
Differential Transcriptome Profiling Unveils Novel Deregulated Gene Signatures Involved in Pathogenesis of Alzheimer’s Disease

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset: NCBI/GEO Database
2.2. Affy Package: Expression Computation
2.3. Reactome FI Cytoscape Plugin: Network-Based Pathway Enrichment Analysis (PEA)
3. Results and Discussion
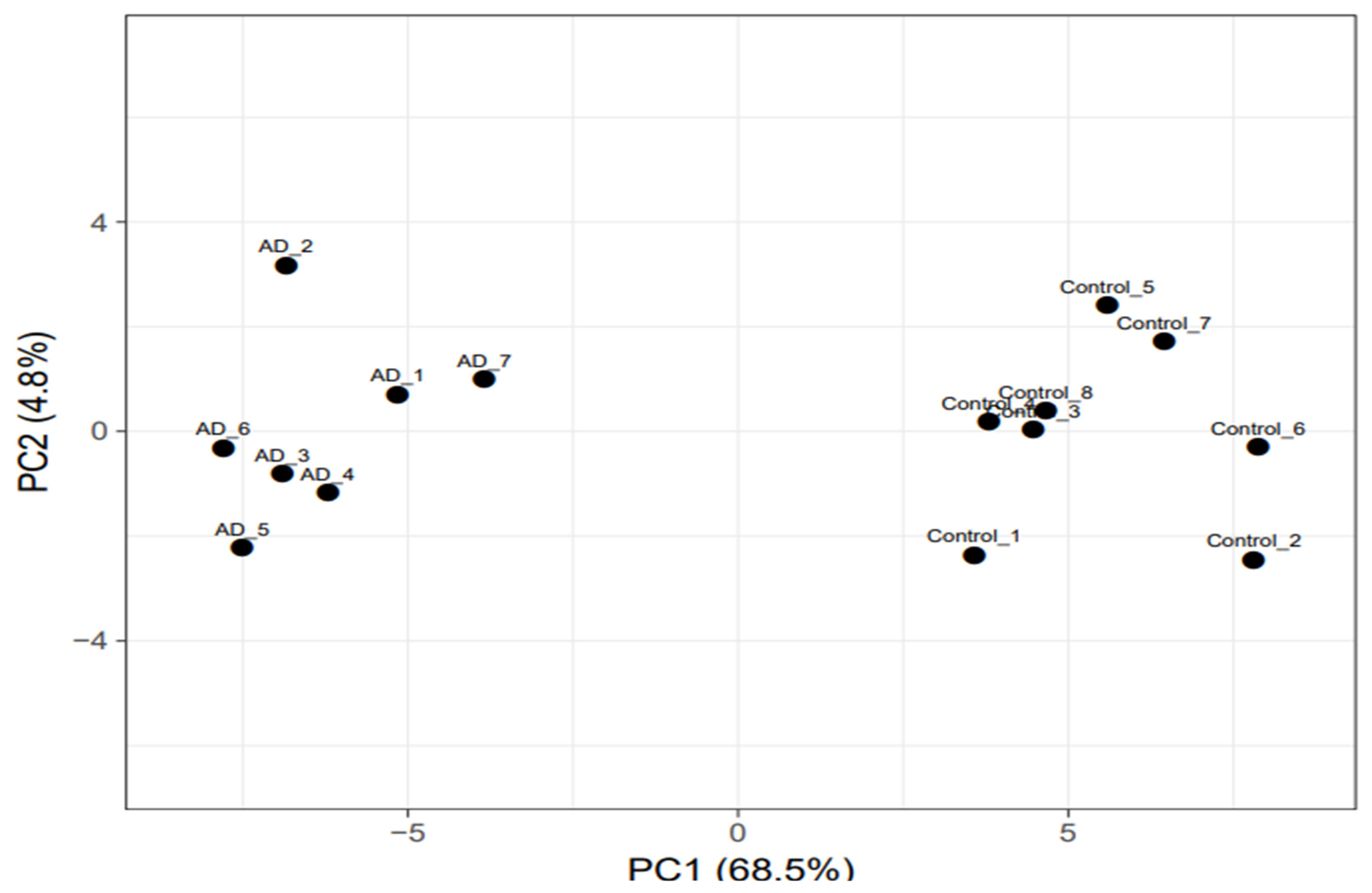
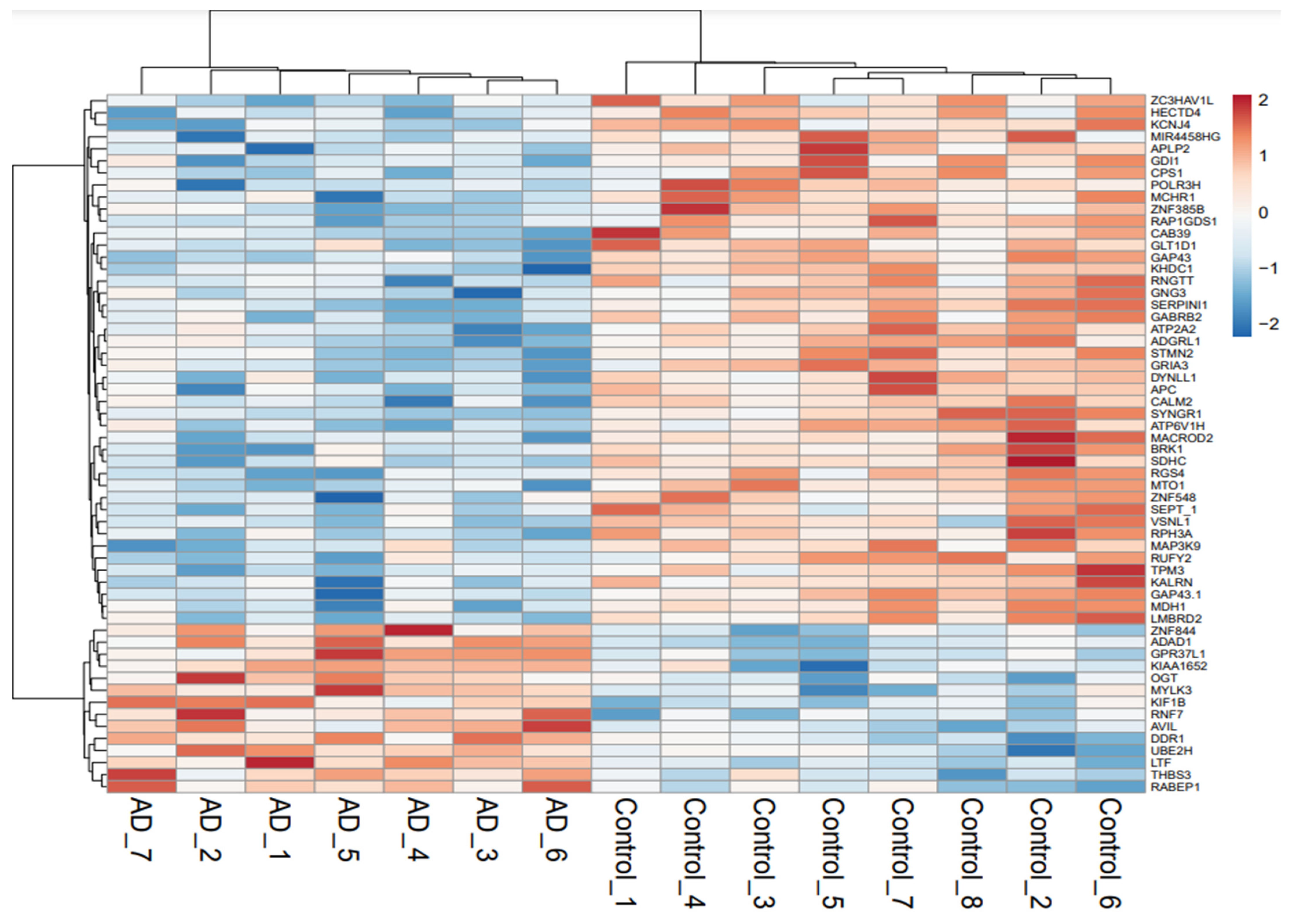
3.1. Identification of Deregulated Genes in AD
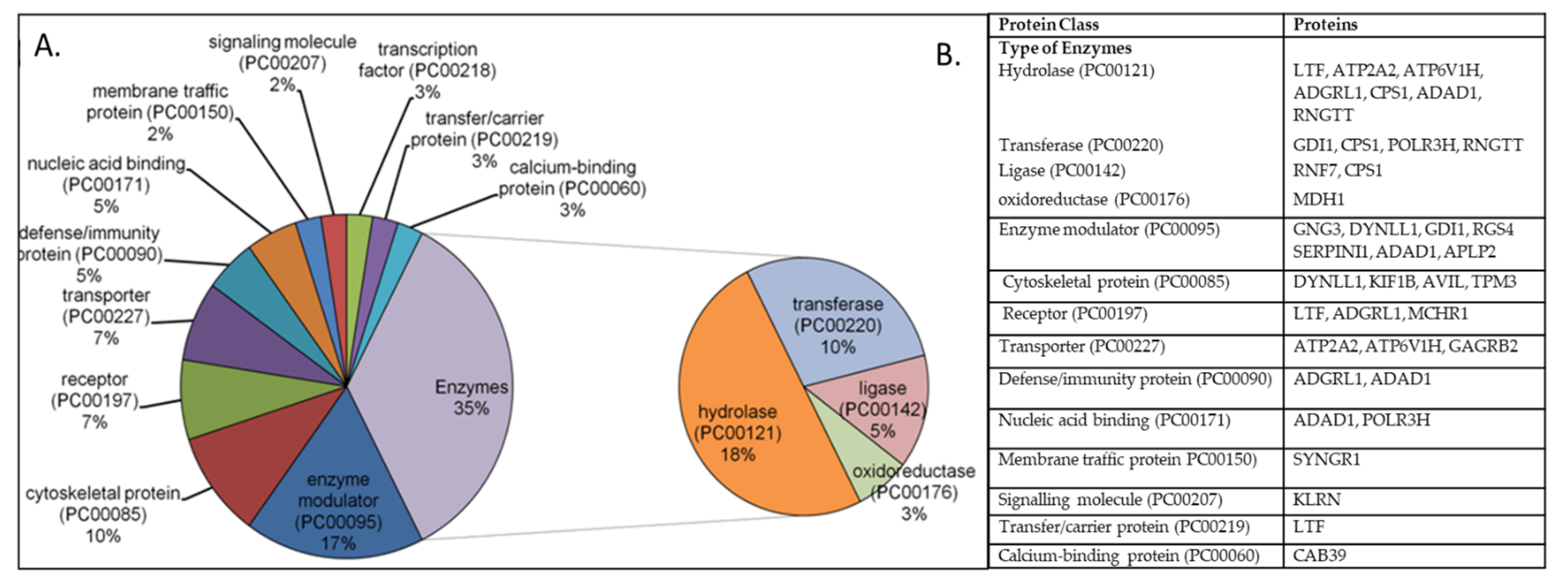
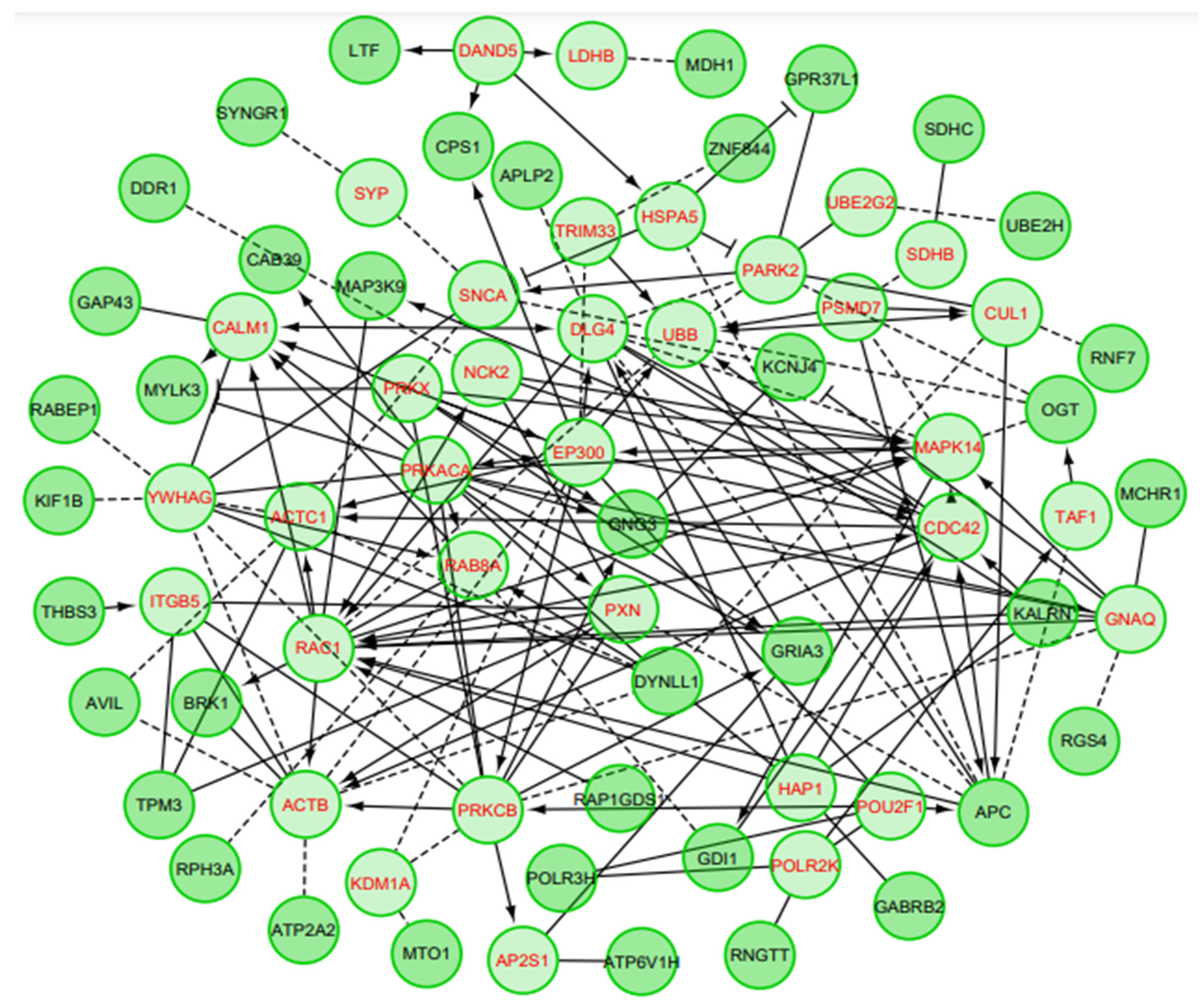
3.2. Gene Set/PEA of DEGs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The public health impact of Alzheimer’s disease, 2000–2050: Potential implication of treatment advances. Annu. Rev. Pub. Health 2002, 23, 213–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isik, A.T. Late onset Alzheimer’s disease in older people. Clin. Interv. Aging 2010, 5, 307–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirtz, D.; Thurman, D.J.; Gwinn-Hardy, K.; Mohamed, M.; Chaudhuri, A.R.; Zalutsky, R. How common are the “Common” neurologic disorders? Neurology 2007, 68, 326–337. [Google Scholar] [CrossRef]
- Giri, M.; Zhang, M.; Lü, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.-I.; Kino, Y.; Niida, S. MicroRNA-seq data analysis pipeline to identify blood biomarkers for Alzheimer’s disease from public data. Biomark. Insights 2015, 10, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from Atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid. Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Chen, Y.; Masiulis, I.; Zhou, L. Expanding functions of lipoprotein receptors. J. Lipid. Res. 2009, 50, S287–S292. [Google Scholar] [CrossRef] [Green Version]
