

Structural and Functional Characterization of Lipoxygenases from Diatoms by Bioinformatics and Modelling Studies
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. LOX Sequences: Detection, Analysis and Clustering Procedure
2.2. 3D Models Construction and Docking Analysis
3. Results
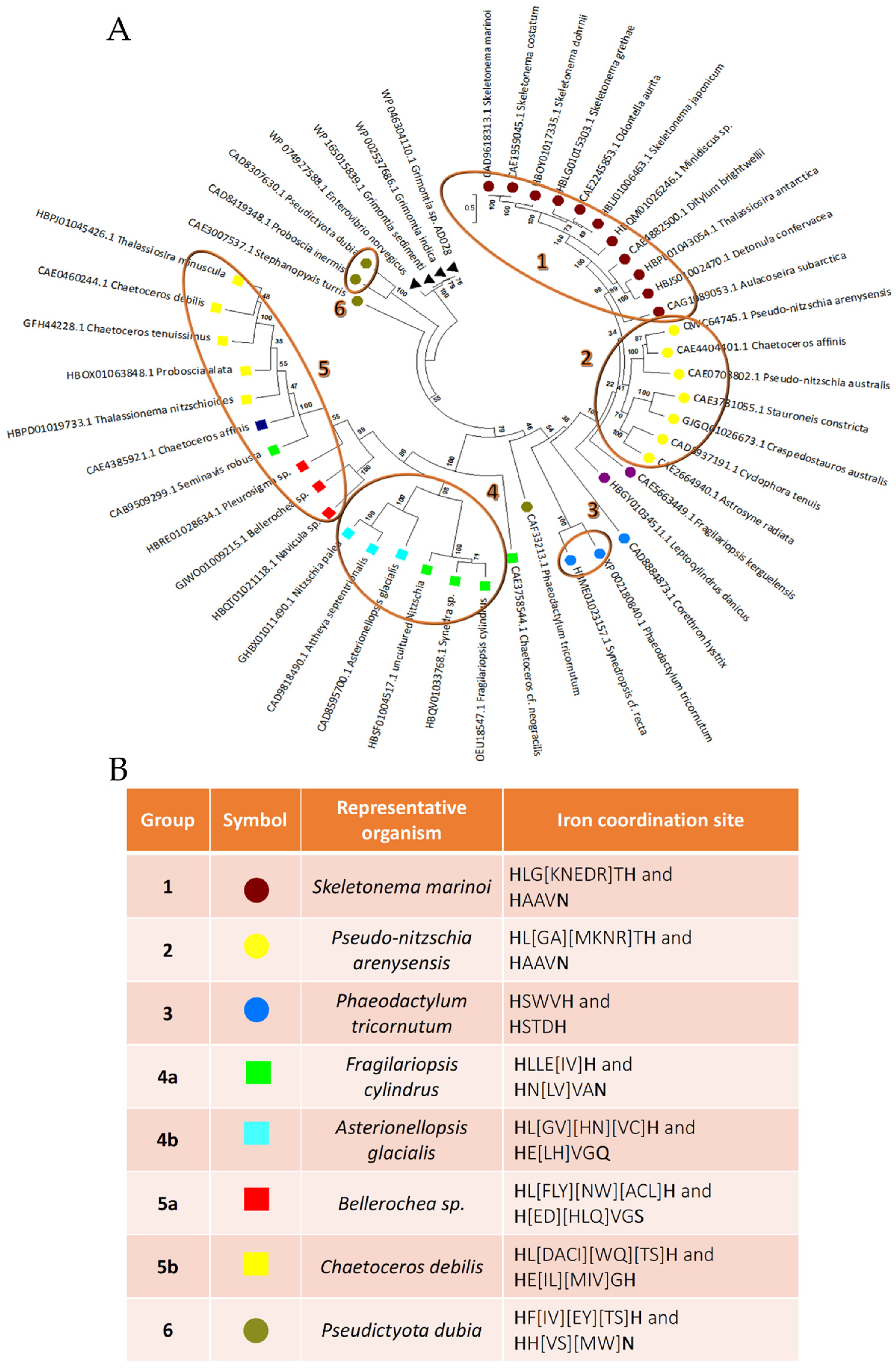
3.1. Putative LOX Sequences Detection, Analysis and Classification
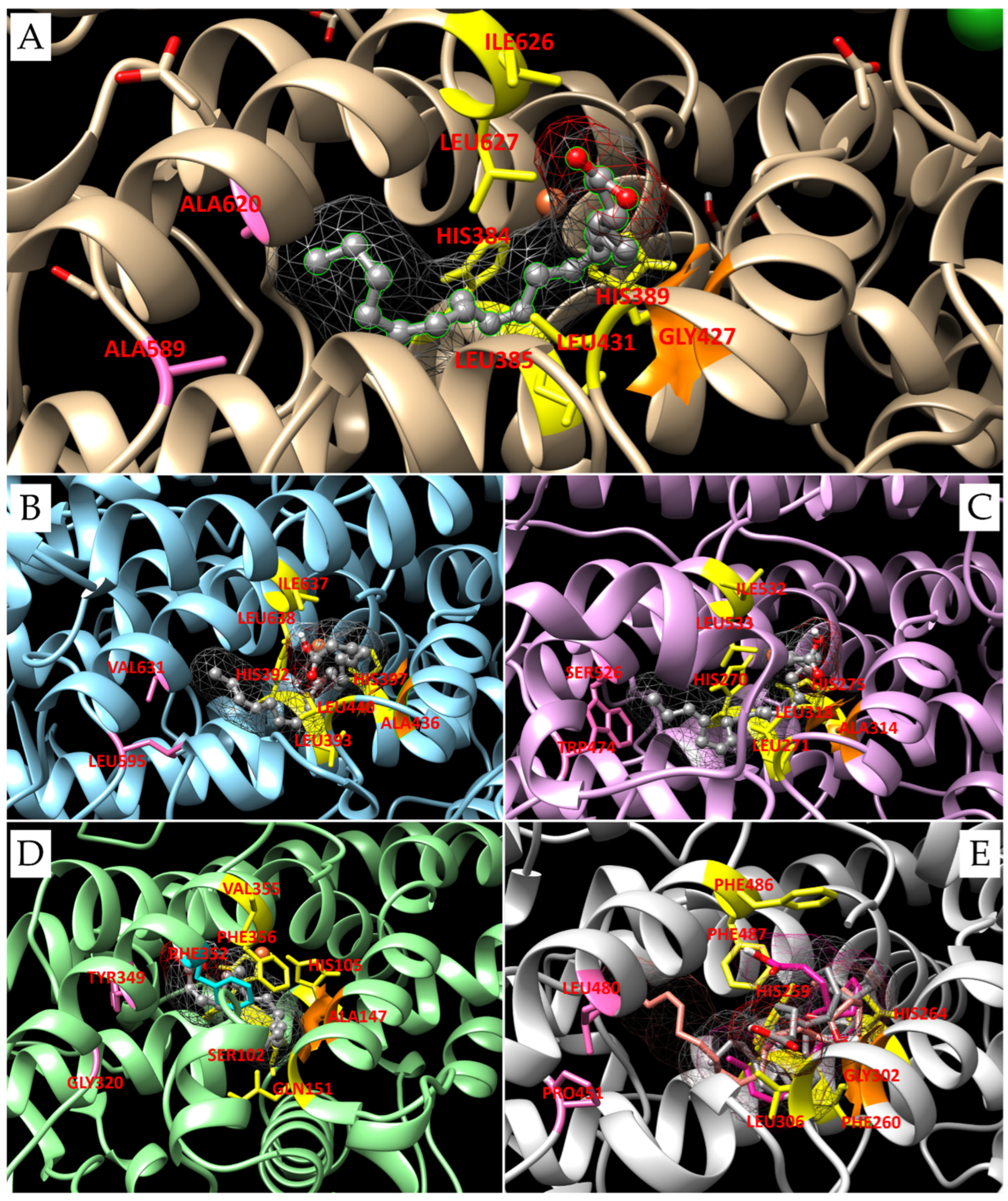
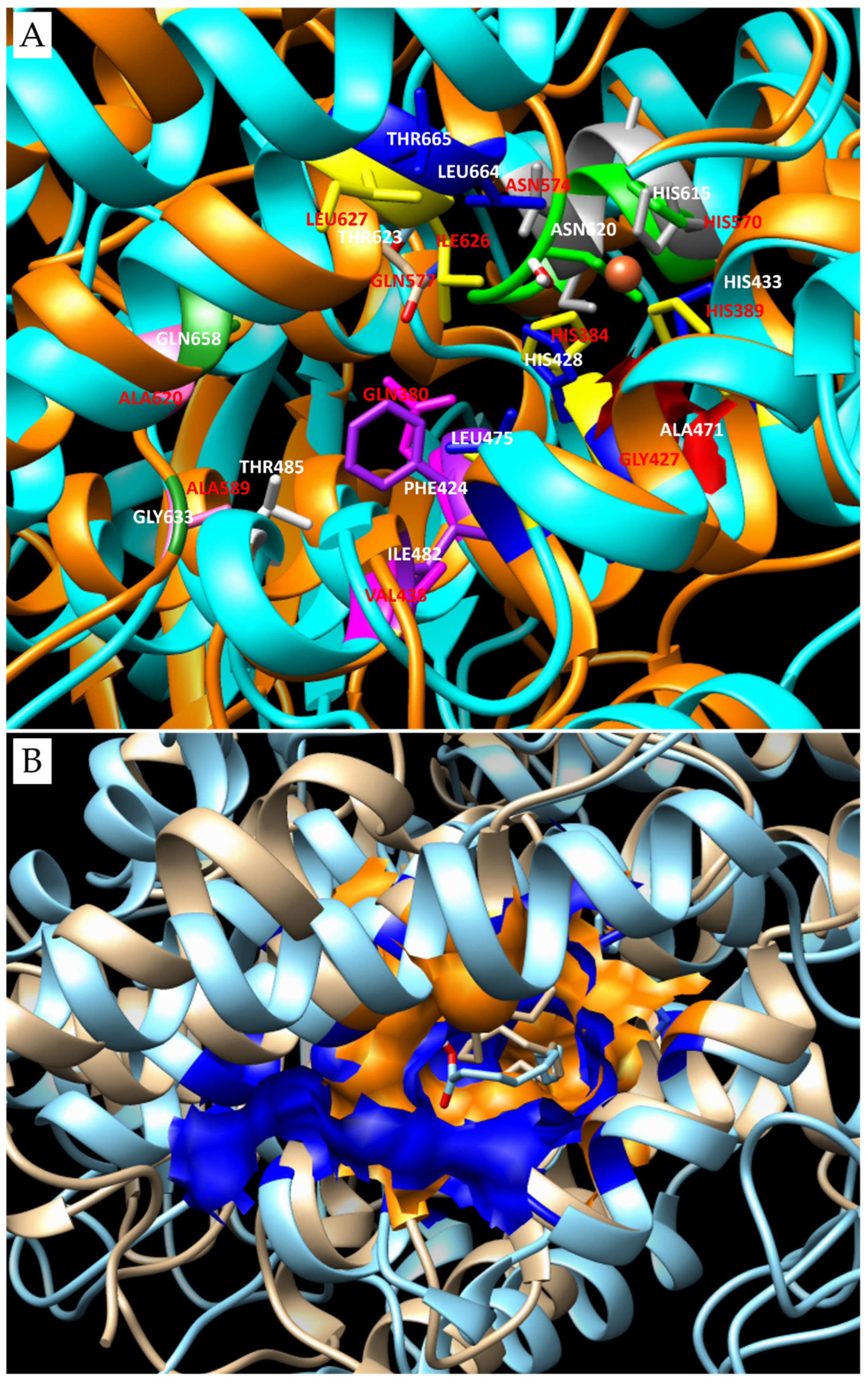
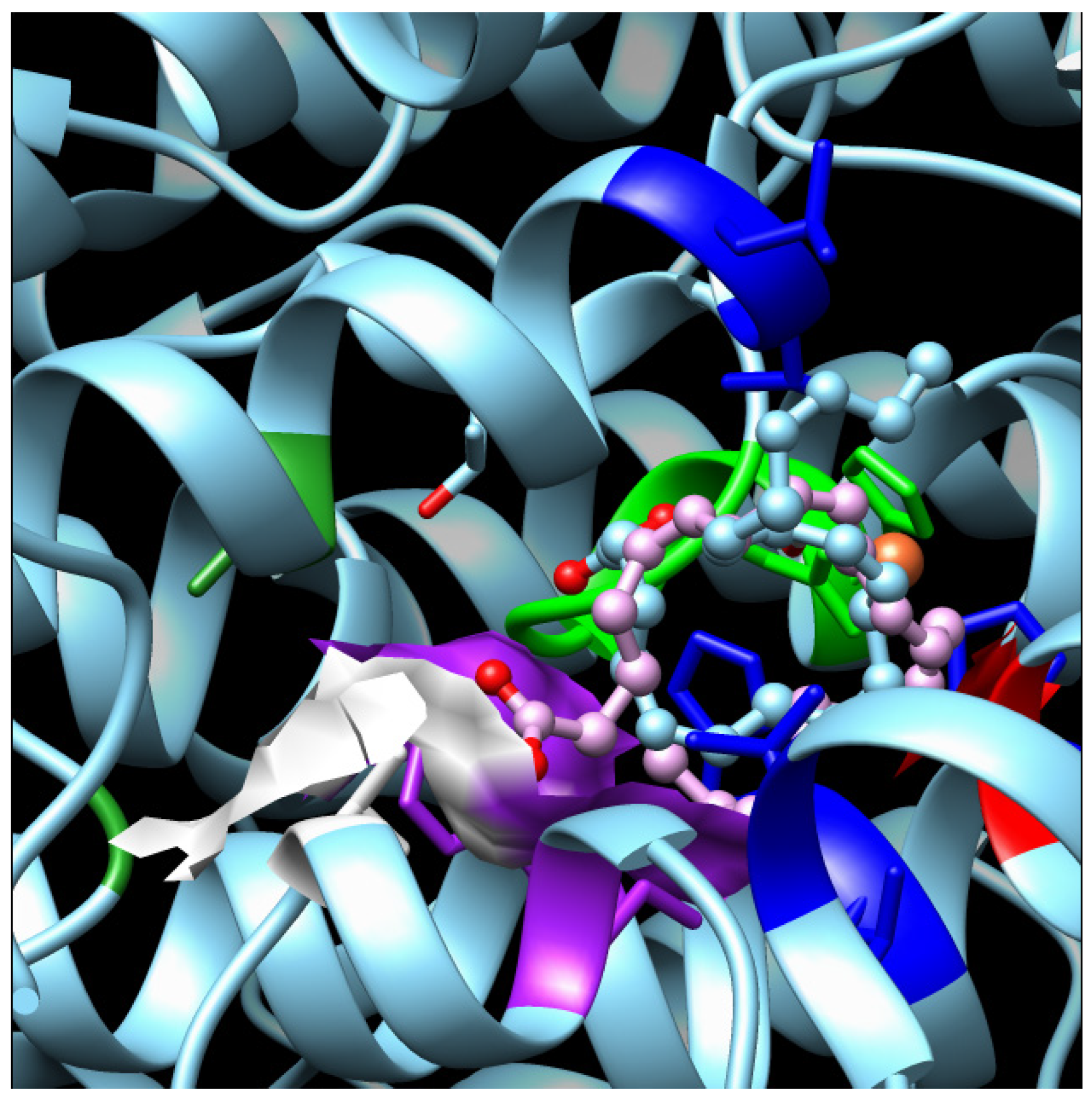
3.2. 3D Models and Docking Analysis
3.3. Docking Analyses for Sequences Similar to LOX from P. arenysensis
3.4. Docking Analyses for Sequences Similar to LOX from F. cylindrus
4. Discussion
Putative LOX Sequences Detection, Analysis and Classification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sardo, A.; Orefice, I.; Balzano, S.; Barra, L.; Romano, G. Mini-Review: Potential of Diatom-Derived Silica for Biomedical Applications. Appl. Sci. 2021, 11, 4533. [Google Scholar] [CrossRef]
- Mallimadugula, B.V.; Hameed, A. Diatom Algae for Carbon Sequestration in Oceans. In: Insights into the World of Diatoms: From Essentials to Applications. In Plant Life and Environment Dynamics; Srivastava, P., Khan, A.S., Verma, J., Dhyani, S., Eds.; Springer: Singapore, 2023. [Google Scholar] [CrossRef]
- D’Ippolito, G.; Nuzzo, G.; Sardo, A.; Manzo, E.; Gallo, C.; Fontana, A. Lipoxygenases and Lipoxygenase Products in Marine Diatoms. Methods Enzymol. 2018, 605, 69–100. [Google Scholar] [CrossRef] [PubMed]
- d’Ippolito, G.; Tucci, S.; Cutignano, A.; Romano, G.; Cimino, G.; Miralto, A.; Fontana, A. The role of complex lipids in the synthesis of bioactive aldehydes of the marine diatom Skeletonema costatum. Biochim. Biophys. Acta 2004, 1686, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; d’Ippolito, G.; Romano, G.; Lamari, N.; Cimino, G.; Febbraio, F.; Fontana, A. Chloroplastic glycolipids fuel aldehyde biosynthesis in the marine diatom Thalassiosira rotula. ChemBioChem 2006, 7, 450–456. [Google Scholar] [CrossRef]
