

Effects of the Exposure of Human Non-Tumour Cells to Sera of Pancreatic Cancer Patients
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Serum Treatments
2.3. Soft Agar Colony Formation Assay
2.4. Wound Healing Migration Assay
2.5. Invasion Assay
2.6. Western Blot
2.7. Chemoresistance
2.8. Whole Genome Sequencing (WGS) and Data Analysis
2.9. Statistical Analyses
3. Results
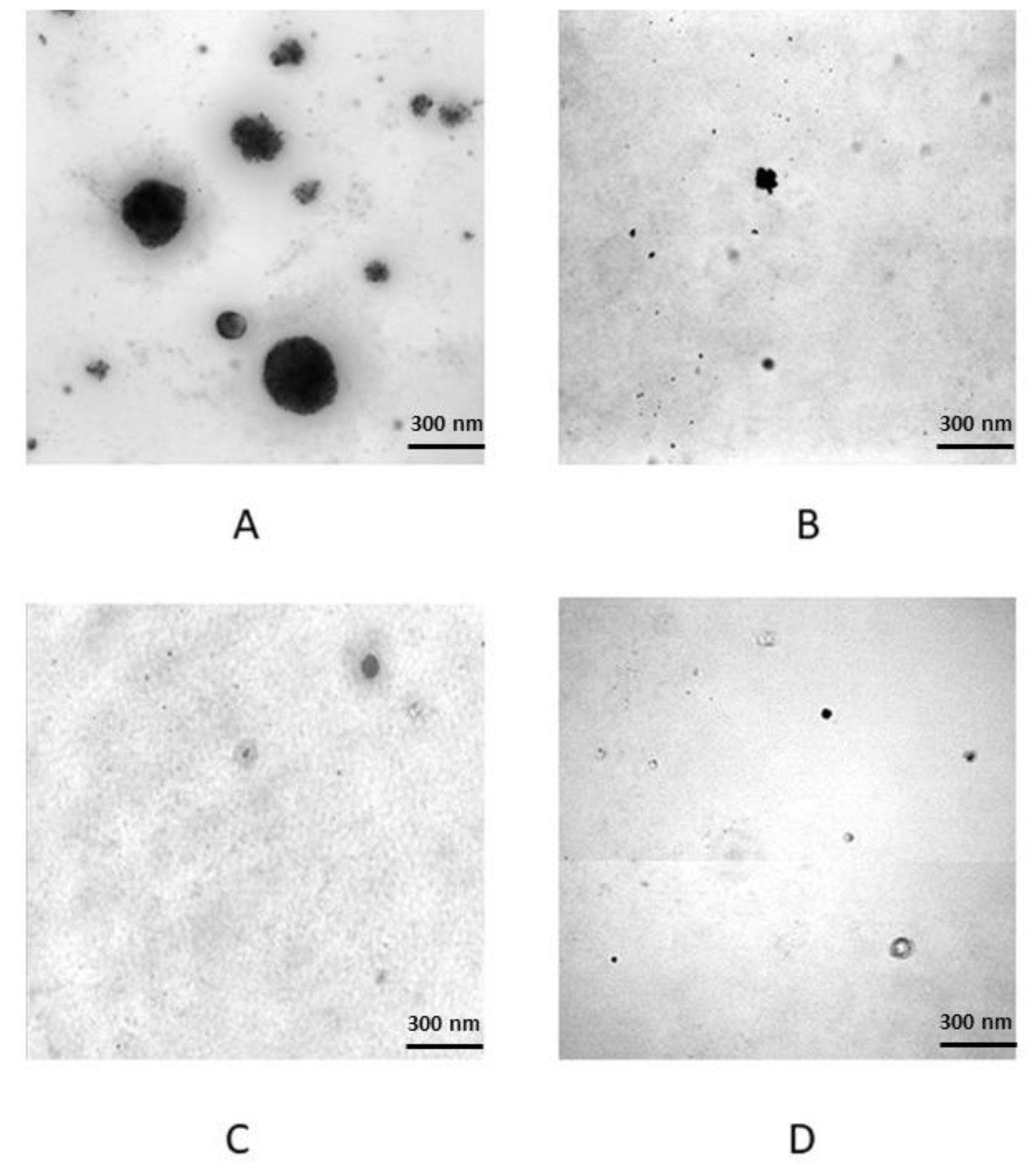
3.1. Serum Treatments and In Vitro Transformation Validation
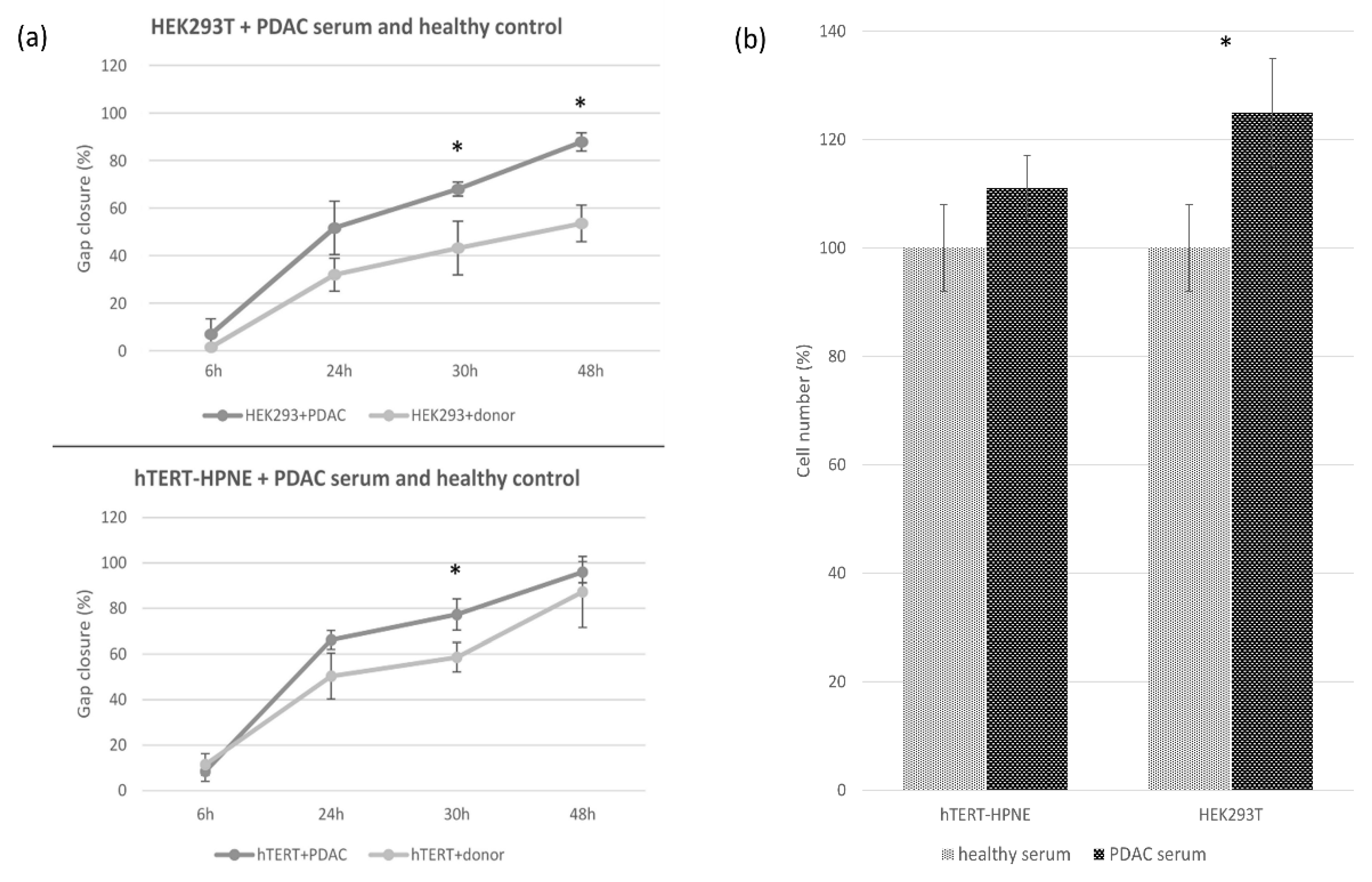
3.2. Cell Migration and Invasion Assay
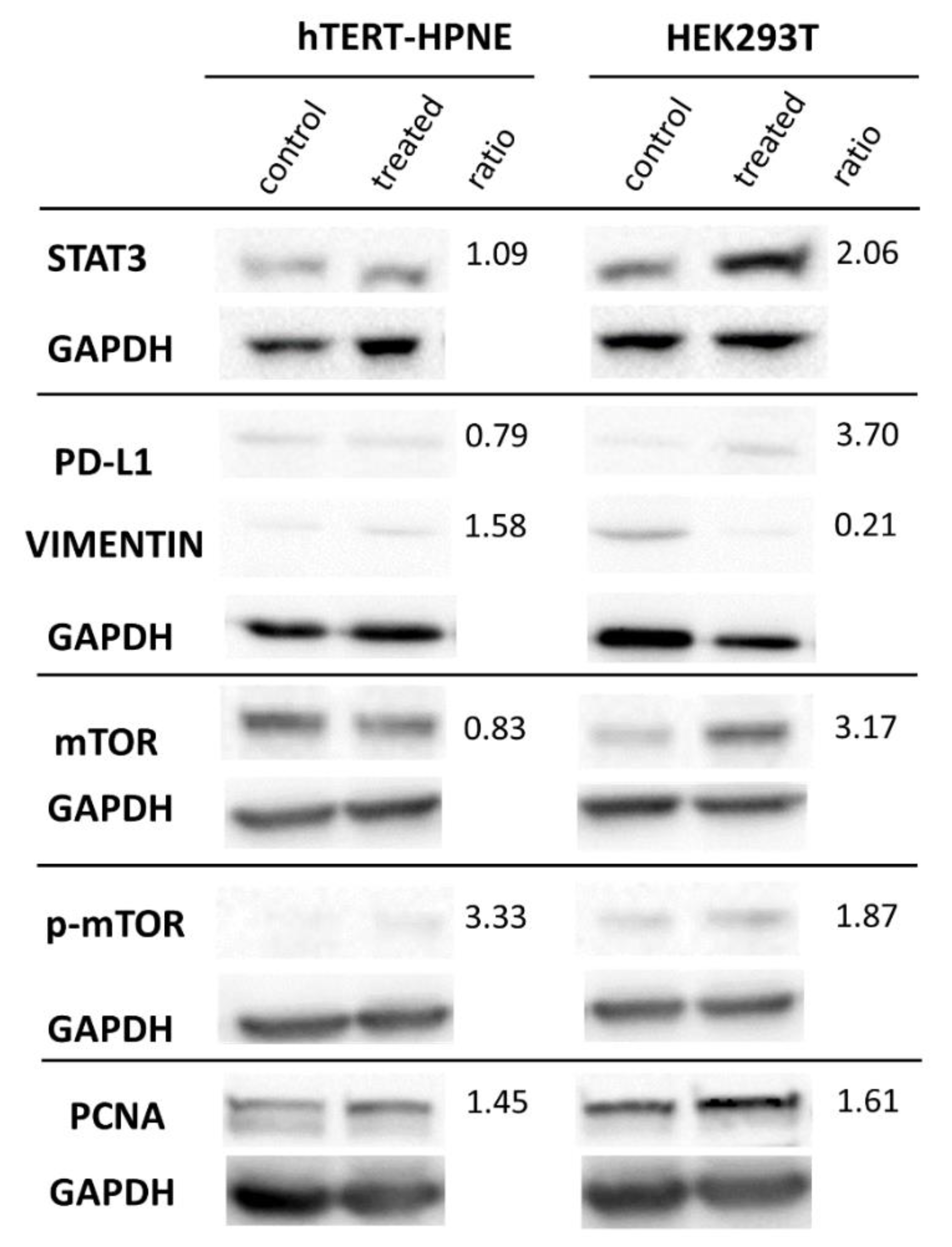
3.3. Protein Expression in Western Blot
3.4. Chemoresistance
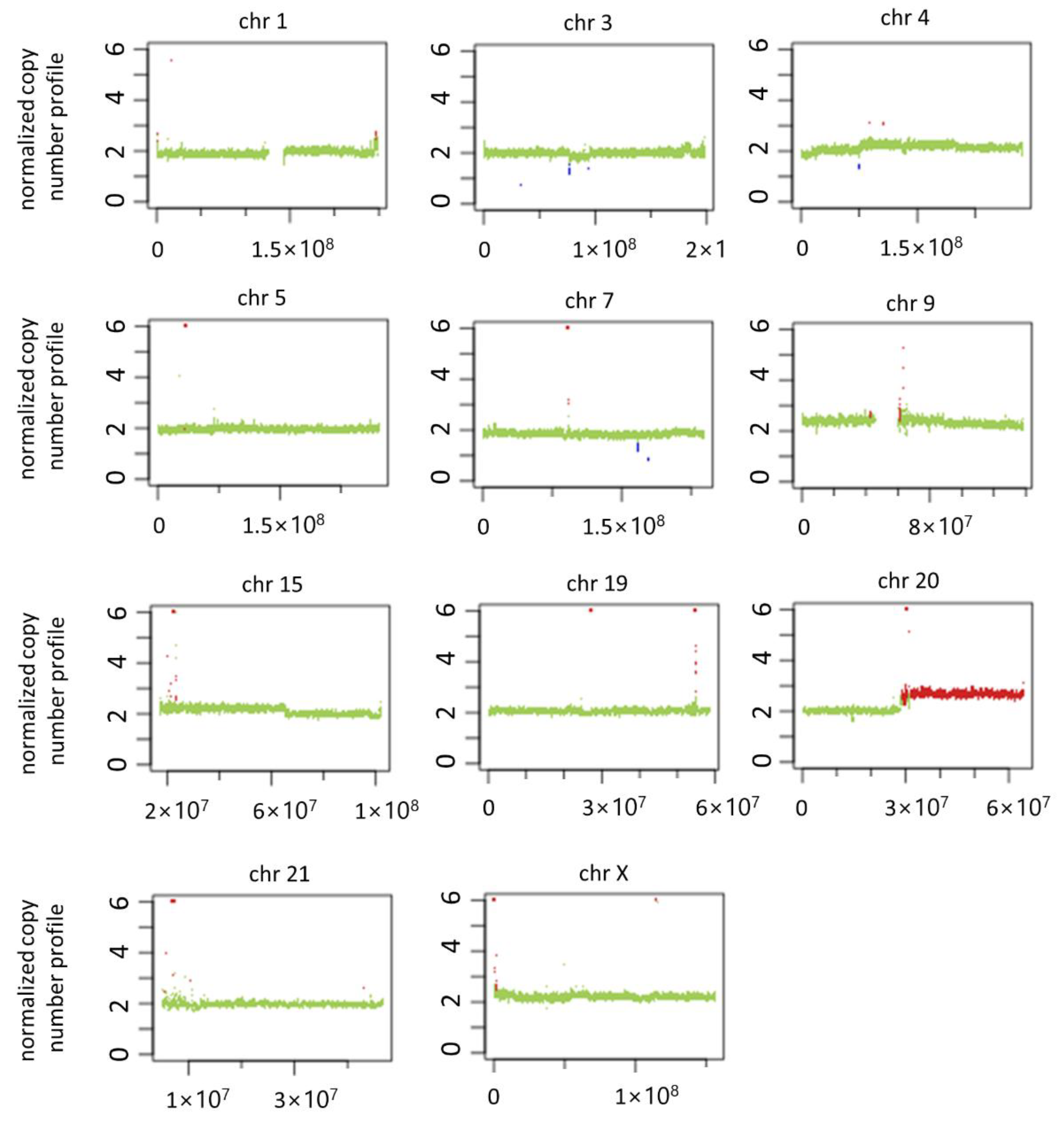
3.5. Genomic Imbalance in PDAC Serum-Treated Cells and Literature Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deramaudt, T.; Rustgi, A.K. Mutant kras in the initiation of pancreatic cancer. Biochim. Biophys. Acta 2005, 1756, 97–101. [Google Scholar] [CrossRef]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Casas, A.; Garcia-Olmo, D.C.; Garcia-Olmo, D. Further the liquid biopsy: Gathering pieces of the puzzle of genometastasis theory. World J. Clin. Oncol. 2017, 8, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [Green Version]
- Arena, G.O.; Arena, V.; Arena, M.; Abdouh, M. Transfer of malignant traits as opposed to migration of cells: A novel concept to explain metastatic disease. Med. Hypotheses 2017, 100, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Olmo, D.C.; Dominguez, C.; Garcia-Arranz, M.; Anker, P.; Stroun, M.; Garcia-Verdugo, J.M.; Garcia-Olmo, D. Cell-free nucleic acids circulating in the plasma of colorectal cancer patients induce the oncogenic transformation of susceptible cultured cells. Cancer Res. 2010, 70, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Olmo, D.; Garcia-Olmo, D.C.; Dominguez-Berzosa, C.; Guadalajara, H.; Vega, L.; Garcia-Arranz, M. Oncogenic transformation induced by cell-free nucleic acids circulating in plasma (genometastasis) remains after the surgical resection of the primary tumor: A pilot study. Expert Opin. Biol. Ther. 2012, 12 (Suppl. 1), S61–S68. [Google Scholar] [CrossRef]
