
The Role of Epigenetics in the Development and Progression of Multiple Myeloma
 ,
,  ,
,  ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
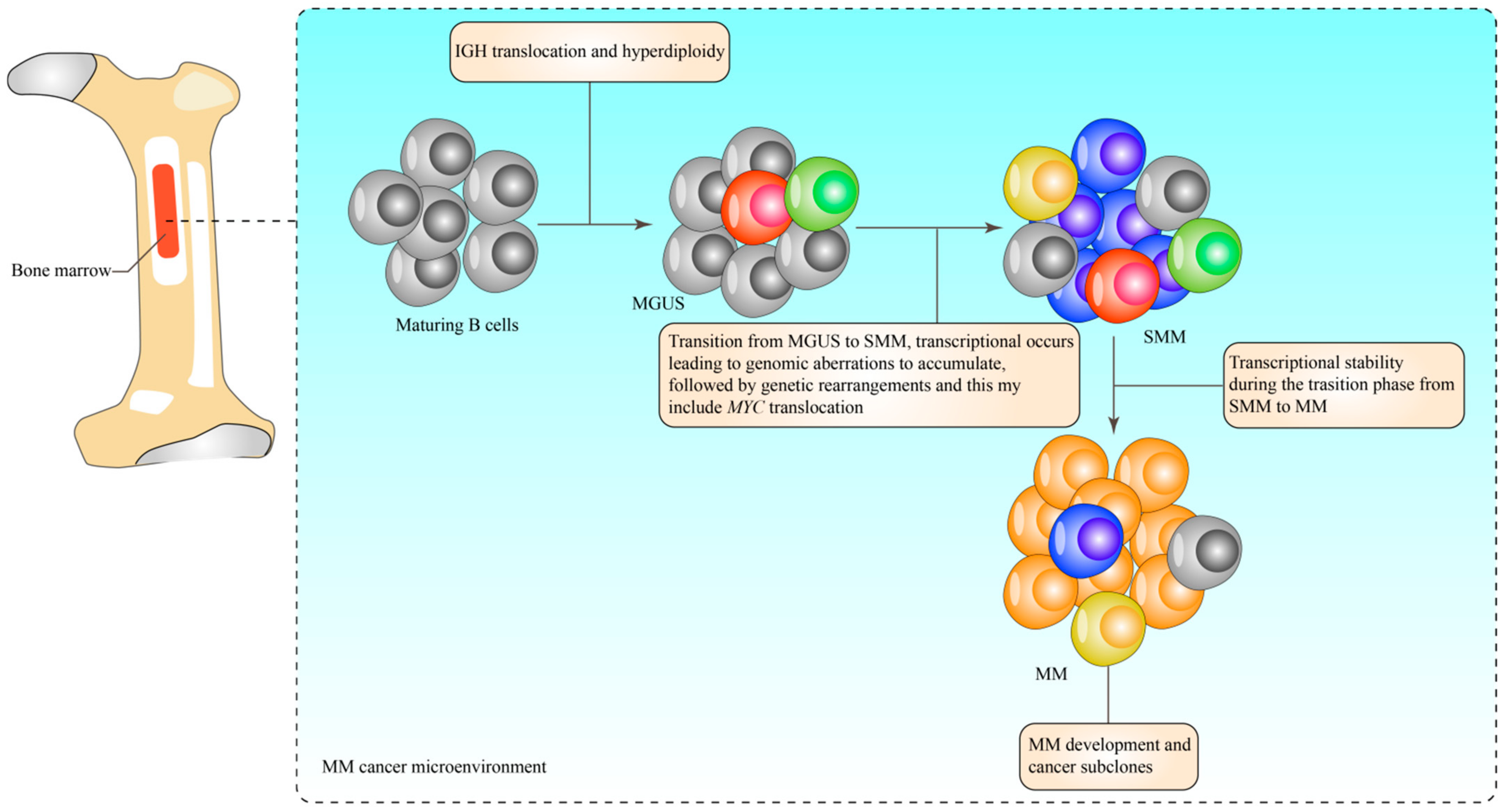
2. Characterisation of Multiple Myeloma
3. Epigenetics
4. DNA Methylation in the Development of Multiple Myeloma
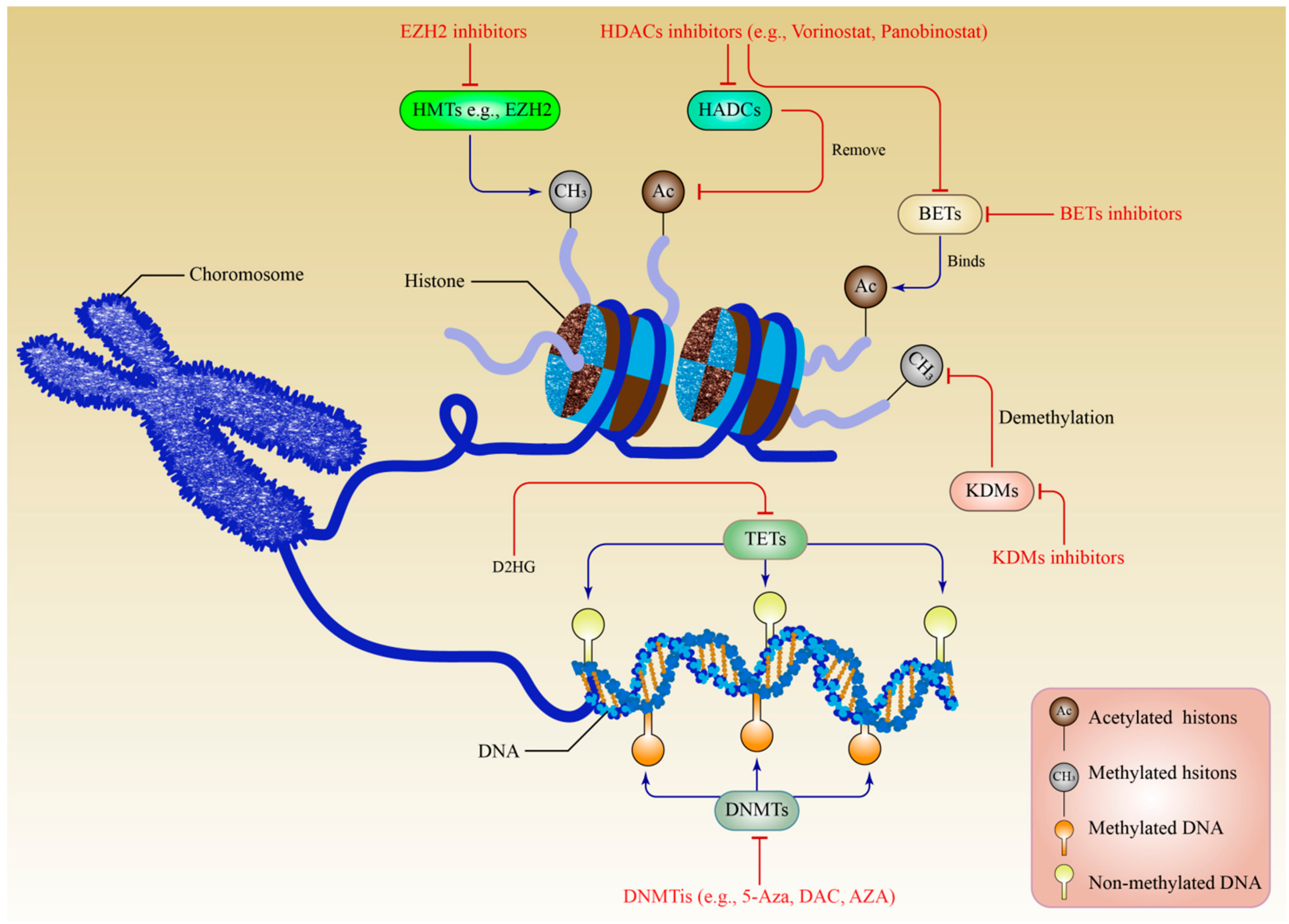
5. Histone Modifications in Multiple Myeloma
5.1. Histone Methylation
5.2. Roles of Histone Methylation Modifiers in Multiple Myeloma
5.3. Histone Acetylation
5.4. Roles of Histone Acetylation Modifiers in Multiple Myeloma
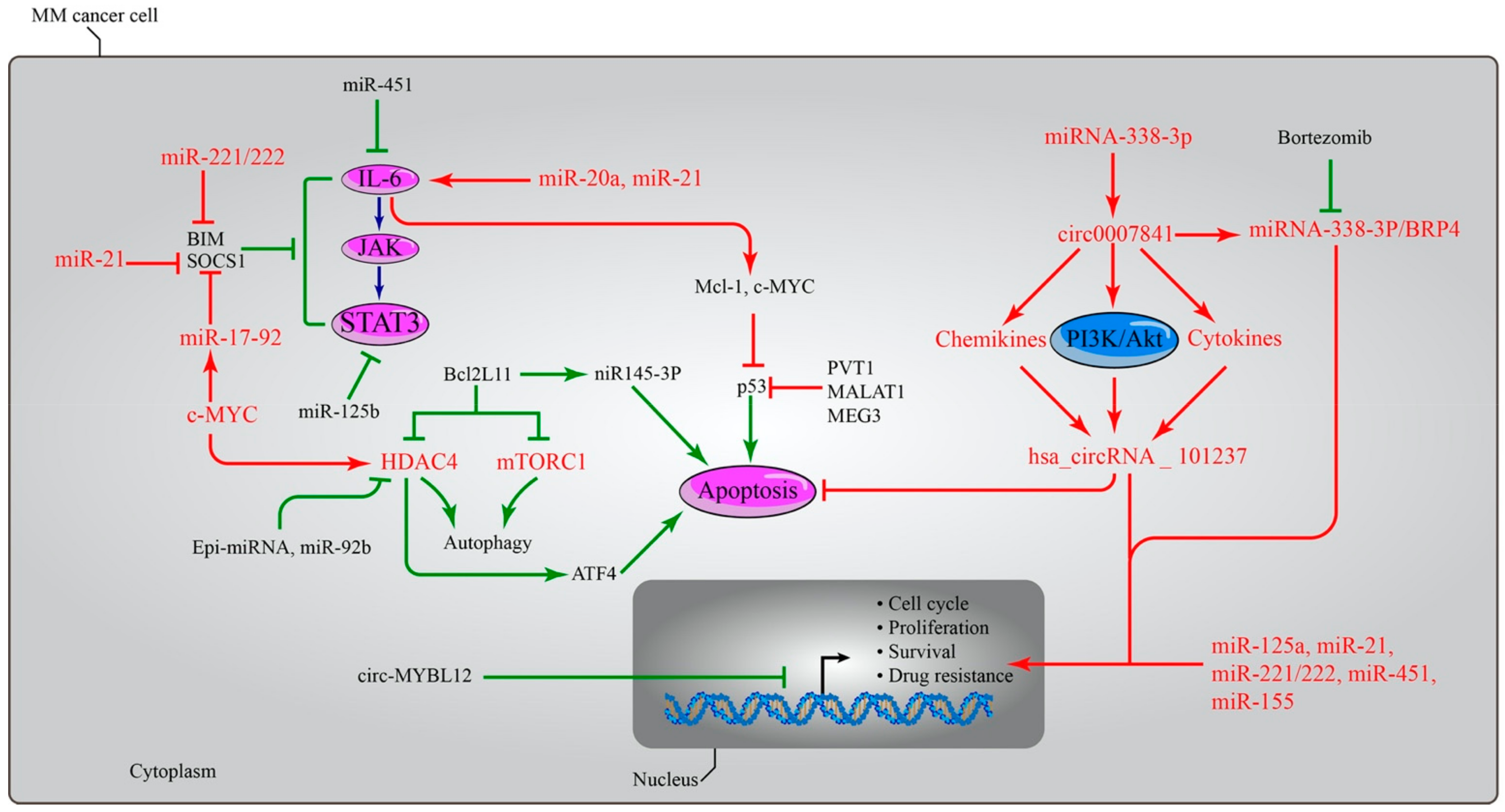
6. Importance of Non-Coding RNA in MM
7. Targeting Epigenetic Mechanisms as Novel Treatment Modalities in MM
7.1. Targeting DNA Methylation
7.2. Targeting Histone Acetylation
7.3. Targeting Histone Methylation
7.4. Targeting MicroRNAs in MM
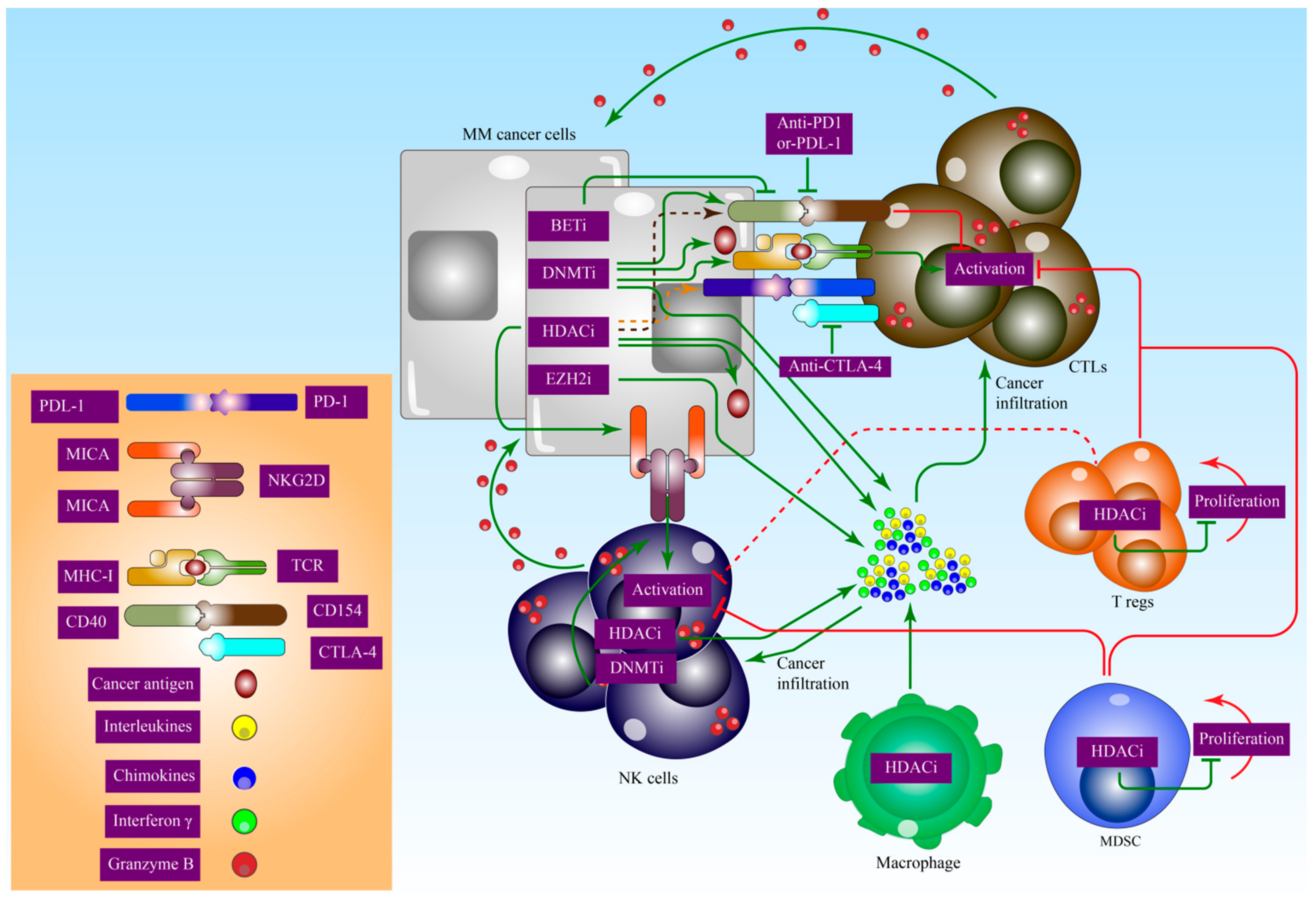
8. Epigenetic Effects on Immunomodulation in MM
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- Talley, P.J.; Chantry, A.D.; Buckle, C.H. Genetics in myeloma: Genetic technologies and their application to screening approaches in myeloma. Br. Med. Bull. 2015, 113, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.S.; Richardson, P.G.; Munshi, N.C.; Anderson, K.C. Multiple myeloma: A prototypic disease model for the characterization and therapeutic targeting of interactions between tumor cells and their local microenvironment. J. Cell Biochem. 2007, 101, 950–968. [Google Scholar] [CrossRef]
- Gundesen, M.T.; Lund, T.; Moeller, H.E.H.; Abildgaard, N. Plasma cell leukemia: Definition, presentation, and treatment. Curr. Oncol. Rep. 2019, 21, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejalvo, M.J.; de la Rubia, J. Which therapies will move to the front line for multiple myeloma? Expert Rev. Hematol. 2017, 10, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37 (Suppl. S1), 62–65. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.H.; Al Hamed, R.; Malard, F.; Harousseau, J.L.; Mohty, M. Relapsed refractory multiple myeloma: A comprehensive overview. Leukemia 2019, 33, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J., 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Sadaf, H.; Hong, H.; Maqbool, M.; Emhoff, K.; Lin, J.; Yan, S.; Anwer, F.; Zhao, J. Multiple myeloma etiology and treatment. J. Transl. Genet. Genom. 2022, 6, 63–83. [Google Scholar] [CrossRef]
- Schmidt, B.; Debold, E.; Frank, M.; Arendt, M.; Dragano, N.; Dürig, J.; Dührsen, U.; Moebus, S.; Erbel, R.; Jöckel, K.H.; et al. Socioeconomic position is positively associated with monoclonal gammopathy of undetermined significance in a population-based cohort study. Ann. Hematol. 2019, 98, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Cleynen, A.; Robiou du Pont, S.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myelome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Tapper, W.J.; Walker, B.A.; Wardell, C.P.; Gregory, W.M.; Szubert, A.J.; et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: Analysis of patients treated in the MRC Myeloma IX trial. Leukemia 2012, 26, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef]
- Rosiñol, L.; Carrió, A.; Bladé, J.; Queralt, R.; Aymerich, M.; Cibeira, M.T.; Esteve, J.; Rozman, M.; Campo, E.; Montserrat, E. Comparative genomic hybridisation identifies two variants of smoldering multiple myeloma. Br. J. Haematol. 2005, 130, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, R.; Dhodapkar, M.; Rosenthal, A.; Heuck, C.; Papanikolaou, X.; Qu, P.; van Rhee, F.; Zangari, M.; Jethava, Y.; Epstein, J.; et al. Four genes predict high risk of progression from smoldering to symptomatic multiple myeloma (SWOG S0120). Haematologica 2015, 100, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Caprio, C.; Sacco, A.; Giustini, V.; Roccaro, A.M. Epigenetic aberrations in multiple myeloma. Cancers 2020, 12, 2996. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Handa, H.; Murakami, Y.; Ishihara, R.; Kimura-Masuda, K.; Masuda, Y. The role and function of microRNA in the pathogenesis of multiple myeloma. Cancers 2019, 11, 1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Juliana, N.; Yazit, N.A.A.; Azmani, S.; Abu, I.F. Multiple Myeloma: Challenges Encountered and Future Options for Better Treatment. Int. J. Mol. Sci. 2022, 23, 1649. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, M.; Qiao, Z.; Kong, Y.; Liang, H.; Sun, Y.; Lu, H.; Xu, Z.; Liu, H. Modulating proteasome inhibitor tolerance in multiple myeloma: An alternative strategy to reverse inevitable resistance. Br. J. Cancer 2021, 124, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.Y.; Chim, C.S. DNA methylation of tumor suppressor protein-coding and non-coding genes in multiple myeloma. Epigenomics 2015, 7, 985–1001. [Google Scholar] [CrossRef]