- Fontana, A.; d’Ippolito, G.; Cutignano, A.; Romano, G.; Lamari, N.; Massa Gallucci, A.; Cimino, G.; Miralto, A.; Ianora, A. LOX-induced lipid peroxidation mechanism responsible for the detrimental effect of marine diatoms on zooplankton grazers. ChemBioChem 2007, 8, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- d’Ippolito, G.; Lamari, N.; Montresor, M.; Romano, G.; Cutignano, A.; Gerecht, A.; Cimino, G.; Fontana, A. 15S-lipoxygenase metabolism in the marine diatom Pseudo-nitzschia delicatissima. New Phytol. 2009, 183, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa, D.; d’Ippolito, G.; Gallo, C.; Zingone, A.; Fontana, A. Oxylipin diversity in the diatom family Leptocylindraceae reveals DHA derivatives in marine diatoms. Mar. Drugs 2014, 12, 368–384. [Google Scholar] [CrossRef]
- Ianora, A.; Miralto, A.; Romano, G. Antipredatory defensive role of planktonic marine natural products. In Handbook of Marine Natural Products; Fattorusso, E., Gerwick, W.H., Taglialatela-Scafati, O., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 711–748. [Google Scholar]
- Russo, E.; Ianora, A.; Carotenuto, Y. Re-shaping marine plankton communities: Effects of diatom oxylipins on copepods and beyond. Mar. Biol. 2019, 166, 9. [Google Scholar] [CrossRef]
- Russo, E.; d’Ippolito, G.; Fontana, A.; Sarno, D.; D’Alelio, D.; Busseni, G.; Ianora, A.; von Elert, E.; Carotenuto, Y. Density-dependent oxylipin production in natural diatom communities: Possible implications for plankton dynamics. ISME J. 2020, 14, 164–177. [Google Scholar] [CrossRef]
- Kâ, S.; Carotenuto, Y.; Romano, G.; Hwang, J.S.; Buttino, I.; Ianora, A. Impact of the diatom-derived polyunsaturated aldehyde 2-trans,4-trans decadienal on the feeding, survivorship and reproductive success of the calanoid copepod Temora stylifera. Mar. Environ. Res. 2014, 93, 31–37. [Google Scholar] [CrossRef]
