Wortel, C.H.; Bauer, K.A.; van Deventer, S.J.; Hack, C.E.; Sauerwein, H.P.; Rosenberg, R.D.; Ten Cate, J.W. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N. Engl. J. Med. 1990, 322, 1622–1627. [Google Scholar] [CrossRef]
- Kishimoto, T.; Akira, S.; Narazaki, M.; Taga, T. Interleukin-6 family of cytokines and gp130. Blood 1995, 86, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Oberholzer, A.; Oberholzer, C.; Moldawer, L.L. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit. Care Med. 2002, 30, S58–S63. [Google Scholar] [CrossRef]
- Sala, A.; Folco, G. Neutrophils, endothelial cells, and cysteinyl leukotrienes: A new approach to neutrophil-dependent inflammation? Biochem. Biophys Res. Commun. 2001, 283, 1003–1006. [Google Scholar] [CrossRef]
- Faix, J.D. Biomarkers of sepsis. Crit. Rev. Clin. Lab. Sci. 2013, 50, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, A.; Carbone, F.; Vecchié, A.; Meessen, J.; Ferraris, S.; Beck, E.; Keim, R.; Minetti, S.; Elia, E.; Ferrara, D.; et al. The role of resistin and myeloperoxidase in severe sepsis and septic shock: Results from the ALBIOS trial. Eur. J. Clin. Investig. 2020, 50, e13333. [Google Scholar] [CrossRef]
- Macdonald, J.; Galley, H.F.; Webster, N.R. Oxidative stress and gene expression in sepsis. Br. J. Anaesth 2003, 90, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, J.; Jacobs, J.D.; Jennings, L.K. Interaction of myeloperoxidase with vascular NAD(P)H oxidase-derived reactive oxygen species in vasculature: Implications for vascular diseases. Am. J. Physiol. Circ. Physiol. 2003, 285, H2563–H2572. [Google Scholar] [CrossRef] [Green Version]
- Ware, L.B.; Fessel, J.P.; May, A.K.; Roberts, L.J. Plasma Biomarkers of Oxidant Stress and Development of Organ Failure in Severe Sepsis. Shock 2011, 36, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Sener, G.; Sehirli, O.; Cetinel, S.; Ercan, F.; Yüksel, M.; Gedik, N.; Yeğen, B.C. Amelioration of sepsis-induced hepatic and ileal injury in rats by the leukotriene receptor blocker montelukast. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 453–462. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Boyce, J.A. Cysteinyl leukotrienes and their receptors: Cellular distribution and function in immune and inflammatory responses. J. Immunol. 2004, 173, 1503–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, M.M.; Fink, M.P.; Marshall, J.C.; Abraham, E.; Angus, D.; Cook, D.; Cohen, J.; Opal, S.M.; Vincent, J.L.; Ramsay, G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit. Care Med. 2003, 31, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Prowle, J.R. Sepsis-Associated AKI. Clin. J. Am. Soc. Nephrol. 