- Salhia, B.; Baker, A.; Ahmann, G.; Auclair, D.; Fonseca, R.; Carpten, J. DNA Methylation Analysis Determines the High Frequency of Genic Hypomethylation and Low Frequency of Hypermethylation Events in Plasma Cell TumorsDNA Methylation Profiling in Multiple Myeloma. Cancer Res. 2010, 70, 6934–6944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuck, C.J.; Mehta, J.; Bhagat, T.; Gundabolu, K.; Yu, Y.; Khan, S.; Chrysofakis, G.; Schinke, C.; Tariman, J.; Vickrey, E.; et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J. Immunol. 2013, 190, 2966–2975. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.-P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Heuck, C.J.; Fazzari, M.J.; Mehta, J.; Singhal, S.; Greally, J.M.; Verma, A. DNA methylation alterations in multiple myeloma as a model for epigenetic changes in cancer. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 654–669. [Google Scholar] [CrossRef]
- Kunwor, R.; Su, Y.; Santucci-Pereira, J.; Russo, J. Present status of epigenetic drugs in cancer treatment. DNA 2015, 3, 5. [Google Scholar]
- Lauta, V.M. A review of the cytokine network in multiple myeloma: Diagnostic, prognostic, and therapeutic implications. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 2440–2452. [Google Scholar] [CrossRef]
- Chim, C.S.; Liang, R.; Leung, M.H.; Yip, S.F.; Kwong, Y.L. Aberrant gene promoter methylation marking disease progression in multiple myeloma. Leukemia 2006, 20, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.-F. DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells 2013, 2, 545–573. [Google Scholar] [CrossRef] [PubMed]
- Amara, K.; Trimeche, M.; Ziadi, S.; Laatiri, A.; Hachana, M.; Korbi, S. Prognostic significance of aberrant promoter hypermethylation of CpG islands in patients with diffuse large B-cell lymphomas. Ann. Oncol. 2008, 19, 1774–1786. [Google Scholar] [CrossRef]
- Chim, C.S.; Fung, T.K.; Cheung, W.C.; Liang, R.; Kwong, Y.L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, H.; Jin, L.; Du, J.; Fan, Z. Genome-Wide DNA Methylation Analysis during Osteogenic Differentiation of Human Bone Marrow Mesenchymal Stem Cells. Stem Cells Int. 2018, 2018, 8238496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Wu, Y.; Zhu, J.; Liu, J.; Tan, S.; Xia, C. Methylation of p16 and p15 genes in multiple myeloma. Chin. Med. Sci. J. 2002, 17, 101–105. [Google Scholar]
- Da Silva, R.A.; Fuhler, G.M.; Janmaat, V.T.; Fernandes, C.J.D.C.; da Silva Feltran, G.; Oliveira, F.A.; Matos, A.A.; Oliveira, R.C.; Ferreira, M.R.; Zambuzzi, W.F.; et al. HOXA cluster gene expression during osteoblast differentiation involves epigenetic control. Bone 2019, 125, 74–86. [Google Scholar] [CrossRef]
- Mateos, M.V.; García-Sanz, R.; López-Pérez, R.; Moro, M.J.; Ocio, E.; Hernández, J.; Megido, M.; Caballero, M.D.; Fernández-Calvo, J.; Bárez, A.; et al. Methylation is an inactivating mechanism of the p16 gene in multiple myeloma associated with high plasma cell proliferation and short survival. Br. J. Haematol. 2002, 118, 1034–1040. [Google Scholar] [CrossRef]
- Ng, M.H.L.; Chung, Y.F.; Lo, K.W.; Wickham, N.W.R.; Lee, J.C.K.; Huang, D.P. Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood 1997, 89, 2500–2506. [Google Scholar] [CrossRef]
- Krämer, A.; Schultheis, B.; Bergmann, J.; Willer, A.; Hegenbart, U.; Ho, A.D.; Goldschmidt, H.; Hehlmann, R. Alterations of the cyclin D1/pRb/p16(INK4A) pathway in multiple myeloma. Leukemia 2002, 16, 1844–1851. [Google Scholar] [CrossRef]
- Hayami, Y.; Iida, S.; Nakazawa, N.; Hanamura, I.; Kato, M.; Komatsu, H.; Miura, I.; Dave, B.J.; Sanger, W.G.; Lim, B.; et al. Inactivation of the E3/LAPTm5 gene by chromosomal rearrangement and DNA methylation in human multiple myeloma. Leukemia 2003, 17, 1650–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, C.S.; Kwong, Y.L.; Fung, T.K.; Liang, R. Methylation profiling in multiple myeloma. Leuk. Res. 2004, 28, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Pang, R.; Fung, T.K.; Choi, C.L.; Liang, R. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia 2007, 21, 2527–2536. [Google Scholar] [CrossRef]
- Rodrigues-Junior, D.M.; Biassi, T.P.; de Albuquerque, G.E.; Carlin, V.; Buri, M.V.; Machado-Junior, J.; Vettore, A.L. Downregulation of DCC sensitizes multiple myeloma cells to bortezomib treatment. Mol. Med. Rep. 2019, 19, 5023–5029. [Google Scholar] [CrossRef] [PubMed]
- Houde, C.; Li, Y.; Song, L.; Barton, K.; Zhang, Q.; Godwin, J.; Nand, S.; Toor, A.; Alkan, S.; Smadja, N.V.; et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood 2004, 104, 3697–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef]
- Wong, K.Y.; Morgan, G.J.; Boyle, E.M.; Cheng, A.S.L.; Yip, K.Y.; Chim, C.S. A proof-of-concept study for the pathogenetic role of enhancer hypomethylation of MYBPHL in multiple myeloma. Sci. Rep. 2021, 11, 7009. [Google Scholar] [CrossRef]
- Ng, M.H.; To, K.W.; Lo, K.W.; Chan, S.; Tsang, K.S.; Cheng, S.H.; Ng, H.K. Frequent death-associated protein kinase promoter hypermethylation in multiple myeloma. Clin. Cancer Res. 2001, 7, 1724–1729. [Google Scholar]