- Abdouh, M.; Zhou, S.; Arena, V.; Arena, M.; Lazaris, A.; Onerheim, R.; Metrakos, P.; Arena, G.O. Transfer of malignant trait to immortalized human cells following exposure to human cancer serum. J. Exp. Clin. Cancer Res. 2014, 33, 86. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Floris, M.; Gao, Z.H.; Arena, V.; Arena, M.; Arena, G.O. Colorectal cancer-derived extracellular vesicles induce transformation of fibroblasts into colon carcinoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 257. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schafer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Kreger, B.T.; Dougherty, A.L.; Greene, K.S.; Cerione, R.A.; Antonyak, M.A. Microvesicle cargo and function changes upon induction of cellular transformation. J. Biol. Chem. 2016, 291, 19774–19785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Stefanius, K.; Servage, K.; de Souza Santos, M.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. elife 2019, 8, e40226. [Google Scholar] [CrossRef]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microrna biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Abdouh, M.; Hamam, D.; Gao, Z.H.; Arena, V.; Arena, M.; Arena, G.O. Exosomes isolated from cancer patients’ sera transfer malignant traits and confer the same phenotype of primary tumors to oncosuppressor-mutated cells. J. Exp. Clin. Cancer Res. 2017, 36, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamam, D.; Abdouh, M.; Gao, Z.H.; Arena, V.; Arena, M.; Arena, G.O. Transfer of malignant trait to brca1 deficient human fibroblasts following exposure to serum of cancer patients. J. Exp. Clin. Cancer Res. 2016, 35, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdouh, M.; Gao, Z.H.; Arena, V.; Arena, M.; Burnier, M.N.; Arena, G.O. Oncosuppressor-mutated cells as a liquid biopsy test for cancer-screening. Sci. Rep. 2019, 9, 2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecati, M.; Giulietti, M.; Righetti, A.; Sabanovic, B.; Piva, F. Effects of cxcl12 isoforms in a pancreatic pre-tumour cellular model: Microarray analysis. World J. Gastroenterol. 2021, 27, 1616–1629. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-freec: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vareedayah, A.A.; Alkaade, S.; Taylor, J.R. Pancreatic adenocarcinoma. Mo Med. 2018, 115, 230–235. [Google Scholar]