- Carotenuto, Y.; Ianora, A.; Miralto, A. Maternal and neonate diatom diets impair development and sex differentiation in the copepod Temora stylifera. J. Exp. Mar. Biol. Ecol. 2011, 396, 99–107. [Google Scholar] [CrossRef]
- Ianora, A.; Bastianini, M.; Carotenuto, Y.; Casotti, R.; Roncalli, V.; Miralto, A.; Romano, G.; Gerecht, A.; Fontana, A.; Turner, J.T. Non-volatile oxylipins can render some diatom blooms more toxic for copepod reproduction. Harmful Algae 2015, 44, 1–7. [Google Scholar] [CrossRef]
- Asai, S.; Sanges, R.; Lauritano, C.; Lindeque, P.K.; Esposito, F.; Ianora, A.; Carotenuto, Y. De Novo Transcriptome Assembly and Gene Expression Profiling of the Copepod Calanus helgolandicus Feeding on the PUA-Producing Diatom Skeletonema marinoi. Mar. Drugs 2020, 18, 392. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Lauritano, C.; d’Ippolito, G.; Fontana, A.; Sarno, D.; von Elert, E.; Ianora, A.; Carotenuto, Y. RNA-Seq and differential gene expression analysis in Temora stylifera copepod females with contrasting non-feeding nauplii survival rates: An environmental transcriptomics study. BMC Genom. 2020, 21, 693. [Google Scholar] [CrossRef] [PubMed]
- Palomera, I.; Olivar, M.P.; Salat, J.; Sabatés, A.; Coll, M.; García, A.; Morales-Nin, B. Small pelagic fish in the NW Mediterranean sea: An ecological review. Prog. Oceanogr. 2007, 74, 377–396. [Google Scholar] [CrossRef]
- Ruocco, N.; Albarano, L.; Esposito, R.; Zupo, V.; Costantini, M.; Ianora, A. Multiple Roles of Diatom-Derived Oxylipins within Marine Environments and Their Potential Biotechnological Applications. Mar. Drugs 2020, 18, 342. [Google Scholar] [CrossRef]
- Linares-Maurizi, A.; Reversat, G.; Awad, R.; Bultel-Poncé, V.; Oger, C.; Galano, J.-M.; Balas, L.; Durbec, A.; Bertrand-Michel, J.; Durand, T.; et al. Bioactive Oxylipins Profile in Marine Microalgae. Mar. Drugs 2023, 21, 136. [Google Scholar] [CrossRef]