- Yuregir, O.O.; Yurtcu, E.; Kizilkilic, E.; Kocer, N.E.; Ozdogu, H.; Sahin, F.I. Detecting methylation patterns of p16, MGMT, DAPK and E-cadherin genes in multiple myeloma patients. Int. J. Lab. Hematol. 2010, 32, 142–149. [Google Scholar] [CrossRef]
- Chim, C.S.; Liang, R.; Fung, T.K.; Choi, C.L.; Kwong, Y.L. Epigenetic dysregulation of the death-associated protein kinase/p14/HDM2/p53/Apaf-1 apoptosis pathway in multiple myeloma. J. Clin. Pathol. 2007, 60, 664–669. [Google Scholar] [CrossRef] [Green Version]
- Barwick, B.G.; Powell, D.R.; Penaherrera, D.; Skerget, S.; Keats, J.J.; Auclair, D.; Lonial, S.; Boise, L.H.; Vertino, P.M. Abstract 839: Whole genome DNA methylation analysis of multiple myeloma identifies pervasive hypomethylation and biomarkers of survival. Cancer Res. 2019, 79 (Suppl. S13), 839. [Google Scholar] [CrossRef]
- Fernández de Larrea, C.; Martín-Antonio, B.; Cibeira, M.T.; Navarro, A.; Tovar, N.; Díaz, T.; Rosiñol, L.; Monzó, M.; Urbano-Ispizua, A.; Bladé, J. Impact of global and gene-specific DNA methylation pattern in relapsed multiple myeloma patients treated with bortezomib. Leuk. Res. 2013, 37, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.F.; Johnson, D.C.; Wu, P.; Walker, B.A.; Brioli, A.; Mirabella, F.; Wardell, C.P.; Melchor, L.; Davies, F.E.; Morgan, G.J. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood J. Am. Soc. Hematol. 2013, 122, 219–226. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Li, T.; de la Calle-Fabregat, C.; Rodríguez-Ubreva, J.; Ciudad, L.; Català-Moll, F.; Godoy-Tena, G.; Martín-Sánchez, M.; San-Segundo, L.; Muntión, S.; et al. Targeting aberrant DNA methylation in mesenchymal stromal cells as a treatment for myeloma bone disease. Nat. Commun. 2021, 12, 421. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The epigenome in multiple myeloma: Impact on tumor cell plasticity and drug response. Front. Oncol. 2018, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Drew, A.E.; Moradei, O.; Jacques, S.L.; Rioux, N.; Boriack-Sjodin, A.P.; Allain, C.; Scott, M.P.; Jin, L.; Raimondi, A.; Handler, J.L.; et al. Identification of a CARM1 inhibitor with potent in vitro and in vivo activity in preclinical models of multiple myeloma. Sci. Rep. 2017, 7, 17993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, K.; Szewczyk, M.M.; Dela Sena, C.; Wu, H.; Dong, A.; Zeng, H.; Li, F.; de Freitas, R.F.; Eram, M.S.; Schapira, M.; et al. TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 2018, 9, 18480. [Google Scholar] [CrossRef] [Green Version]
- Ohguchi, H.; Hideshima, T.; Anderson, K.C. The biological significance of histone modifiers in multiple myeloma: Clinical applications. Blood Cancer J. 2018, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Gullà, A.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Kemal Samur, M.; Qi, J.; Tai, Y.T.; Harada, T.; Morelli, E.; Amodio, N.; et al. Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 2018, 32, 996–1002. [Google Scholar] [CrossRef]
- Xia, T.; Liu, M.; Zhao, Q.; Ouyang, J.; Xu, P.; Chen, B. PRMT5 regulates cell pyroptosis by silencing CASP1 in multiple myeloma. Cell Death Dis. 2021, 12, 851. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Trojer, P.; Xu, C.F.; Cheung, P.; Kuo, A.; Drury, W.J., 3rd; Qiao, Q.; Neubert, T.A.; Xu, R.M.; Gozani, O.; et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t (4; 14) multiple myeloma cells. Blood J. Am. Soc. Hematol. 2011, 117, 211–220. [Google Scholar]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The role of nuclear receptor–binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef] [Green Version]
- Alzrigat, M.; Jernberg-Wiklund, H.; Licht, J.D. Targeting EZH2 in Multiple Myeloma-Multifaceted Anti-Tumor Activity. Epigenomes 2018, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Majello, B.; Gorini, F.; Sacca, C.D.; Amente, S. Expanding the Role of the Histone Lysine-Specific Demethylase LSD1 in Cancer. Cancers 2019, 11, 324. [Google Scholar] [CrossRef] [Green Version]
- Ohguchi, H.; Hideshima, T.; Bhasin, M.K.; Gorgun, G.T.; Santo, L.; Cea, M.; Samur, M.K.; Mimura, N.; Suzuki, R.; Tai, Y.-T.; et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat. Commun. 2016, 7, 10258. [Google Scholar] [CrossRef]
- Ikeda, S.; Kitadate, A.; Abe, F.; Takahashi, N.; Tagawa, H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv. 2018, 2, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Ezponda, T.; Dupéré-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef] [Green Version]