- Lin, Y.C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.; et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014, 5, 4767. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, D.J.; Adelaide, J.; Mamessier, E.; Finetti, P.; Lagarde, A.; Monges, G.; Viret, F.; Goncalves, A.; Turrini, O.; Delpero, J.R.; et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer 2011, 50, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Tabach, Y.; Kogan-Sakin, I.; Buganim, Y.; Solomon, H.; Goldfinger, N.; Hovland, R.; Ke, X.S.; Oyan, A.M.; Kalland, K.H.; Rotter, V.; et al. Amplification of the 20q chromosomal arm occurs early in tumorigenic transformation and may initiate cancer. PLoS ONE 2011, 6, e14632. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.S.; Im, K.; Lee, D.S.; Kwon, W.; Kim, J.R.; Han, Y.; Kim, S.W.; Jang, J.Y. The implication of cytogenetic alterations in pancreatic ductal adenocarcinoma and intraductal papillary mucinous neoplasm identified by fluorescence in situ hybridization and their potential diagnostic utility. Gut Liver 2020, 14, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlamaki, E.H.; Barlund, M.; Tanner, M.; Gorunova, L.; Hoglund, M.; Karhu, R.; Kallioniemi, A. Frequent amplification of 8q24, 11q, 17q, and 20q-specific genes in pancreatic cancer. Genes Chromosomes Cancer 2002, 35, 353–358. [Google Scholar] [CrossRef]
- Eichler, M.; Distler, U.; Nasrullah, U.; Krishnan, A.; Kaulich, M.; Husnjak, K.; Eberhardt, W.; Rajalingam, K.; Tenzer, S.; Pfeilschifter, J.; et al. The caspase-2 substrate p54nrb exhibits a multifaceted role in tumor cell death susceptibility via gene regulatory functions. Cell Death Dis. 2022, 13, 386. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Decker, C.C.; Zechner, L.; Krstin, S.; Wink, M. In vitro wound healing of tumor cells: Inhibition of cell migration by selected cytotoxic alkaloids. BMC Pharmacol. Toxicol. 2019, 20, 4. [Google Scholar] [CrossRef] [Green Version]
- Pijuan, J.; Barcelo, C.; Moreno, D.F.; Maiques, O.; Siso, P.; Marti, R.M.; Macia, A.; Panosa, A. In vitro cell migration, invasion, and adhesion assays: From cell imaging to data analysis. Front. Cell Dev. Biol. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between stat3 and p53/ras signaling controls cancer cell metastasis and cisplatin resistance via the slug/mapk/pi3k/akt-mediated regulation of emt and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. Stat3 plays a critical role in kras-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Lin, I.Y.; Pan, M.H.; Lai, C.S.; Lin, T.T.; Chen, C.T.; Chung, T.S.; Chen, C.L.; Lin, C.H.; Chuang, W.C.; Lee, M.C.; et al. Ccm111, the water extract of antrodia cinnamomea, regulates immune-related activity through stat3 and nf-kappab pathways. Sci. Rep. 2017, 7, 4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capiralla, H.; Vingtdeux, V.; Venkatesh, J.; Dreses-Werringloer, U.; Zhao, H.; Davies, P.; Marambaud, P. Identification of potent small-molecule inhibitors of stat3 with anti-inflammatory properties in raw 264.7 macrophages. FEBS J. 2012, 279, 3791–3799. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated stat3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, S. Role of mtor signaling in tumor cell motility, invasion and metastasis. Curr. Protein. Pept. Sci. 2011, 12, 30–42. [Google Scholar]
- Liu, L.; Li, F.; Cardelli, J.A.; Martin, K.A.; Blenis, J.; Huang, S. Rapamycin inhibits cell motility by suppression of mtor-mediated s6k1 and 4e-bp1 pathways. Oncogene 2006, 25, 7029–7040. [Google Scholar] [CrossRef] [Green Version]
- Buenafe, A.C.; Dorrell, C.; Reddy, A.P.; Klimek, J.; Marks, D.L. Proteomic analysis distinguishes extracellular vesicles produced by cancerous versus healthy pancreatic organoids. Sci. Rep. 2022, 12, 3556. [Google Scholar] [CrossRef]
- Tani, T.; Lumme, A.; Linnala, A.; Kivilaakso, E.; Kiviluoto, T.; Burgeson, R.E.; Kangas, L.; Leivo, I.; Virtanen, I. Pancreatic carcinomas deposit laminin-5, preferably adhere to laminin-5, and migrate on the newly deposited basement membrane. Am. J. Pathol. 1997, 151, 1289–1302. [Google Scholar]
- Wang, G.; Zhang, Q.; Song, Y.; Wang, X.; Guo, Q.; Zhang, J.; Li, J.; Han, Y.; Miao, Z.; Li, F. Pak1 regulates rufy3-mediated gastric cancer cell migration and invasion. Cell Death Dis. 2015, 6, e1682. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Estrella, J.S.; Katz, M.H.; Kim, M.; Rashid, A.; Lee, J.E.; Maitra, A.; Wistuba, I.I.; Wolff, R.A.; Varadhachary, G.R.; et al. Expression of epithelial-mesenchymal transition markers in treated pancreatic ductal adenocarcinoma. Pancreas 2019, 48, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Maehira, H.; Miyake, T.; Iida, H.; Tokuda, A.; Mori, H.; Yasukawa, D.; Mukaisho, K.I.; Shimizu, T.; Tani, M. Vimentin expression in tumor microenvironment predicts survival in pancreatic ductal adenocarcinoma: Heterogeneity in fibroblast population. Ann. Surg. Oncol. 