- Shipelin, V.A.; Sidorova, Y.S. Oxylipins—Biologically active substances of food. Vopr. Pitan. 2020, 89, 6–13. [Google Scholar] [CrossRef]
- Shearer, G.C.; Walker, R. An overview of the biologic effects of omega-6 oxylipins in humans. Prostaglandins Leukot. Essent. Fat. Acids 2018, 137, 26–38. [Google Scholar] [CrossRef]
- Miralto, A.; Barone, G.; Romano, G.; Poulet, S.A.; Ianora, A.; Russo, G.L.; Buttino, I.; Mazzarella, G.; Laabir, M.; Cabrini, M.; et al. The insidious effect of diatoms on copepod reproduction. Nature 1999, 402, 173–176. [Google Scholar] [CrossRef]
- Sansone, C.; Braca, A.; Ercolesi, E.; Romano, G.; Palumbo, A.; Casotti, R.; Francone, M.; Ianora, A. Diatom-derived polyunsaturated aldehydes activate cell death in human cancer cell lines but not normal cells. PLoS ONE 2014, 9, e101220. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, M.E.; Brash, A.R. The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 2015, 24, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.; Ivanov, I.; Myagkova, G.; Kuhn, H. Alterations of lipoxygenase specificity by targeted substrate modification and site-directed mutagenesis. Chem. Biol. 2001, 8, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Tanaka, T. Molecular Insights into Lipoxygenases in Diatoms Based on Structure Prediction: A Pioneering Study on Lipoxygenases Found in Pseudo-nitzschia arenysensis and Fragilariopsis cylindrus. Mar. Biotechnol. 2022, 24, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Neau, D.B.; Bender, G.; Boeglin, W.E.; Bartlett, S.G.; Brash, A.R.; Newcomer, M.E. Crystal structure of a lipoxygenase in complex with substrate: The arachidonic acid-binding site of 8R-lipoxygenase. J. Biol. Chem. 2014, 289, 31905–31913. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.; Boguski, M.; Gish, W.; Wootton, J.C. Issues in searching molecular sequence databases. Nat. Genet. 1994, 6, 119–129. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Giordano, D.; Facchiano, A. Classification of microbial transglutaminases by evaluation of evolution trees, sequence motifs, secondary structure topology and conservation of potential catalytic residues. Biochem. Biophys. Res. Commun. 2019, 509, 506–513. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- De Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetic Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Facchiano, A.; Minasi, P.; D’Agostino, N.; Parisi, M.; Carbone, V. Phenolic Compounds and Capsaicinoids in Three Capsicum annuum Varieties: From Analytical Characterization to In Silico Hypotheses on Biological Activity. Molecules 2023, 28, 6772. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.Y.; Sali, A. Protein Structure Modeling with MODELLER. In Structural Proteomics. Methods in Molecular Biology™; Kobe, B., Guss, M., Huber, T., Eds.; Humana Press: Totowa, NJ, USA, 2008; Volume 426. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. App. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Xu, S.; Mueser, T.C.; Marnett, L.J.; Funk, M.O., Jr. Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure 2012, 20, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Coffa, G.; Brash, A.R. A single active site residue directs oxygenation stereospecificity in lipoxygenases: Stereocontrol is linked to the position of oxygenation. Proc. Natl. Acad. Sci. USA 2004, 101, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, V.; Orefice, I.; Marotta, P.; Ambrosino, L.; Chiusano, M.L.; d’Ippolito, G.; Romano, G.; Fontana, A.; Ferrante, M.I. Silencing of a Pseudo-nitzschia arenysensis lipoxygenase transcript leads to reduced oxylipin production and impaired growth. New Phytol. 2022, 233, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Batna, A.; Scheinkönig, J.; Spiteller, G. The occurrence of furan fatty acids in Isochrysis sp. and Phaeodactylum tricornutum. Biochim. Et Biophys. Acta 1993, 1166, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, N.; Nuzzo, G.; d’Ippolito, G.; Manzo, E.; Sardo, A.; Ianora, A.; Romano, G.; Iuliano, A.; Zupo, V.; Costantini, M.; et al. Lipoxygenase Pathways in Diatoms: Occurrence and Correlation with Grazer Toxicity in Four Benthic Species. Mar. Drugs 2020, 18, 66. [Google Scholar] [CrossRef]