- Gajera, C.R.; Emich, H.; Lioubinski, O.; Christ, A.; Beckervordersandforth-Bonk, R.; Yoshikawa, K.; Bachmann, S.; Christensen, E.I.; Götz, M.; Kempermann, G.; et al. LRP2 in ependymal cells regulates BMP signaling in the adult neurogenic niche. J. Cell. Sci. 2010, 123, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Sando, S.B.; Melquist, S.; Cannon, A.; Hutton, M.L.; Sletvold, O.; Saltvedt, I.; White, L.R.; Lydersen, S.; Aasly, J.O. APOE Ε4 lowers age at onset and is a high risk factor for Alzheimer’s disease; A case control study from central Norway. BMC Neurol. 2008, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Ann. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikai, S.; Sasaki, H.; Doh-ura, K.; Furuya, H.; Sakaki, Y. Genomic organization of the human amyloid beta-protein precursor gene. Gene 1990, 87, 257–263. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Bentahir, M.; Nyabi, O.; Verhamme, J.; Tolia, A.; Horré, K.; Wiltfang, J.; Esselmann, H.; De Strooper, B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 2006, 96, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H. Uncovering gamma-secretase. Curr Alzheimer Res. 2004, 1, 175–181. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Park, K.H.; Noh, Y.; Choi, E.J.; Kim, H.; Chun, S.; Son, Y.D. Functional connectivity of the hippocampus in early- and vs. late-onset Alzheimer’s disease. J. Clin. Neurol. 2017, 13, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of affymetrix geneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- An, L.; Sato, H.; Konishi, Y.; Walker, D.G.; Beach, T.G.; Rogers, J.; Tooyama, I. Expression and localization of lactotransferrin MRNA in the cortex of Alzheimer’s disease. Neurosci. Lett. 2009, 452, 277–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Acute Phase Protein Lactoferrin Is a Key Feature of Alzheimer’s Disease and Predictor of Aβ Burden through Induction of APP Amyloidogenic Processing—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/34400772/ (accessed on 9 January 2022).