2019, 26, 4791–4804. [Google Scholar] [CrossRef]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000 Res. 2018, 7, F1000 Faculty Rev-1796. [Google Scholar] [CrossRef] [PubMed]
- Inada, M.; Izawa, G.; Kobayashi, W.; Ozawa, M. 293 cells express both epithelial as well as mesenchymal cell adhesion molecules. Int. J. Mol. Med. 2016, 37, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanani, K.C.H.; Samson, W.J.; Edkins, A.L. Fibronectin is a stress responsive gene regulated by hsf1 in response to geldanamycin. Sci. Rep. 2017, 7, 17617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, E.; Doi, R.; Koizumi, M.; Kami, K.; Ito, D.; Mori, T.; Fujimoto, K.; Nakajima, S.; Wada, M.; Imamura, M. Analysis of e-, n-cadherin, alpha-, beta-, and gamma-catenin expression in human pancreatic carcinoma cell lines. Pancreas 2005, 30, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Kirkbride, K.C.; How, T.; Blobe, G.C. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2009, 30, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Zhang, S.; Gao, S.; Shang, J.; Li, Y.; Cui, N.; Zhao, Q. Mrgbp as a potential biomarker for the malignancy of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 64224–64236. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhao, J.; Lu, J.; Wang, P.; Feng, H.; Zong, Y.; Ou, B.; Zheng, M.; Lu, A. Cadherin-12 enhances proliferation in colorectal cancer cells and increases progression by promoting emt. Tumour Biol. 2016, 37, 9077–9088. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, P.; Feng, H.; Wang, P.; Zong, Y.; Ma, J.; Zhang, Z.; Chen, X.; Zheng, M.; Zhu, Z.; et al. Cadherin-12 contributes to tumorigenicity in colorectal cancer by promoting migration, invasion, adhersion and angiogenesis. J. Transl. Med. 2013, 11, 288. [Google Scholar] [CrossRef] [Green Version]