- Tachihana, S.; Nagao, N.; Katayama, T.; Hirahara, M.; Yusoff, F.M.; Banerjee, S.; Shariff, M.; Kurosawa, N.; Toda, T.; Furuya, K. High Productivity of Eicosapentaenoic Acid and Fucoxanthin by a Marine Diatom Chaetoceros gracilis in a Semi-Continuous Culture. Front. Bioeng. Biotechnol. 2020, 11, 602721. [Google Scholar] [CrossRef]
- Gorina, S.S.; Egorova, A.M.; Lantsova, N.V.; Toporkova, Y.Y.; Grechkin, A.N. Discovery of α-Linolenic Acid 16(S)-Lipoxygenase: Cucumber (Cucumis sativus L.) Vegetative Lipoxygenase 3. Int. J. Mol. Sci. 2023, 24, 12977. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Accession Number | Annotation | Organism | Template 1 | Template 2 | Sequence Identity |
|---|---|---|---|---|---|
| OEU18547.1 | lipoxygenase | Fragilariopsis cylindrus | SWISS-MODEL model | 3FG1 | 20.88% |
| QWC64745.1 | lipoxygenase | Pseudo-nitzschia arenysensis | SWISS-MODEL model | 1HU9 | 26.64% |
| CAD9618313.1 | unnamed protein product | Skeletonema marinoi | AlphaFold model | LOX from P. arenysensis model | 45.32% |
| XP_002180840.1 | predicted protein | Phaeodactylum tricornutum | AlphaFold model | LOX from P. arenysensis model | 32.10% |
| CAD8595700.1 | unnamed protein product | Asterionellopsis glacialis | AlphaFold model | LOX from F. cylindrus model | 26.22% |
| GJWO01009215.1 | transcribed RNA sequence | Bellerochea sp. | AlphaFold model | LOX from F. cylindrus model | 22.62% |
| CAE0460244.1 | unnamed protein product | Chaetoceros debilis | AlphaFold model | LOX from F. cylindrus model | 23.61% |
| CAD8307630.1 | unnamed protein product | Pseudictyota dubia | AlphaFold model | LOX from P. arenysensis model | 22.30% |