- Nakajima, K.; Ishiwata, M.; Weitemier, A.Z.; Shoji, H.; Monai, H.; Miyamoto, H.; Yamakawa, K.; Miyakawa, T.; McHugh, T.J.; Kato, T. Brain-specific heterozygous loss-of-function of ATP2A2, endoplasmic reticulum Ca2+ pump responsible for Darier’s disease, causes behavioral abnormalities and a hyper-dopaminergic state. Hum. Mol. Genet. 2021, 30, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bai, J.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Downregulation of ATP6V1A involved in Alzheimer’s disease via synaptic vesicle cycle, phagosome, and oxidative phosphorylation. Oxid. Med. Cell. Longev. 2021, 2021, 5555634. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.F.; Qu, H.-Q.; Rentfro, A.R.; Nair, S.; Lu, Y.; Hanis, C.L.; McCormick, J.B.; Fisher-Hoch, S.P. Decreased expression of ATP6V1H in type 2 diabetes: A pilot report on the diabetes risk study in Mexican Americans. Biochem. Biophys. Res. Commun. 2011, 412, 728–731. [Google Scholar] [CrossRef] [Green Version]
- Geyer, M.; Fackler, O.T.; Peterlin, B.M. Subunit H of the V-ATPase involved in endocytosis shows homology to β-adaptins. Mol. Biol. Cell. 2002, 13, 2045–2056. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification—The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Leite Góes Gitai, D.; de Andrade, T.G.; dos Santos, Y.D.R.; Attaluri, S.; Shetty, A.K. Chronobiology of limbic seizures: Potential mechanisms and prospects of chronotherapy for mesial temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2019, 98, 122–134. [Google Scholar] [CrossRef]
- Schwindinger, W.F.; Mirshahi, U.L.; Baylor, K.A.; Sheridan, K.M.; Stauffer, A.M.; Usefof, S.; Stecker, M.M.; Mirshahi, T.; Robishaw, J.D. Synergistic roles for G-protein Γ3 and Γ7 subtypes in seizure susceptibility as revealed in double knock-out mice. J. Biol. Chem. 2012, 287, 7121–7133. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.-L.; Yang, F.; Frautschy, S.A.; Cole, G.M. PAK in Alzheimer disease, huntington disease and X-linked mental retardation. Cell. Logist. 