2018, 13, 339–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urushihara, M.; Kondo, S.; Kagami, S.; Kobori, H. Urinary angiotensinogen accurately reflects intrarenal Renin-Angiotensin system activity. Am. J. Nephrol. 2010, 31, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Navar, L.G. Urinary Angiotensinogen as a Novel Biomarker of Intrarenal Renin-Angiotensin System in Chronic Kidney Disease. Int. Rev. Thromb. 2011, 6, 108–116. [Google Scholar]
- Ohashi, N.; Aoki, T.; Matsuyama, T.; Ishigaki, S.; Isobe, S.; Katahashi, N.; Sato, T.; Fujikura, T.; Kato, A.; Yasuda, H. The Urinary Angiotensinogen to Urinary Albumin Ratio Reflects Whether the Renin-angiotensin System in the Kidney Is Activated due to Filtration of Plasma Angiotensinogen through the Damaged Glomeruli or the Production of Angiotensinogen in the Proximal Tubules. Intern. Med. 2020, 59, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Ba Aqeel, S.H.; Sanchez, A.; Batlle, D. Angiotensinogen as a biomarker of acute kidney injury. Clin. Kidney J. 2017, 10, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Alge, J.L.; Karakala, N.; Neely, B.A.; Janech, M.G.; Velez, J.C.; Arthur, J.M. Urinary angiotensinogen predicts adverse outcomes among acute kidney injury patients in the intensive care unit. Crit. Care 2013, 17, R69. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yin, Y.; Yao, Y. Advances in sepsis-associated liver dysfunction. Burn. Trauma 2014, 2, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Hofer, S.; Brenner, T.; Bopp, C.; Steppan, J.; Lichtenstern, C.; Weitz, J.; Bruckner, T.; Martin, E.; Hoffmann, U.; Weigand, M.A. Cell death serum biomarkers are early predictors for survival in severe septic patients with hepatic dysfunction. Crit. Care 2009, 13, R93. [Google Scholar] [CrossRef] [Green Version]
- Bakker, J.; Gris, P.; Coffernils, M.; Kahn, R.J.; Vincent, J.L. Serial blood lactate levels can predict the development of multiple organ failure following septic shock. Am. J. Surg. 1996, 171, 221–226. [Google Scholar] [CrossRef]
- Jansen, T.C.; van Bommel, J.; Woodward, R.; Mulder, P.G.; Bakker, J. Association between blood lactate levels, Sequential Organ Failure Assessment subscores, and 28-day mortality during early and late intensive care unit stay: A retrospective observational study. Crit. Care Med. 2009, 37, 2369–2374. [Google Scholar] [CrossRef]