- Ohguchi, H.; Harada, T.; Sagawa, M.; Kikuchi, S.; Tai, Y.T.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 2017, 31, 2661–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.; Tsai, L.H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.; Chen, T.; Liu, J.; Dowling, P.; Hideshima, T.; Zhang, L.; Morelli, E.; Camci-Unal, G.; Wu, X.; Tai, Y.T.; et al. Targeting histone deacetylase 3 (HDAC3) in the bone marrow microenvironment inhibits multiple myeloma proliferation by modulating exosomes and IL-6 trans-signaling. Leukemia 2020, 34, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Suzuki, R.; Ohguchi, H.; Yoshida, Y.; Lu, D.; Cottini, F.; Jakubikova, J.; Bianchi, G.; Harada, T.; Gorgun, G.; et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia 2015, 29, 1918–1927. [Google Scholar] [CrossRef]
- Amodio, N.; Stamato, M.A.; Gullà, A.M.; Morelli, E.; Romeo, E.; Raimondi, L.; Pitari, M.R.; Ferrandino, I.; Misso, G.; Caraglia, M.; et al. Therapeutic Targeting of miR-29b/HDAC4 Epigenetic Loop in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1364–1375. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, S.T.; Carew, J.S.; Maclean, K.H.; Courage, J.F.; Huang, P.; Houghton, J.A.; Cleveland, J.L.; Giles, F.J.; McConkey, D.J. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood 2008, 112, 2917–2926. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.-T.; Acharya, C.; Zhong, M.; Hideshima, T.; Gobbi, M.; Munshi, N.; Chauhan, D.; et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostofa, A.; Distler, A.; Meads, M.B.; Sahakian, E.; Powers, J.J.; Achille, A.; Noyes, D.; Wright, G.; Fang, B.; Izumi, V.; et al. Plasma cell dependence on histone/protein deacetylase 11 reveals a therapeutic target in multiple myeloma. JCI Insight 2021, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Nieto, Y.; Valdez, B.C.; Thall, P.F.; Jones, R.B.; Wei, W.; Myers, A.; Hosing, C.; Ahmed, S.; Popat, U.; Shpall, E.J.; et al. Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer 2016, 122, 2680–2688. [Google Scholar] [CrossRef] [Green Version]
- Ronchetti, D.; Todoerti, K.; Tuana, G.; Agnelli, L.; Mosca, L.; Lionetti, M.; Fabris, S.; Colapietro, P.; Miozzo, M.; Ferrarini, M.; et al. The expression pattern of small nucleolar and small Cajal body-specific RNAs characterizes distinct molecular subtypes of multiple myeloma. Blood Cancer J. 2012, 2, e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccaro, A.M.; Sacco, A.; Thompson, B.; Leleu, X.; Azab, A.K.; Azab, F.; Runnels, J.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood J. Am. Soc. Hematol. 2009, 113, 6669–6680. [Google Scholar]
- Gao, Y.N.; Ma, Y.P. Expression Analysis and Epigenetics of MicroRNA-28-5p in Multiple Myeloma. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2019, 27, 1540–1547. [Google Scholar]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6–dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood J. Am. Soc. Hematol. 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Kumar, R.; Seth, T.; Garg, B.; Sati, H.C.; Sharma, A. Clinical significance of circulatory microRNA-203 in serum as novel potential diagnostic marker for multiple myeloma. J. Cancer Res. Clin. Oncol. 2019, 145, 1601–1611. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, H.; Chan, M.T.; Wu, W.K. Modulation of chemoresponsiveness to platinum-based agents by microRNAs in cancer. Am. J. Cancer Res. 2017, 7, 1769. [Google Scholar]
- Di Martino, M.T.; Gullà, A.; Cantafio, M.E.; Lionetti, M.; Leone, E.; Amodio, N.; Guzzi, P.H.; Foresta, U.; Conforti, F.; Cannataro, M.; et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget 2013, 4, 242. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wu, X.; Dai, L.; Ge, Z.; Gao, C.; Zhang, H.; Wang, F.; Zhang, X.; Chen, B. Gambogenic acid exerts antitumor activity in hypoxic multiple myeloma cells by regulation of miR-21. J. Cancer 2017, 8, 3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Li, X.; Wang, W.; He, W.; Wang, J.; Wang, Y. Expression of peripheral blood miRNA-720 and miRNA-1246 can be used as a predictor for outcome in multiple myeloma patients. Clin. Lymphoma Myeloma Leuk. 2017, 17, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenart, P.; Novak, J.; Bienertova-Vasku, J. PIWI-piRNA pathway: Setting the pace of aging by reducing DNA damage. Mech. Ageing Dev. 2018, 173, 29–38. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, W.; Qi, R.; Mao, Z.W.; Shen, H. Engineering functional inorganic–organic hybrid systems: Advances in siRNA therapeutics. Chem. Soc. Rev. 2018, 47, 1969–1995. [Google Scholar] [CrossRef]
- Yan, H.; Wu, Q.L.; Sun, C.Y.; Ai, L.S.; Deng, J.; Zhang, L.; Chen, L.; Chu, Z.B.; Tang, B.; Wang, K.; et al. piRNA-823 contributes to tumorigenesis by regulating de novo DNA methylation and angiogenesis in multiple myeloma. Leukemia 2015, 29, 196–206. [Google Scholar] [CrossRef]