2012, 2, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, A.; Garcia, P.; Kollmus, H.; Schughart, K.; Del Sol, A.; Buttini, M.; Glaab, E. Absence of regulator of G-protein signaling 4 does not protect against dopamine neuron dysfunction and injury in the mouse 6-hydroxydopamine lesion model of Parkinson’s Disease. Neurobiol. Aging 2017, 58, 30–33. [Google Scholar] [CrossRef] [Green Version]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnham, K.J.; McKinstry, W.J.; Multhaup, G.; Galatis, D.; Morton, C.J.; Curtain, C.C.; Williamson, N.A.; White, A.R.; Hinds, M.G.; Norton, R.S.; et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 2003, 278, 17401–17407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashisht, R.; Mondal, A.K.; Jain, A.; Shah, A.; Vishnoi, P.; Priyadarshini, P.; Bhattacharyya, K.; Rohira, H.; Bhat, A.G.; Passi, A.; et al. Crowd sourcing a new paradigm for interactome driven drug target identification in mycobacterium tuberculosis. PLoS ONE 2012, 7, e39808. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association 2011 Alzheimer’s Disease Facts and Figures. Alzheimers Dement 2011, 7, 208–244. [CrossRef] [PubMed]
- Novak, M.J.U.; Tabrizi, S.J. Huntington’s disease. BMJ 2010, 340, c3109. [Google Scholar] [CrossRef] [Green Version]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.J.; Lovinger, D.M. Developmental alteration of endocannabinoid retrograde signaling in the hippocampus. J. Neurophysiol. 2010, 103, 1123–1129. [Google Scholar] [CrossRef] [Green Version]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134, 1041–1060. [Google Scholar] [CrossRef]
- Greenberg, D.A.; Jin, K. From angiogenesis to neuropathology. Nature 2005, 438, 954–959. [Google Scholar] [CrossRef]
- Martin, L.; Bouvet, P.; Chounlamountri, N.; Watrin, C.; Besançon, R.; Pinatel, D.; Meyronet, D.; Honnorat, J.; Buisson, A.; Salin, P.-A.; et al. VEGF counteracts amyloid-β-induced synaptic dysfunction. Cell. Rep. 2021, 35, 109121. [Google Scholar] [CrossRef]