- Bakker, J.; Nijsten, M.W.; Jansen, T.C. Clinical use of lactate monitoring in critically ill patients. Ann. Intensive Care 2013, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Jansen, T.C.; van Bommel, J.; Schoonderbeek, F.J.; Sleeswijk Visser, S.J.; van der Klooster, J.M.; Lima, A.P.; Willemsen, S.P.; Bakker, J. Early lactate-guided therapy in intensive care unit patients: A multicenter, open-label, randomized controlled trial. Am. J. Respir. Crit. Care Med. 2010, 182, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Gotmaker, R.; Peake, S.L.; Forbes, A.; Bellomo, R.; Investigators, A. Mortality is Greater in Septic Patients With Hyperlactatemia Than With Refractory Hypotension. Shock 2017, 48, 294–300. [Google Scholar] [CrossRef]
- Bakker, J.; Postelnicu, R.; Mukherjee, V. Lactate: Where Are We Now? Crit. Care Clin. 2020, 36, 115–124. [Google Scholar] [CrossRef]
- Hernandez, G.; Ospina-Tascon, G.A.; Damiani, L.P.; Estenssoro, E.; Dubin, A.; Hurtado, J.; Friedman, G.; Castro, R.; Alegria, L.; Teboul, J.L.; et al. Effect of a Resuscitation Strategy Targeting Peripheral Perfusion Status vs. Serum Lactate Levels on 28-Day Mortality Among Patients With Septic Shock: The ANDROMEDA-SHOCK Randomized Clinical Trial. JAMA 2019, 321, 654–664. [Google Scholar] [CrossRef]
- Hernandez, G.; Bellomo, R.; Bakker, J. The ten pitfalls of lactate clearance in sepsis. Intensive Care Med. 2019, 45, 82–85. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. Role of the Heart in Lactate Shuttling. Front. Nutr. 2021, 8, 663560. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Hernandez, G.; Boerma, E.C.; Dubin, A.; Bruhn, A.; Koopmans, M.; Edul, V.K.; Ruiz, C.; Castro, R.; Pozo, M.O.; Pedreros, C.; et al. Severe abnormalities in microvascular perfused vessel density are associated to organ dysfunctions and mortality and can be predicted by hyperlactatemia and norepinephrine requirements in septic shock patients. J. Crit. Care 2013, 28, 538.e9–538.e14. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.C.; van Bommel, J.; Bakker, J. Blood lactate monitoring in critically ill patients: A systematic health technology assessment. Crit. Care Med. 2009, 37, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Regueira, T.; Bruhn, A.; Castro, R.; Rovegno, M.; Fuentealba, A.; Veas, E.; Berrutti, D.; Florez, J.; Kattan, E.; et al. Relationship of systemic, hepatosplanchnic, and microcirculatory perfusion parameters with 6-hour lactate clearance