- Song, T.L.; Nairismagi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the stat3 pathway drives pd-l1 expression in natural killer/t-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Faried, L.S.; Faried, A.; Kanuma, T.; Nakazato, T.; Tamura, T.; Kuwano, H.; Minegishi, T. Inhibition of the mammalian target of rapamycin (mtor) by rapamycin increases chemosensitivity of caski cells to paclitaxel. Eur. J. Cancer 2006, 42, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Masuda, T.; Fujii, A.; Shimizu, D.; Sato, K.; Kitagawa, A.; Tobo, T.; Ozato, Y.; Saito, H.; Kuramitsu, S.; et al. Mitotic checkpoint regulator rae1 promotes tumor growth in colorectal cancer. Cancer Sci. 2021, 112, 3173–3189. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Furukawa, T.; Sunamura, M.; Egawa, S.; Motoi, F.; Ohmura, N.; Marumoto, T.; Saya, H.; Horii, A. Rna interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005, 65, 2899–2905. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Zhang, P.; Yu, S.; Tang, C.; Wang, Y.; Shen, Z.; Chen, W.; Liu, T.; Cui, Y. Targeting zfp64/gal-1 axis promotes therapeutic effect of nab-paclitaxel and reverses immunosuppressive microenvironment in gastric cancer. J. Exp. Clin. Cancer Res. 2022, 41, 14. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, N.; Wei, Y.; Zhou, D.; Lin, R.; Wang, X.; Shi, B. Anticancer effects of mir-124 delivered by bm-msc derived exosomes on cell proliferation, epithelial mesenchymal transition, and chemotherapy sensitivity of pancreatic cancer cells. Aging 2020, 12, 19660–19676. [Google Scholar] [CrossRef]
- Li, L.; Zhang, J.W.; Jenkins, G.; Xie, F.; Carlson, E.E.; Fridley, B.L.; Bamlet, W.R.; Petersen, G.M.; McWilliams, R.R.; Wang, L. Genetic variations associated with gemcitabine treatment outcome in pancreatic cancer. Pharm. Genom. 2016, 26, 527–537. [Google Scholar] [CrossRef]
- Ono, H.; Basson, M.D.; Ito, H. Ptk6 potentiates gemcitabine-induced apoptosis by prolonging s-phase and enhancing DNA damage in pancreatic cancer. Mol. Cancer Res. 2015, 13, 1174–1184. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Huang, H.; Feng, M.; Yang, G.; Zheng, S.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; Zhao, Y. Mir-10a-5p targets tfap2c to promote gemcitabine resistance in pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 76. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | HEK293T (Control) | HEK293T (Treated) | p-Value |
|---|---|---|---|
| Gemcitabine 13 uM | 42% | 16% | 0.009 |
| Doxorubicin 100 nM | 53% | 30% | 0.001 |
| Paclitaxel 100 nM | 51% | 91% | 0.001 |