- Okabe, K.; Fukada, H.; Tai-Nagara, I.; Ando, T.; Honda, T.; Nakajima, K.; Takeda, N.; Fong, G.-H.; Ema, M.; Kubota, Y. Neuron-derived VEGF contributes to cortical and hippocampal development independently of VEGFR1/2-mediated neurotrophism. Dev. Biol. 2020, 459, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.Y.; Keene, C.D.; Jayadev, S.; Bird, T. The Co-occurrence of Alzheimer’s disease and Huntington’s disease: A neuropathological study of 15 elderly Huntington’s disease subjects. J. Huntingtons Dis. 2014, 3, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Peavy, G.M.; Jacobson, M.W.; Goldstein, J.L.; Hamilton, J.M.; Kane, A.; Gamst, A.C.; Lessig, S.L.; Lee, J.C.; Corey-Bloom, J. Cognitive and functional decline in Huntington’s disease: Dementia criteria revisited. Mov. Disord. 2010, 25, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, M.K.; Hokom, M.; Eakin, C.; Zhou, L.; Deshpande, M.; Baker, M.P.; Goletz, T.J.; Kerwin, B.A.; Chirmule, N.; Narhi, L.O.; et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 2012, 287, 25266–25279. [Google Scholar] [CrossRef] [Green Version]
- Price, J.L.; Ko, A.I.; Wade, M.J.; Tsou, S.K.; McKeel, D.W.; Morris, J.C. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch. Neurol. 2001, 58, 1395–1402. [Google Scholar] [CrossRef]
- Hanna Al-Shaikh, F.S.; Duara, R.; Crook, J.E.; Lesser, E.R.; Schaeverbeke, J.; Hinkle, K.M.; Ross, O.A.; Ertekin-Taner, N.; Pedraza, O.; Dickson, D.W.; et al. Selective vulnerability of the nucleus basalis of meynert among neuropathologic subtypes of Alzheimer disease. JAMA Neurol. 2020, 77, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, H.B.; Raymond, L.A. NMDA receptors and Huntington’s disease. In Biology of the NMDA Receptor; Van Dongen, A.M., Ed.; Frontiers in Neuroscience, CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; ISBN 978-1-4200-4414-0. [Google Scholar]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.-Q.; et al. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-L.; Liu, G.; Li, Y.-C.; Gao, W.-J.; Huang, Y.-Q. Dopamine D1 Receptor-mediated NMDA receptor insertion depends on fyn but not src kinase pathway in prefrontal cortical neurons. Mol. Brain 2010, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Hennigan, A.; O’Callaghan, R.M.; Kelly, A.M. Neurotrophins and their receptors: Roles in plasticity, neurodegeneration and neuroprotection. Biochem. Soc. Trans. 2007, 35, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Peethumnongsin, E.; Yang, L.; Kallhoff-Muñoz, V.; Hu, L.; Takashima, A.; Pautler, R.G.; Zheng, H. Convergence of presenilin- and tau-mediated pathways on axonal trafficking and neuronal function. J. Neurosci. 2010, 30, 13409–13418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.L.; Fan, Y.; Li, S.; Li, X.-J.; Zhou, X.-F. Huntingtin-associated protein-1 interacts with pro-brain-derived neurotrophic factor and mediates its transport and release. J. Biol. Chem. 2010, 285, 5614–5623. [Google Scholar] [CrossRef] [Green Version]
- Youn, H.; Jeoung, M.; Koo, Y.; Ji, H.; Markesbery, W.R.; Ji, I.; Ji, T.H. Kalirin is under-expressed in Alzheimer’s disease Hippocampus. J. Alzheimers Dis. 2007, 11, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Puigdellívol, M.; Cherubini, M.; Brito, V.; Giralt, A.; Suelves, N.; Ballesteros, J.; Zamora-Moratalla, A.; Martín, E.D.; Eipper, B.A.; Alberch, J.; et al. A role for kalirin-7 in corticostriatal synaptic dysfunction in Huntington’s disease. Hum. Mol. Genet. 2015, 24, 7265–7285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridolfsson, H.N.; Ly, N.; Meyerzon, M.; Starr, D.A. UNC-83 Coordinates kinesin-1 and dynein activities at the nuclear envelope during nuclear migration. Dev. Biol. 2010, 338, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Guedes-Dias, P.; Holzbaur, E.L.F. Axonal transport: Driving synaptic function. Science 2019, 366, eaaw9997. [Google Scholar] [CrossRef]