- He, Q.; Liu, Y.; Sun, W. Statistical analysis of non-coding RNA data. Cancer Lett. 2018, 417, 161–167. [Google Scholar] [CrossRef]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear long noncoding RNAs: Key regulators of gene expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Saltarella, I.; Apollonio, B.; Lamanuzzi, A.; Desantis, V.; Mariggiò, M.A.; Desaphy, J.F.; Vacca, A.; Frassanito, M.A. The Landscape of lncRNAs in Multiple Myeloma: Implications in the “Hallmarks of Cancer”, Clinical Perspectives and Therapeutic Opportunities. Cancers 2022, 14, 1963. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood J. Am. Soc. Hematol. 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Lyu, H.; Huang, J.; Wang, S.; Lee, C.K.; Gao, C.; Liu, B. Combination of bendamustine and entinostat synergistically inhibits proliferation of multiple myeloma cells via induction of apoptosis and DNA damage response. Cancer Lett. 2013, 335, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richon, V. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, S2–S6. [Google Scholar] [CrossRef]
- Maes, K.; De Smedt, E.; Kassambara, A.; Hose, D.; Seckinger, A.; Van Valckenborgh, E.; Menu, E.; Klein, B.; Vanderkerken, K.; Moreaux, J.; et al. In vivo treatment with epigenetic modulating agents induces transcriptional alterations associated with prognosis and immunomodulation in multiple myeloma. Oncotarget 2015, 6, 3319. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, D.; DeSimone, J.; Hankewych, M.; Kousnetzova, T.; Chen, Y.-H. Decitabine induces cell cycle arrest at the G1 phase via p21WAF1 and the G2/M phase via the p38 MAP kinase pathway. Leuk. Res. 2003, 27, 999–1007. [Google Scholar] [CrossRef]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef]
- Nojima, M.; Maruyama, R.; Yasui, H.; Suzuki, H.; Maruyama, Y.; Tarasawa, I.; Sasaki, Y.; Asaoku, H.; Sakai, H.; Hayashi, T.; et al. Genomic screening for genes silenced by DNA methylation revealed an association between RASD1 inactivation and dexamethasone resistance in multiple myeloma. Clin. Cancer Res. 2009, 15, 4356–4364. [Google Scholar] [CrossRef] [Green Version]
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: Molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537. [Google Scholar] [CrossRef] [Green Version]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656. [Google Scholar] [CrossRef] [Green Version]
- Maes, K.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Vanderkerken, K.; De Bruyne, E. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers 2013, 5, 430–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.M.; Basar, A.P.; Reyes, L.; Duncan, R.M.; Li, H.; Dolloff, N.G. PDI inhibitor LTI6426 enhances panobinostat efficacy in preclinical models of multiple myeloma. Cancer Chemother. Pharmacol. 2022, 89, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. PANORAMA 2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337. [Google Scholar] [CrossRef]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a inhibition suppresses multiple myeloma cell proliferation by regulating the interferon signal and IRF4-MYC axis. Cell Death Discov. 2021, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marango, J.; Shimoyama, M.; Nishio, H.; Meyer, J.A.; Min, D.-J.; Sirulnik, A.; Martinez-Martinez, Y.; Chesi, M.; Bergsagel, P.L.; Zhou, M.-M.; et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood J. Am. Soc. Hematol. 2008, 111, 3145–3154. [Google Scholar] [CrossRef] [Green Version]
- Di Luccio, E. Inhibition of nuclear receptor binding SET domain 2/multiple myeloma SET domain by LEM-06 implication for epigenetic cancer therapies. J. Cancer Prev. 2015, 20, 113. [Google Scholar] [CrossRef]
- Oka, S.; Ono, K.; Nohgawa, M. Successful treatment with azacitidine for the simultaneous occurrence of multiple myeloma and acute myeloid leukemia with concomitant del (5q) and the JAK2 V617F mutation. Ann. Hematol. 2017, 96, 1411–1413. [Google Scholar] [CrossRef] [Green Version]
- Tian, E.; Tang, H.; Xu, R.; Liu, C.; Deng, H.; Wang, Q. Azacytidine induces necrosis of multiple myeloma cells through oxidative stress. Proteome Sci. 2013, 11, 24. [Google Scholar] [CrossRef]
- Maes, K.; De Smedt, E.; Lemaire, M.; De Raeve, H.; Menu, E.; Van Valckenborgh, E.; McClue, S.; Vanderkerken, K.; De Bruyne, E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014, 5, 3115. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Lyu, H.; Pei, S.; Song, D.; Ni, J.; Liu, B. Cladribine in combination with entinostat synergistically elicits anti-proliferative/anti-survival effects on multiple myeloma cells. Cell Cycle 2018, 17, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Gao, L.; Wang, S.; Lee, C.K.; Ordentlich, P.; Liu, B. HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res. 2009, 69, 8403–8411. [Google Scholar] [CrossRef] [Green Version]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics combination therapies: An overview of the role of HDACs in cancer immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Yee, A.J. Do We Need Another HDAC Inhibitor in Multiple Myeloma? Hematologist 2015, 12, 4155. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, J.; Wada, T.; Shimizu, R.; Izumi, T.; Akutsu, M.; Mitsunaga, K.; Noborio-Hatano, K.; Nobuyoshi, M.; Ozawa, K.; Kano, Y.; et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood J. Am. Soc. Hematol. 2010, 116, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; McMullan, C.; Poulaki, V.; Fanourakis, G.; Schlossman, R.; Chauhan, D.; Munshi, N.C.; Hideshima, T.; et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood J. Am. Soc. Hematol. 2003, 101, 4055–4062. [Google Scholar] [CrossRef]