| Group | Organism of Origin | Substrate | Binding Energy (Kcal/mol) | N° of Poses in Cluster | Ki (µM) | Residues of Interaction (H-bonds are underlined) | LOX Type |
|---|---|---|---|---|---|---|---|
| control | Plexaura homomalla | AA | −6.95 (l.b.e.= −7.40) | 15 | 3.74 | Tyr178-Arg182-Gln380-His384-Leu385-His389-Gly427-Leu431-Ser441-Leu444-Thr623-Val624-Leu627-Fe2+-H2O | LOX8 |
| 1 | Skeletonema marinoi | EPA | −5.46 (l.b.e.= −7.04) | 11 | 6.93 | Gln266-Trp267-His270-Leu271-His275-Ser317-Leu318-Phe326-Gln330-Met529-Leu533-Ile599 | LOX9 |
| HTrA | −5.52 (l.b.e.= −5.79) | 11 | 56.56 | Ala213-Gln266-Leu271-His275-Ala314-Leu318-Phe326-Gln330-Ser526-Met529-Val530-Leu533 | LOX9 | ||
| HTrA | −4.89 (l.b.e.= −5.46) | 4 | 334.45 | Gln266-His270-Leu271-His275-Ala313-Ala314-Ser317-Leu318-Phe326-Gln330-Met529-Leu533 | LOX9 | ||
| 2 | Pseudo-nitzschia arenysensis | EPA | −7.59 (l.b.e.= −7.59) | 1 | 2.75 | Glu388-Trp389-Leu393-Leu432-Thr439-Leu440-Phe448-Gln583-Ile634-Ile637-Leu638-Ile704-Fe2+-H2O | LOX12 |
| 3 | Phaeodactylum tricornutum | 9,12-hexa-decadienoic acid | −5.62 | 1 | 75.75 | Ala97-His101-His105-Ser102-Phe352-Ala353-Phe356-Val357-Ala147-Val150-Gln151-Phe198- Trp418-Fe2+ | LOX12 |
| 4a | Fragilariopsis cylindrus | EPA | −5.63 (l.b.e.= −5.81) | 2 | 54.97 | Phe160-His164-Leu165-Ile203-Ala207-Leu211-Ala355-Asn356-Val397-Leu400-Thr401-Fe2+-H2O | LOX8 |
| −5.51 | 1 | 91.52 | Phe160-His164-Leu165-His169-Ile203-Ala207-Leu211-Ile218-Thr221-Val397-Thr401-Leu478-Fe2+ | LOX15 | |||
| 4b | Asterionellopsis glacialis | EPA | −5.25 | 1 | 141.17 | Thr459-His463-Leu464-Val503-Ser506-Ala507-Ile510-Leu511-Phe517-Ile518-Trp522-Leu663-Leu690-Thr697-Fe2+ | LOX9 |
| DHA | −4.77 (l.b.e.= −5.60) | 4 | 78.95 | His463-Leu464-Leu511-Phe517-Ile518-Trp522-Leu690-Leu693-Val694-Ser696-Thr697-Leu77-Fe2+ | LOX14 | ||
| Octacosa-octaenoic acid | −6.50 (l.b.e.= −6.93) | 3 | 17.41 | Phe-455-Thr459-His463-Leu464-Val503-Ser506-Ala507-Leu511-Phe517-Ile518-Val521-Trp522-Leu690-Leu693-Val694-Thr697-Ser766-Leu767 | LOX20 | ||