| 5-FU 5 uM | 46% | 17% | 0.001 |
| Coordinates (Chromosome: Start–End) | Status (Copy Number) | p-Value (Wilcoxon Rank Sum; Kolmogorov–Smirnov) | Genes |
|---|---|---|---|
| 1: 246,640,000–246,749,999 | gain (3) | 4.34 × 10−2; 2.55 × 10−1 | CNST, SCCPDH |
| 3: 33,290,000–33,379,999 | loss (1) | 1.11 × 10−4; 9.40 × 10−5 | FBXL2 |
| 3: 76,610,000–76,719,999 | loss (1) | 6.26 × 10−6; 2.23 × 10−6 | ROBO2 |
| 3: 93,430,000–93,519,999 | loss (1) | 1.40 × 10−4; 1.09 × 10−4 | none |
| 4: 49,660,000–49,759,999 | loss (1) | 2.91 × 10−5; 1.55 × 10−5 | none |
| 4: 70,690,000–70,779,999 | gain (3) | 1.37 × 10−3; 5.15 × 10−4 | RUFY3, UTP3 |
| 5: 22,290,000–22,379,999 | gain (6) | 2.76 × 10−2; 4.79 × 10−3 | CDH12 |
| 7: 111,400,000–111,579,999 | loss (1) | 1.46 × 10−10; 2.54 × 10−12 | IMMP2L |
| 7: 119,650,000–119,739,999 | loss (1) | 1.15 × 10−4; 9.62 × 10−5 | LINC02476 |
| 9: 61,000,000–61,509,999 | gain (3) | 2.23 × 10−6; 4.67 × 10−6 | SPATA31A7, FAM74A4, CNTNAP3C |
| 15: 23,180,000–23,239,999 | gain (3) | 3.63 × 10−2; 6.56 × 10−2 | none |
| 19: 54,730,000–54,779,999 | gain (4) | 4.25 × 10−4; 3.21 × 10−4 | KIR3DL3, KIR2DL1 |
| 19: 54,740,000–54,789,999 | gain (38) | 3.71 × 10−4; 3.21 × 10−4 | KIR3DL3, KIR2DL1 |
| 19: 54,750,000–54,839,999 | gain (4) | 2.93 × 10−6; 4.53 × 10−6 | KIR3DL4, KIR3DL3, KIR2DL1 |
| 19: 54,800,000–54,869,999 | gain (8) | 1.77 × 10−3; 6.15 × 10−3 | KIR2DL4, KIR3DL1, KIR2DS4, KIR3DL2 |
| 20: 29,120,000–30,079,999 | gain (3) | 8.02 × 10−22; 0.00 | FAM242B, FRG1EP, FRG2EP |
| 20: 30,040,000–30,089,999 | gain (114) | 2.14 × 10−3; 3.09 × 10−3 | none |
| 20: 30,050,000–30,359,999 | gain (3) | 1.01 × 10−2; 2.62 × 10−2 | FAM242A, LINC01597 |
| 20: 51,700,000–52,099,999 | gain (3) | 6.01 × 10−20; 0.00 | ATP9A, SALL4, LINC01429, ZFP64 |
| 20: 52,100,000–52,989,999 | gain (3) | 1.01 × 10−5; 2.39 × 10−8 | ZFP64, LINC01524, TSHZ2 |
| 20: 52,990,000–53,889,999 | gain (3) | 4.21 × 10−24; 0.00 | TSHZ2, ZNF217, SUMO1P1 |
| 20: 53,890,000–61,649,999 | gain (3) | 7.00 × 10−3; 1.17 × 10−3 | ANKRD60, APCDD1L, APCDD1L-DT, ATP5F1E, AURKA, BCAS1, BMP7, BMP7-AS1, C20orf85, CASS4, CBLN4, CDH26, CDH4, CSTF1, CTCFL, CTSZ, CYP24A1, DOK5, EDN3, FAM209A, FAM209B, FAM210B, FAM217B, GCNT7, GNAS, GNAS-AS1, LINC01440, LINC01441, LINC01711, LINC01716, LINC01718, LINC01742, LINC02910, MC3R, MIR296, MIR298, MIR4325, MIR4533, MIR4756, MIR548AG2, MIR646, MIR646HG, MTRNR2L3, NELFCD, NKILA, NPEPL1, PCK1, PFDN4, PHACTR3, PHACTR3-AS1, PMEPA1, PPP1R3D, PPP4R1L, PRELID3B, RAB22A, RAE1, RBM38, RBM38-AS1, RTF2, SLMO2-ATP5E, SPO11, STX16, SYCP2, TFAP2C, TUBB1, VAPB, ZBP1, ZNF831 |