- Dickinson, M.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors: Potential targets responsible for their anti-cancer effect. Investig. New Drugs 2010, 28, 3–20. [Google Scholar] [CrossRef]
- Khan, S.B.; Maududi, T.; Barton, K.; Ayers, J.; Alkan, S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br. J. Haematol. 2004, 125, 156–161. [Google Scholar] [CrossRef]
- Ocio, E.M.; San Miguel, J.F. The DAC system and associations with multiple myeloma. Investig. New Drugs 2010, 28, 28–35. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, e0173507. [Google Scholar] [CrossRef] [Green Version]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, e184–e366. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple MyelomaRicolinostat, Bortezomib, and Dexamethasone for Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreaux, J.; Reme, T.; Leonard, W.; Veyrune, J.L.; Requirand, G.; Goldschmidt, H.; Hose, D.; Klein, B. Gene expression-based prediction of myeloma cell sensitivity to histone deacetylase inhibitors. Br. J. Cancer 2013, 109, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fandy, T.E.; Srivastava, R.K. Trichostatin A sensitizes TRAIL-resistant myeloma cells by downregulation of the antiapoptotic Bcl-2 proteins. Cancer Chemother. Pharmacol. 2006, 58, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Schmidt, W.M.; Ziegler, B.; Holzer, S.; Müllauer, L.; Bilban, M.; Zielinski, C.C.; Drach, J.; Zöchbauer-Müller, S. Genome-wide transcriptional response to 5-aza-2′-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008, 68, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Yang, X.; Liu, M.; Zhang, Z.; Xing, E. Roles of miRNA dysregulation in the pathogenesis of multiple myeloma. Cancer Gene Ther. 2021, 28, 1256–1268. [Google Scholar] [CrossRef]
- Kumar, D.; Gokhale, P.; Broustas, C.; Chakravarty, D.; Ahmad, I.; Kasid, U. Expression of SCC-S2, an antiapoptotic molecule, correlates with enhanced proliferation and tumorigenicity of MDA-MB 435 cells. Oncogene 2004, 23, 612–616. [Google Scholar] [CrossRef]
- Kumar, D.; Whiteside, T.L.; Kasid, U. Identification of a novel tumor necrosis factor-α-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein. J. Biol. Chem. 2000, 275, 2973–2978. [Google Scholar] [CrossRef] [Green Version]
- Rastgoo, N.; Wu, J.; Liu, A.; Pourabdollah, M.; Atenafu, E.G.; Reece, D.; Chen, W.; Chang, H. Targeting CD47/TNFAIP8 by miR-155 overcomes drug resistance and inhibits tumor growth through induction of phagocytosis and apoptosis in multiple myeloma. Haematologica 2020, 105, 2813. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Tang, H.; Liu, J.; Wang, X. hsa_circRNA_101237: A novel diagnostic and prognostic biomarker and potential therapeutic target for multiple myeloma. Cancer Manag. Res. 2020, 12, 2109. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Liu, C.; Yang, Q.; Xin, C.; Du, J.; Sun, F.; Zhou, L. MIR145-3p promotes autophagy and enhances bortezomib sensitivity in multiple myeloma by targeting HDAC4. Autophagy 2020, 16, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ai, L.; Wei, W.; Pan, J. circRNA circ-MYBL2 is a novel tumor suppressor and potential biomarker in multiple myeloma. Hum. Cell 2021, 34, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lin, Q.; Song, C.; Ma, R.; Li, X. Circ_0007841 promotes the progression of multiple myeloma through targeting miR-338-3p/BRD4 signaling cascade. Cancer Cell Int. 2020, 20, 383. [Google Scholar] [CrossRef]
- Petersen, S.; Wilson, A.J.; Hirst, J.; Roby, K.F.; Fadare, O.; Crispens, M.A.; Beeghly-Fadiel, A.; Khabele, D. CCNE1 and BRD4 co-amplification in high-grade serous ovarian cancer is associated with poor clinical outcomes. Gynecol. Oncol. 2020, 157, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.; Rao, S. Epigenetics and immunotherapy: The current state of play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16ink4a expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Critical roles for immunoglobulin translocations and cyclin D dysregulation in multiple myeloma. Immunol. Rev. 2003, 194, 96–104. [Google Scholar] [CrossRef]
- Wong, K.Y.; Yim, R.L.H.; So, C.C.; Jin, D.-Y.; Liang, R.; Chim, C.S. Epigenetic inactivation of the MIR34B/C in multiple myeloma. Blood J. Am. Soc. Hematol. 2011, 118, 5901–5904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichiorri, F.; Suh, S.-S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, K.; Gimsing, P.; Grønbæk, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.Y.; She, X.M.; Qin, Y.; Chu, Z.B.; Chen, L.; Ai, L.S.; Zhang, L.; Hu, Y. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 2013, 34, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Martello, G.; Zacchigna, L.; Inui, M.; Montagner, M.; Adorno, M.; Mamidi, A.; Morsut, L.; Soligo, S.; Tran, U.; Dupont, S.; et al. MicroRNA control of Nodal signalling. Nature 2007, 449, 183–188. [Google Scholar] [CrossRef]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D., Jr. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef]