in hyperdynamic septic shock patients: An acute, clinical-physiological, pilot study. Ann. Intensive Care 2012, 2, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmon, C.T. Crosstalk between inflammation and thrombosis. Maturitas 2004, 47, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken barriers: A new take on sepsis pathogenesis. Sci. Transl. Med. 2011, 3, 88ps25. [Google Scholar] [CrossRef]
- Gourdin, M.J.; Bree, B.; De Kock, M. The impact of ischaemia-reperfusion on the blood vessel. Eur. J. Anaesthesiol. 2009, 26, 537–547. [Google Scholar] [CrossRef]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kellum, J.A.; Pike, F.; Yealy, D.M.; Huang, D.T.; Shapiro, N.I.; Angus, D.C.; the Protocol-Based Care for Early Septic Shock Investigators (ProCESS) Investigators. Relationship Between Alternative Resuscitation Strategies, Host Response and Injury Biomarkers, and Outcome in Septic Shock: Analysis of the Protocol-Based Care for Early Septic Shock Study. Crit. Care Med. 2017, 45, 438–445. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic, Clinical, and Biochemical Parameters | Controls (n = 22) | SS patients (n = 13) |
|---|---|---|
| Age (Years) | 56 ± 1 | 62 ± 4 |
| Gender: men, n (%) | 13 (59) | 5 (38) |
| Gender: women, n (%) | 9 (41) | 8 (62) |
| APACHE II score | n/a | 26 ± 2 |
| SAPS II score | n/a | 55 ± 4 |
| SOFA score VV-ECMO,n(%) | n/an/a | 9 (7; 12) 1 (8) |
| Lactate (mmol/L) normal: <2 | n.d. | 2.6 (1.3; 3.7) |
| Source of sepsis, n(%) | ||
| Respiratory tract infection | n/a | 2 (15) |
| Renal and genitourinary tract infection | n/a | 10 (77) |
| Skin and cutaneous tissue infection | n/a | 1(8) |
| Microbiology,n(%) | ||
| Gram-negative | n/a | 7 (54) |
| Other | n/a | 2 (15) |
| Unidentified | n/a | 4 (31) |
| Comorbidities,n(%) | ||
| Diabetes mellitus type 2 | n.d. | 3 (23) |
| Arterial hypertension | n.d. | 5 (38) |
| Atrial fibrillation | n.d. | 3 (23) |
| Dyslipidemia | n.d. | 3 (23) |
| Cardiovascular disease | n.d. | 1 (8) |
| Obesity | n.d. | 2 (15) |
| Alcohol abuse | n.d. | 1 (8) |
| Cardiac biomarkers | ||
| BNP (pg/mL) normal: <100 | n.d. | 382 (315; 526) |
| hsTnI (ng/L) normal: <34 (men); <16 (women) | n.d. | 295 (74; 2251) |
| Renal function | ||
| eGFR (mL/min per 1.73 m2) normal: ≥90; mildly reduced: 60–89 | 77 ± 4 | 36 ± 6 |
| Urea (mg/dL) normal: 10–50 mg/dL | n.d | 93 ± 13 |
| Urine protein/creatinine (mg/mg) normal: <0.2 | n.d. | 2.0 ± 0.4 |
| Respiratory parameters | ||
| PaO2/FiO2 (P/F) ratio normal: ≥300 | n/a | 288 ± 31 |