| 20: 61,650,000–63,469,999 | gain (3) | 6.32 × 10−37; 0.00 | ADRM1, ARFGAP1, BHLHE23, BIRC7, CABLES2, CDH4, CHRNA4, COL20A1, COL9A3, DIDO1, GATA5, GID8, HAR1A, HAR1B, HRH3, KCNQ2, KCNQ2-AS1, LAMA5, LAMA5-AS1, LINC00029, LINC00659, LINC01056, LINC01749, LSM14B, MIR1-1, MIR1-1HG, MIR1-1HG-AS1, MIR124-3, MIR1257, MIR133A2, MIR3195, MIR3196, MIR4326, MIR4758, MRGBP, MTG2, NKAIN4, NTSR1, OGFR, OGFR-AS1, OSBPL2, PSMA7, RBBP8NL, RPS21, SLC17A9, SLCO4A1, SLCO4A1-AS1, SNORA117, SS18L1, TAF4, TCFL5, WI2-87327B8.2, YTHDF1 |
| 20: 63,470,000–64,444,167 | gain (3) | 2.66 × 10−4; 3.03 × 10−4 | ABHD16B, ARFRP1, C20orf181, C20orf204, DNAJC5, EEF1A2, FNDC11, GMEB2, HELZ2, KCNQ2, LIME1, LINC00266-1, LKAAEAR1, MHENCR, MIR1914, MIR647, MIR6813, MIR941-1, MIR941-2, MIR941-3, MIR941-4, MIR941-5, MYT1, NPBWR2, OPRL1, PCMTD2, PPDPF, PRPF6, PTK6, RGS19, RTEL1, SAMD10, SLC2A4RG, SOX18, SRMS, STMN3, TCEA2, TNFRSF6B, TPD52L2, UCKL1, UCKL1-AS1, ZBTB46, ZBTB46-AS1, ZGPAT, ZNF512B |
| 21: 10,170,000–10,319,999 | gain (3) | 2.41 × 10−3; 7.19 × 10−4 | none |
| X: 1–229,999 | gain (7) | 6.45 × 10−6; 2.95 × 10−6 | none |
| X: 1,800,000–1,979,999 | gain (3) | 3.95 × 10−4; 8.63 × 10−5 | none |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabanovic, B.; Giulietti, M.; Cecati, M.; Spolverato, G.; Benna, C.; Pucciarelli, S.; Piva, F. Effects of the Exposure of Human Non-Tumour Cells to Sera of Pancreatic Cancer Patients. Biomedicines 2022, 10, 2588. https://doi.org/10.3390/biomedicines10102588
Sabanovic B, Giulietti M, Cecati M, Spolverato G, Benna C, Pucciarelli S, Piva F. Effects of the Exposure of Human Non-Tumour Cells to Sera of Pancreatic Cancer Patients. Biomedicines. 2022; 10(10):2588. https://doi.org/10.3390/biomedicines10102588
Chicago/Turabian StyleSabanovic, Berina, Matteo Giulietti, Monia Cecati, Gaya Spolverato, Clara Benna, Salvatore Pucciarelli, and Francesco Piva. 2022. "Effects of the Exposure of Human Non-Tumour Cells to Sera of Pancreatic Cancer Patients" Biomedicines 10, no. 10: 2588. https://doi.org/10.3390/biomedicines10102588