| 5a | Bellerochea sp. | DHA | −5.20 (l.b.e.= −6.29) | 6 | 25.02 | His279-Thr275-Leu280-Thr322-Ala323-Ala326-Leu327-Leu333-Phe337-Glu461-Thr471-Ser488-Leu491-Met492-Leu494-Val558-H2O | LOX11 |
| Octacosa-octaenoic acid | −5.80 (l.b.e.= −7.51) | 6 | 3.14 | Val271-Thr275-Ile319-Thr322-Ala323-Leu327-Leu333-Glu461-Thr464-Ser465-Ala469-Gly470-Trp485-Ser488-Leu491-Met492-Leu494-Val558-Fe2+-H2O | LOX11 | ||
| 5b | Chaetoceros debilis | EPA | −5.78 (l.b.e.= −5.87) | 6 | 49.54 | Thr337-His341-Leu342-His346-Ser388-Leu389-Thr395-Ile396-Ala399-Phe538-Ile559-Thr562-Thr563-Fe2+-H2O | LOX9 |
| 6 | Pseudictyota dubia | EPA | −5.64 (l.b.e.= −5.89) | 9 | 47.91 | Gln256-Met257-Phe260-His264-Asn298-Gly302-Leu306-Tyr313-Phe482-Leu483-Phe486-Phe487-H2O | LOX8 |
| DHA | −6.55 (l.b.e.= −7.35) | 6 | 4.08 | Phe82-Gln256-Phe260-His264-Asn298-Ala301-Gly302-Leu306-Tyr313-Trp317-Gly439-Ser443-Ile444-Leu483-Thr484-Phe487 | LOX4/7 | ||
| Octacosa-octaenoic acid | −6.53 (l.b.e.= −7.97) | 19 | 1.43 | Phe82-Gln256-Met257-Phe260-His264-His259-Asn298-Ala301-Gly305-Leu306-Thr311-Thr312-Tyr313-Trp317-Leu483-Phe487 | LOX16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, D.; Bonora, S.; D’Orsi, I.; D’Alelio, D.; Facchiano, A. Structural and Functional Characterization of Lipoxygenases from Diatoms by Bioinformatics and Modelling Studies. Biomolecules 2024, 14, 276. https://doi.org/10.3390/biom14030276
Giordano D, Bonora S, D’Orsi I, D’Alelio D, Facchiano A. Structural and Functional Characterization of Lipoxygenases from Diatoms by Bioinformatics and Modelling Studies. Biomolecules. 2024; 14(3):276. https://doi.org/10.3390/biom14030276
Chicago/Turabian StyleGiordano, Deborah, Simone Bonora, Ilenia D’Orsi, Domenico D’Alelio, and Angelo Facchiano. 2024. "Structural and Functional Characterization of Lipoxygenases from Diatoms by Bioinformatics and Modelling Studies" Biomolecules 14, no. 3: 276. https://doi.org/10.3390/biom14030276