| Hepatic biomarkers | ||
| AST (U/L) normal: 10–37 | n.d. | 56 (29; 98) |
| ALT (U/L) normal: 10–37 | n.d. | 35 (18; 98) |
| ALP (U/L) normal: 30–120 | n.d. | 69 (65; 106) |
| GGT (U/L) normal: 10–49 | n.d. | 37 (19;68) |
| Total bilirubin (mg/dL) normal: <1.2 | n.d. | 0.9 (0.5; 3.1) |
| PT (s) normal: 9.6–13.6 | n.d. | 14.3 (13.4; 18.0) |
| Therapeutics at admission | ||
| Norepinephrine dose (µg/kg/min) | n/a | 0.76 ± 0.15 |
| Use of renal replacement therapy, n (%) | n/a | 2 (15) |
| Antibiotic therapy, n (%) | ||
| Ceftriaxone | n/a | 9 (69) |
| Piperacillin/tazobactam | n/a | 4 (31) |
| Dual or triple combination | n/a | 4 (31) |
| Antiviral therapy, n (%) | n/a | 1 (8) |
| Outcomes | ||
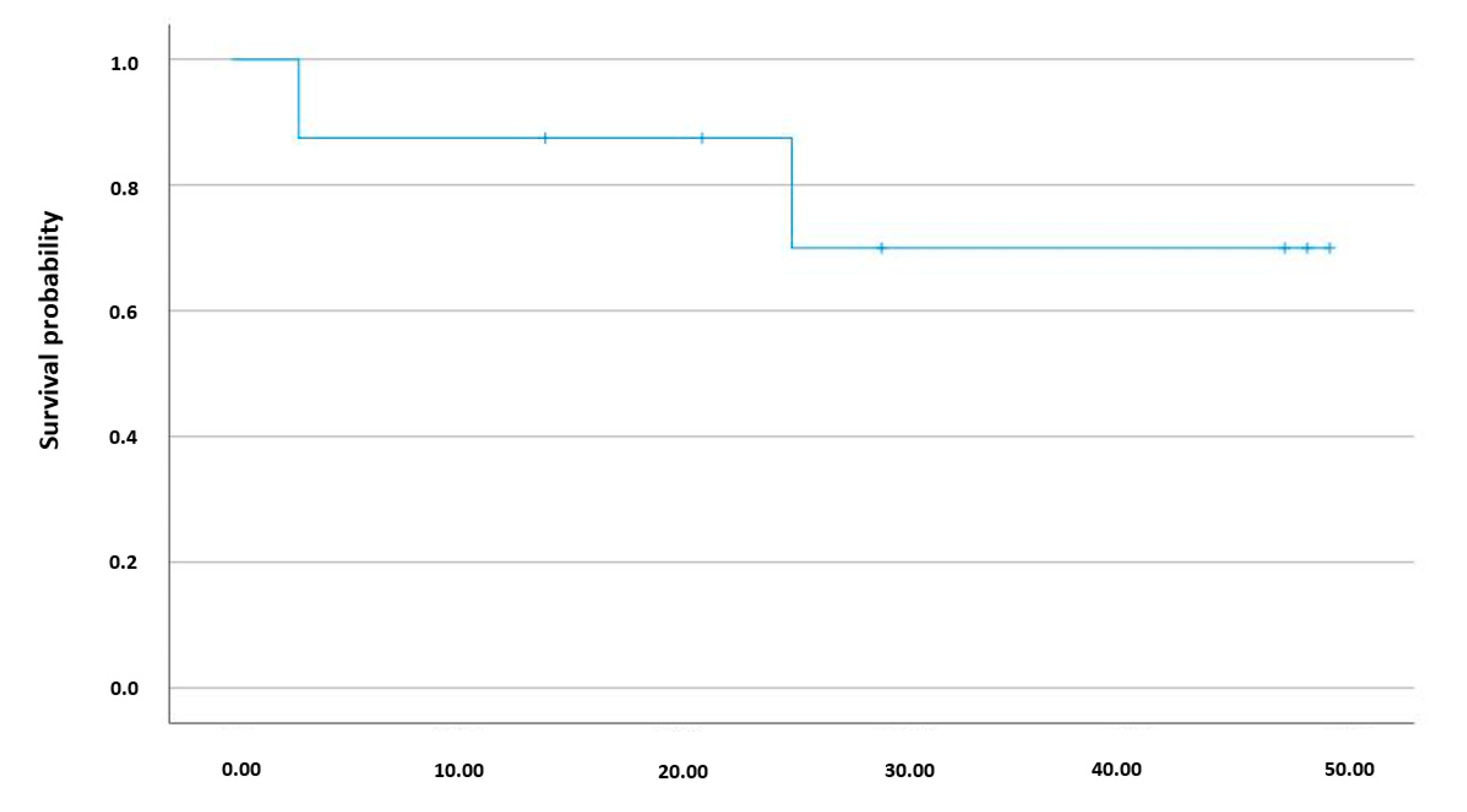
| ICU length of stay (days) | n/a | 6 (4; 9) |
| Total hospital length of stay (days) | n/a | 7 (5; 11) |
| In-hospital mortality, n (%) | n/a | 1 (8) |
| Mortality within 12 months, n (%) | n/a | 1 (8) |
| Biomarkers | Controls | n | SS | n | p-Value |
|---|---|---|---|---|---|
| Endothelial activation | |||||
| s-ICAM-1 (ng/mL) | 474.9 ± 70.7 | 22 | 694.5 ± 75.3 | 13 | 0.038 |
| s-VCAM-1 (ng/mL) | 705.1 (516.2; 827.7) | 22 | 3810.0 (2201.0; 5232.0) | 13 | <0.001 |
| s-E-Selectin (ng/mL) | 27.7 (22.6; 34.1) | 22 | 216.4 (169.1; 334.5) | 13 | <0.001 |
| Inflammatory status | |||||
| s-TNF-α (pg/mL) | 4.8 (3.6; 6.2) | 22 | 36.3 (16.6; 49.3) | 13 | <0.001 |
| s-IL-1β (pg/mL) | 0.0 (0.0; 0.2) | 22 | 0.2 (0.1; 0.5) | 13 | 0.010 |
| s-IL-6 (pg/mL) | 0.7 (0.0; 2.4) | 22 | 31.0 (12.9; 293.2) | 13 | <0.001 |
| s-IL-10 (pg/mL) | 0.0 (0.0; 1.2) | 22 | 49.1 (19.5; 147.6) | 12 | <0.001 |
| s-MPO (ng/mL) | 116 (76; 149) | 22 | 746 (475; 1162) | 13 | <0.001 |
| s-CRP (mg/L) | n.d. | - | 225.3 ± 26.4 | 13 | n/a |
| Total leukocytes (×109/L) | n.d. | - | 19.2 ± 2.1 | 13 | n/a |
| Neutrophils (×109/L) | n.d. | - | 17.1 ± 1.9 | 13 | n/a |
| Monocytes (×109/L) | n.d. | - | 0.6 ± 0.1 | 13 | n/a |
| Lymphocytes (×109/L) | n.d. | - | 0.6 (0.5; 0.8) | 13 | n/a |
| NLR | n.d. | - | 23.3 (14.2; 45.7) | 13 | n/a |
| NMR | n.d. | - | 23.3 (15.3; 58.4) | 13 | n/a |
| u-CysLT (pg/mg creatinine) | 495 (373; 639) | 22 | 1242 (599; 1583) | 13 | 0.003 |
| Oxidative stress | |||||
| u-Isop (ng/mg creatinine) | 1.7 (1.0; 2.7) | 22 | 3.4 (2.8; 5.2) | 13 | 0.002 |
| Systemic and intrarenal RAAS | |||||
| p-AGT (μg/mL) | 19.6 (17.5; 24.4) | 22 | 35.3 (21.4; 46.4) | 13 | 0.003 |
| u-AGT (ng/mg creatinine) | 3 (2; 7) | 22 | 256 (45; 549) | 13 | <0.001 |