- Karunakaran, K.B.; Chaparala, S.; Lo, C.W.; Ganapathiraju, M.K. Cilia interactome with predicted protein–protein interactions reveals connections to Alzheimer’s disease, aging and other neuropsychiatric processes. Sci. Rep. 2020, 10, 15629. [Google Scholar] [CrossRef]
- King, A.; Li, L.; Wong, D.M.; Liu, R.; Bamford, R.; Strasser, A.; Tarlinton, D.M.; Heierhorst, J. Dynein light chain regulates adaptive and innate B cell development by distinctive genetic mechanisms. PLoS Genet. 2017, 13, e1007010. [Google Scholar] [CrossRef] [Green Version]
- Corneveaux, J.J.; Liang, W.S.; Reiman, E.M.; Webster, J.A.; Myers, A.J.; Zismann, V.L.; Joshipura, K.D.; Pearson, J.V.; Hu-Lince, D.; Craig, D.W.; et al. Evidence for an association between KIBRA and late-onset Alzheimer’s disease. Neurobiol. Aging 2010, 31, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Eschbach, J.; Sinniger, J.; Bouitbir, J.; Fergani, A.; Zoll, J.; Geny, B.; Rene, F.; Larmet, Y.; Baloh, R.H.; Harms, M.B.; et al. Dynein mutations associated with hereditary motor neuropathies impair mitochondrial morphology and function with age. Neurobiol. Dis. 2013, 58, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabiner, C.A.; Mains, R.E.; Eipper, B.A. Kalirin: A dual rho guanine nucleotide exchange factor that is so much more than the sum of its many parts. Neuroscientist 2005, 11, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Mandela, P.; Ma, X.-M. Kalirin, a key player in synapse formation, is implicated in human diseases. Neural. Plast. 2012, 2012, 728161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.; Metzger, S.; Riess, O.; Soehn, A.S.; Nguyen, H.P. Genetic analysis of polymorphisms in the kalirin gene for association with age-at-onset in European Huntington Disease patients. BMC Med. Genet. 2012, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-C.; Riess, O.; Soehn, A.S.; Nguyen, H.P. The guanine nucleotide exchange factor kalirin-7 is a novel synphilin-1 interacting protein and modifies synphilin-1 aggregate transport and formation. PLoS ONE 2012, 7, e51999. [Google Scholar] [CrossRef]