- Cha, Y.H.; Kim, N.H.; Park, C.; Lee, I.; Kim, H.S.; Yook, J.I. MiRNA-34 intrinsically links p53 tumor suppressor and Wnt signaling. Cell Cycle 2012, 11, 1273–1281. [Google Scholar] [CrossRef]
- Wong, K.Y.; Liang, R.; So, C.C.; Jin, D.Y.; Costello, J.F.; Chim, C.S. Epigenetic silencing of MIR203 in multiple myeloma. Br. J. Haematol. 2011, 154, 569–578. [Google Scholar] [CrossRef]
- Chi, J.; Ballabio, E.; Chen, X.H.; Kušec, R.; Taylor, S.; Hay, D.; Tramonti, D.; Saunders, N.J.; Littlewood, T.; Pezzella, F.; et al. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol. Direct 2011, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Zhang, J.-X.; Han, L.; You, Y.-P.; Jiang, T.; Pu, P.-Y.; Kang, C.-S. MicroRNA roles in beta-catenin pathway. Mol. Cancer 2010, 9, 252. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, C.S.; Liang, R.; Leung, M.H.; Yip, S.F.; Kwong, Y.L. Aberrant gene methylation implicated in the progression of monoclonal gammopathy of undetermined significance to multiple myeloma. J. Clin. Pathol. 2007, 60, 104–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, C.; Ju, S.; Wang, Y.; Wang, H.; Zhong, R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk. Lymphoma 2011, 52, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Zhong, Q.; Zhang, J.; Yang, M.; Li, C.; Zheng, P.; Bi, L.J.; Ge, F. Identification of novel miR-21 target proteins in multiple myeloma cells by quantitative proteomics. J. Proteome Res. 2012, 11, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Martoriati, A.; Doumont, G.; Alcalay, M.; Bellefroid, E.; Pelicci, P.G.; Marine, J.C. dapk1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 2005, 24, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Pichiorri, F.; Suh, S.S.; Rocci, A.; De Luca, L.; Taccioli, C.; Santhanam, R.; Zhou, W.; Benson, D.M., Jr.; Hofmainster, C.; Alder, H.; et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010, 18, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Símová, J.; Polláková, V.; Indrová, M.; Mikyšková, R.; Bieblová, J.; Stěpánek, I.; Bubeník, J.; Reiniš, M. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. Br. J. Cancer 2011, 105, 1533–1541. [Google Scholar] [CrossRef]
- Serrano, A.; Castro-Vega, I.; Redondo, M. Role of gene methylation in antitumor immune response: Implication for tumor progression. Cancers 2011, 3, 1672–1690. [Google Scholar] [CrossRef]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef]
- Heninger, E.; Krueger, T.E.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 2015, 6, 29. [Google Scholar]
- Costantini, B.; Kordasti, S.Y.; Kulasekararaj, A.G.; Jiang, J.; Seidl, T.; Abellan, P.P.; Mohamedali, A.; Thomas, N.S.; Farzaneh, F.; Mufti, G.J. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica 2013, 98, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Amoozgar, Z.; Huang, J.; Saleh, M.H.; Xing, D.; Orsulic, S.; Goldberg, M.S. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol. Res. 2015, 3, 1030–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [Green Version]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2017, 169, 361. [Google Scholar] [CrossRef] [Green Version]
- Terranova-Barberio, M.; Thomas, S.; Munster, P.N. Epigenetic modifiers in immunotherapy: A focus on checkpoint inhibitors. Immunotherapy 2016, 8, 705–719. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, P.; Tonelli, R.; Camerin, C.; Purgato, S.; Fronza, R.; Bianucci, F.; Guerra, F.; Pession, A.; Ferreri, A.M. p21Waf1/Cip1 is a common target induced by short-chain fatty acid HDAC inhibitors (valproic acid, tributyrin and sodium butyrate) in neuroblastoma cells. Oncol. Rep. 2005, 13, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Smyth, M.J.; Johnstone, R.W. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology 2014, 3, e27414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Malamas, A.S.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget 2016, 7, 7390–7402. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, T.; Towatari, M.; Kosugi, H.; Saito, H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood 2000, 96, 3847–3856. [Google Scholar] [CrossRef]
- Chou, S.D.; Khan, A.N.; Magner, W.J.; Tomasi, T.B. Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int. Immunol. 2005, 17, 1483–1494. [Google Scholar] [CrossRef]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Skov, S.; Pedersen, M.T.; Andresen, L.; Straten, P.T.; Woetmann, A.; Odum, N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005, 65, 11136–11145. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Methylation/Gene | Effects | References |