| Biomarkers/Parameters | Days 1–2 | n | Days 3–4 | n | Days 5–8 | n |
|---|---|---|---|---|---|---|
| Endothelial activation | ||||||
| s-ICAM-1 (ng/mL) | 694.5 ± 75.3 | 13 | 559.0 ± 92.1 * | 8 | 553.3 ± 85.3 | 6 |
| s-VCAM-1 (ng/mL) | 3855.0 ± 488.7 | 13 | 2559.0 ± 479.3 | 8 | 1912.0 ± 488.8 * | 6 |
| s-E-Selectin (ng/mL) | 240.9 ± 30.1 | 13 | 137.1 ± 23.2 ** | 8 | 138.2 ± 24.1 ** | 6 |
| Inflammatory status | ||||||
| s-TNF-α (pg/mL) | 36.3 (16.6; 49.3) | 13 | 13.3 (5.9; 18.9) * | 8 | 14.2 (8.9; 38.8) | 6 |
| s-IL-1β (pg/mL) | 0.21 (0.07; 0.48) | 13 | 0.03 (0.01; 0.08) * | 8 | 0.15 (0.00; 0.68) | 6 |
| s-IL-6 (pg/mL) | 31.0 (12.9; 293.2) | 13 | 23.2 (5.6; 54.4) | 8 | 11.4 (6.8; 25.3) | 6 |
| s-IL-10 (pg/mL) | 49.1 (19.5; 147.6) | 12 | 18.5 (1.7; 70.9) | 8 | 5.4 (2.4; 39.7) | 5 |
| s-MPO (mg/mL) | 746 (475; 1162) | 13 | 548 (301; 688) | 8 | 227 (192; 590) | 6 |
| s-CRP (mg/L) | 225.3 ± 26.4 | 13 | 162.0 ± 43.6* | 8 | 91.5 ± 19.2 * | 5 |
| Total leukocytes (×109/L) | 19.2 ± 2.1 | 13 | 18.0 ± 3.7 | 8 | 12.1 ± 3.8 | 6 |
| Neutrophils (×109/L) | 17.1 ± 1.9 | 13 | 14.8 ± 3.4 | 8 | 8.8 ± 2.8 | 6 |
| Monocytes (×109/L) | 0.6 ± 0.1 | 13 | 0.8 ± 0.1 | 8 | 0.9 ± 0.2 | 6 |
| Lymphocytes (×109/L) | 0.6 (0.5; 0.8) | 13 | 1.3 (0.9; 2.2) | 8 | 1.3 (0.9; 1.8) | 6 |
| NLR | 23.3 (14.2; 45.7) | 13 | 7.3 (5.8; 18.7) ** | 8 | 6.6 (3.4; 8.6) * | 6 |
| NMR | 23.3 (15.3; 58.4) | 13 | 19.10 (8.4; 29.6) ** | 8 | 9.9 (5.4; 13.5) * | 6 |
| u-CysLT (ng/mg creatinine) | 1242 (599; 1583) | 13 | 873 (758; 1842) | 7 | 1332 (484; 2368) | 5 |
| Oxidative stress | ||||||
| u-Isop (ng/mg creatinine) | 3.4 (2.8; 5.2) | 13 | 4.2 (2.7; 10.1) | 7 | 3.6 (2.2; 7.7) | 5 |
| Systemic and intrarenal RAAS | ||||||
| p-AGT (μg/mL) | 35.4 ± 4.1 | 13 | 36.5 ± 4.8 | 8 | 36.6 ± 6.8 | 6 |
| u-AGT (ng/mg creatinine) | 256 (45; 549) | 13 | 104 (57; 361) | 8 | 316 (143; 1735) | 6 |
| Parameters | Days 1–2 | n | Days 3–4 | n | Days 5–8 | n |
|---|---|---|---|---|---|---|
| Lactate (mmol/L) | 2.6 (1.3; 3.7) | 13 | 1.3 (1.1; 1.6) * | 8 | 1.5 (1.1; 1.7) | 4 |
| SOFA score | 9 (7; 12) | 13 | 6 (2; 11) | 8 | 6 (4; 7) * | 4 |
| Cardiac biomarkers | ||||||
| BNP (pg/mL) | 382 (315; 526) | 8 | n.d. | - | n.d. | - |
| hsTnI (ng/L) | 295 (74; 2251) | 13 | 128 (32; 5460) | 6 | 10 (5; 495) | 4 |
| Renal function | ||||||
| eGFR (mL/min per 1.73 m2) | 36 ± 6 | 13 | 62 ± 12 * | 8 | 82 ± 12 ** | 5 |
| Urea (mg/dL) | 79 (59; 124) | 13 | 55 (44; 82) * | 8 | 71 (46; 125) | 6 |
| Urine protein/creatinine (mg/mg) | 2.0 ± 0.4 | 10 | 2.0 ± 0.5 | 6 | 1.0 ± 0.3 | 3 |