- Ratovitski, E.A.; Alam, M.R.; Quick, R.A.; McMillan, A.; Bao, C.; Kozlovsky, C.; Hand, T.A.; Johnson, R.C.; Mains, R.E.; Eipper, B.A.; et al. Kalirin inhibition of inducible nitric-oxide synthase. J. Biol. Chem. 1999, 274, 993–999. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Ratio of Protein in GeneSet | Number of Protein in GeneSet | Protein from Network | p-Value | FDR | Nodes |
|---|---|---|---|---|---|---|
| Huntington disease (P) | 0.0124 | 121 | 9 | 1.03 × 10−7 | 5.17 × 10−5 | ACTB, EP300, RAC1, DYNLL1, HAP1, KLRN, CDC42, ACTC1, DLG4 |
| Retrograde endocannabinoid signaling (K) | 0.0106 | 103 | 8 | 4.04 × 10−7 | 5.17 × 10−5 | GABRB2, PRKACA, PRKCB, PRKX, GNG3, GRIA3, MAPK14, GNAQ |
| VEGF Signaling (R) | 0.0106 | 103 | 8 | 4.04 × 10−7 | 5.17 × 10−5 | BRK1, NCK2, RAC1, PRKCB, PXN, CDC42, MAPK14, CALM1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, H.N.; Swarup, V.; Dubey, N.K.; Jha, N.K.; Singh, A.K.; Lo, W.-C.; Kumar, S. Differential Transcriptome Profiling Unveils Novel Deregulated Gene Signatures Involved in Pathogenesis of Alzheimer’s Disease. Biomedicines 2022, 10, 611. https://doi.org/10.3390/biomedicines10030611
Singh HN, Swarup V, Dubey NK, Jha NK, Singh AK, Lo W-C, Kumar S. Differential Transcriptome Profiling Unveils Novel Deregulated Gene Signatures Involved in Pathogenesis of Alzheimer’s Disease. Biomedicines. 2022; 10(3):611. https://doi.org/10.3390/biomedicines10030611
Chicago/Turabian StyleSingh, Himanshu Narayan, Vishnu Swarup, Navneet Kumar Dubey, Niraj Kumar Jha, Anjani Kumar Singh, Wen-Cheng Lo, and Sanjay Kumar. 2022. "Differential Transcriptome Profiling Unveils Novel Deregulated Gene Signatures Involved in Pathogenesis of Alzheimer’s Disease" Biomedicines 10, no. 3: 611. https://doi.org/10.3390/biomedicines10030611