|---|---|---|
| Promoter/hypermethylation CDKN2A and CDKN2B | Activates cell cycle Enhances cell proliferation and disease progression | [49,50,51] |
| LAPTm5 gene | Loss of E3 gene activity Crucial in MM progression | [52] |
| CHD1 | Suppresses cell adhesion Increases cell motility Promotes metastasis | [53,54] |
| Promoter/hypermethylation WNT signalling pathway inhibitor genes (SFR1, SFR2, SFR4, SFR5, APC, WIF1, and DKK3) | Activates WNT signal | [55,56] |
| Promoter/hypermethylation DCC | Involved in cell migration Improves MM cells’ sensitivity to bortezomib | [56] |
| Promoter/hypomethylation Notch ligand JAG2 | Increases expression growth factor IL-6 Enhances cell proliferation | [57] |
| Hypomethylation ATP binding cassette transporter ABCG2-hypomethylation | Involved in drug resistance | [58] |
| Promoter/hypomethylation CpG1 within the enhancer of MYBPHL | Promotes myelomagenesis | [59] |
| Promoter/hypermethylation DAPK1 | Involved in early event MM pathogenesis Worst therapeutic response and short survival | [60,61,62] |
| Reduced DNA methylation Gene bodies at the loci of PRKCE, MGMT, FHIT, and WWOX | Poor survival Enhances MAF expression | [63] |
| Low methylation CXCR4 and NFKB1 | Prolongs PFS and OS in relapsed patients following bortezomib treatment | [64] |
| Hypermethylation GPX3, RBP1, SPARC, and TGFB1 | Aggressive phenotype of MM cells Extremely short OS | [65] |
| Homeobox genes | Osteogenic differentiation MM bone disease | [42] |
| Epigenetic Inhibitors | Mechanisms | References |
|---|---|---|
| DNMT inhibitors | ||
| Azacytidine | Destructs proteosome DNMT and decondenses chromatin Enhances necrosis via oxidative stress | [137] [138] |
| 5-aza-2′deoxycytidine/decitabine | Damages DNA via gamma-H2AX foci formation G0/G1- or G2/M-phase arrest and caspase-mediated apoptosis Activates DDR | [139] |
| HDAC inhibitor | ||
| Entinostat | HDACi Class I, effectively inhibits HDAC1 and HDAC3 Induces apoptosis via the downregulation of erbB3 expression Loosens the chromatin conformation and exposes DNA structure to destructive agents | [140] [141] [114] |
| Panobinostat (LBH-589) | HDACi Class I, II, and IV Dysregulates canonical Wnt signalling and key player β-catenin Reactivates cancer-suppressed genes and promotes cell death Significant toxicity across all HDAC classes | [142] [143] [144] |
| Vorinostat | HDACi class I and II Enhances cancer cell-cycle arrest and apoptosis Upregulates the E-cadherin gene and is less toxic than monotherapy or combination therapy | [115] [145] |
| Romidepsin | Cyclic tetrapeptide HDAC inhibitor HDACi Class 1, Class 2, and Class 6 Enhances cancer suppressor genes p21 and p53 Suppresses antiapoptotic molecules, e.g., Bcl-2, Bcl-XL, BAX, and MCL-1 Initiates apoptosis in a dose-dependent fashion Enhances p53, agitates the function of HSP90, tubulin, and the endoplasmic reticulum, and forms aggresomes Induces cell-cycle arrest (via the p21 and AKT pathways) | [146] [147] [148,149] [150] |
| ACY-241 (citarinostat) | Second-generation selective HDAC6 inhibitor More selective (13 to 18-fold) HDAC6 in comparison to HDAC1-3 Promotes higher serum concentrations Higher rating of HDAC inhibition including HDAC1/2 Alternative for potent and well-tolerated oral HDAC inhibitor | [151] [152] |
| ACY-1215 (ricolinostat) | Enhances α-tubulin acetylation and accumulation of ubiquitinated proteins Reduces the inhibition of class I HDACs Lower toxicity than nonselective HDAC inhibitors | [152,153] |
| Trichostatin A (TSA) | HDACi class I and II Effectively inhibits cell proliferation and initiates cell cycle arrest and apoptosis Sensitises TNF-related apoptosis-inducing factor (TRAIL)-resistant cells by suppressing the antiapoptotic BCL2 proteins Decreases expression of MM proliferation-associated factors | [154] [155] [156] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, N.H.; Mussa, A.; Zakaria, N.A.; Al-Khreisat, M.J.; Zahidin, M.A.; Ramli, N.N.; Mohammad, S.N.N.A.; Hassan, R.; Mohd Noor, N.H.; Iberahim, S.; et al. The Role of Epigenetics in the Development and Progression of Multiple Myeloma. Biomedicines 2022, 10, 2767. https://doi.org/10.3390/biomedicines10112767
Ismail NH, Mussa A, Zakaria NA, Al-Khreisat MJ, Zahidin MA, Ramli NN, Mohammad SNNA, Hassan R, Mohd Noor NH, Iberahim S, et al. The Role of Epigenetics in the Development and Progression of Multiple Myeloma. Biomedicines. 2022; 10(11):2767. https://doi.org/10.3390/biomedicines10112767
Chicago/Turabian StyleIsmail, Nor Hayati, Ali Mussa, Nur Atikah Zakaria, Mutaz Jamal Al-Khreisat, Muhamad Aidil Zahidin, Noor Nabila Ramli, Siti Nur Nabeela A’ifah Mohammad, Rosline Hassan, Noor Haslina Mohd Noor, Salfarina Iberahim, and et al. 2022. "The Role of Epigenetics in the Development and Progression of Multiple Myeloma" Biomedicines 10, no. 11: 2767. https://doi.org/10.3390/biomedicines10112767