| Respiratory parameters | ||||||
| PaO2/FiO2 (P/F) ratio | 288 ± 31 | 12 | 282 ± 43 | 8 | 223 ± 12 | 5 |
| Hepatic biomarkers | ||||||
| AST (U/L) | 56 (29; 98) | 13 | 61 (17; 236) | 6 | 86 (41; 476) | 5 |
| ALT (U/L) | 35 (18; 98) | 13 | 90 (42; 309) | 6 | 119 (49; 454) | 5 |
| ALP (U/L) | 69 (65; 106) | 13 | 125 (92; 214) * | 6 | 227 (108; 297) | 5 |
| GGT (U/L) | 37 (19;68) | 13 | 81 (47; 89) * | 6 | 135 (77; 217) | 4 |
| Total bilirubin (mg/dL) | 0.9 (0.5; 3.1) | 13 | 0.9 (0.5; 3.6) | 6 | 1.9 (0.6; 8.8) | 5 |
| PT (s) | 14.3 (13.4; 18.0) | 12 | 13.6 (12.7; 14.5) * | 8 | 12.9 (11.0; 15.2) | 5 |
| u-CysLT (pg/mg Creatinine) | Adjusted β | 95% CI | p-Value |
|---|---|---|---|
| Model 1 | |||
| s-ICAM-1 (ng/mL) | 1.760 | 0.281–3.238 | 0.020 |
| Model 2 | |||
| s-VCAM-1 (ng/mL) | 0.268 | 0.043–0.492 | 0.019 |
| Model 3 | |||
| s-IL-6 (pg/mL) | 0.219 | 0.068–0.369 | 0.004 |
| Model 4 | |||
| u-Isop (ng/mg creatinine) | 299 | 185–414 | <0.001 |
| Model 5 | |||
| u-protein/creatinine (mg/mg) | 434 | 226–642 | <0.001 |
| Model 6 | |||
| AST (U/L) | 1.992 | 0.936–3.048 | <0.001 |
| Model 7 | |||
| ALT (U/L) | 1.523 | 0.719–2.326 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reina-Couto, M.; Santos-Oliveira, M.; Pereira-Terra, P.; Silva-Pereira, C.; Quelhas-Santos, J.; Duarte, Á.; Martins, S.; Serrão, P.; Dias, C.C.; Morato, M.; et al. Urinary Cysteinyl Leukotrienes as Biomarkers of Endothelial Activation, Inflammation and Oxidative Stress and Their Relationship with Organ Dysfunction in Human Septic Shock. Biomedicines 2022, 10, 2845. https://doi.org/10.3390/biomedicines10112845
Reina-Couto M, Santos-Oliveira M, Pereira-Terra P, Silva-Pereira C, Quelhas-Santos J, Duarte Á, Martins S, Serrão P, Dias CC, Morato M, et al. Urinary Cysteinyl Leukotrienes as Biomarkers of Endothelial Activation, Inflammation and Oxidative Stress and Their Relationship with Organ Dysfunction in Human Septic Shock. Biomedicines. 2022; 10(11):2845. https://doi.org/10.3390/biomedicines10112845
Chicago/Turabian StyleReina-Couto, Marta, Marisa Santos-Oliveira, Patrícia Pereira-Terra, Carolina Silva-Pereira, Janete Quelhas-Santos, Álvaro Duarte, Sandra Martins, Paula Serrão, Cláudia Camila Dias, Manuela Morato, and et al. 2022. "Urinary Cysteinyl Leukotrienes as Biomarkers of Endothelial Activation, Inflammation and Oxidative Stress and Their Relationship with Organ Dysfunction in Human Septic Shock" Biomedicines 10, no. 11: 2845. https://doi.org/10.3390/biomedicines10112845