
Syntheses of 25-Adamantyl-25-alkyl-2-methylidene-1α,25-dihydroxyvitamin D3 Derivatives with Structure–Function Studies of Antagonistic and Agonistic Active Vitamin D Analogs

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
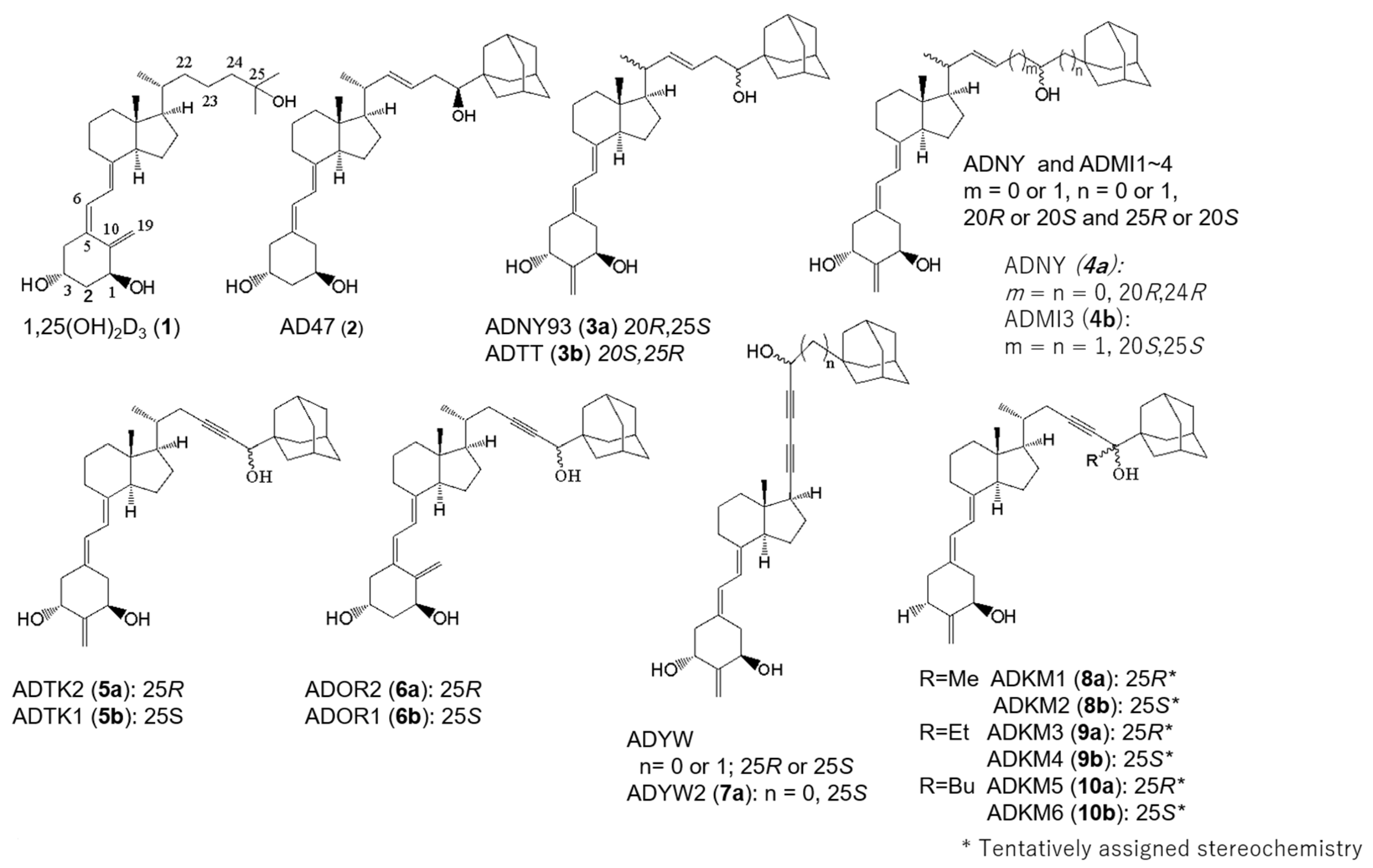
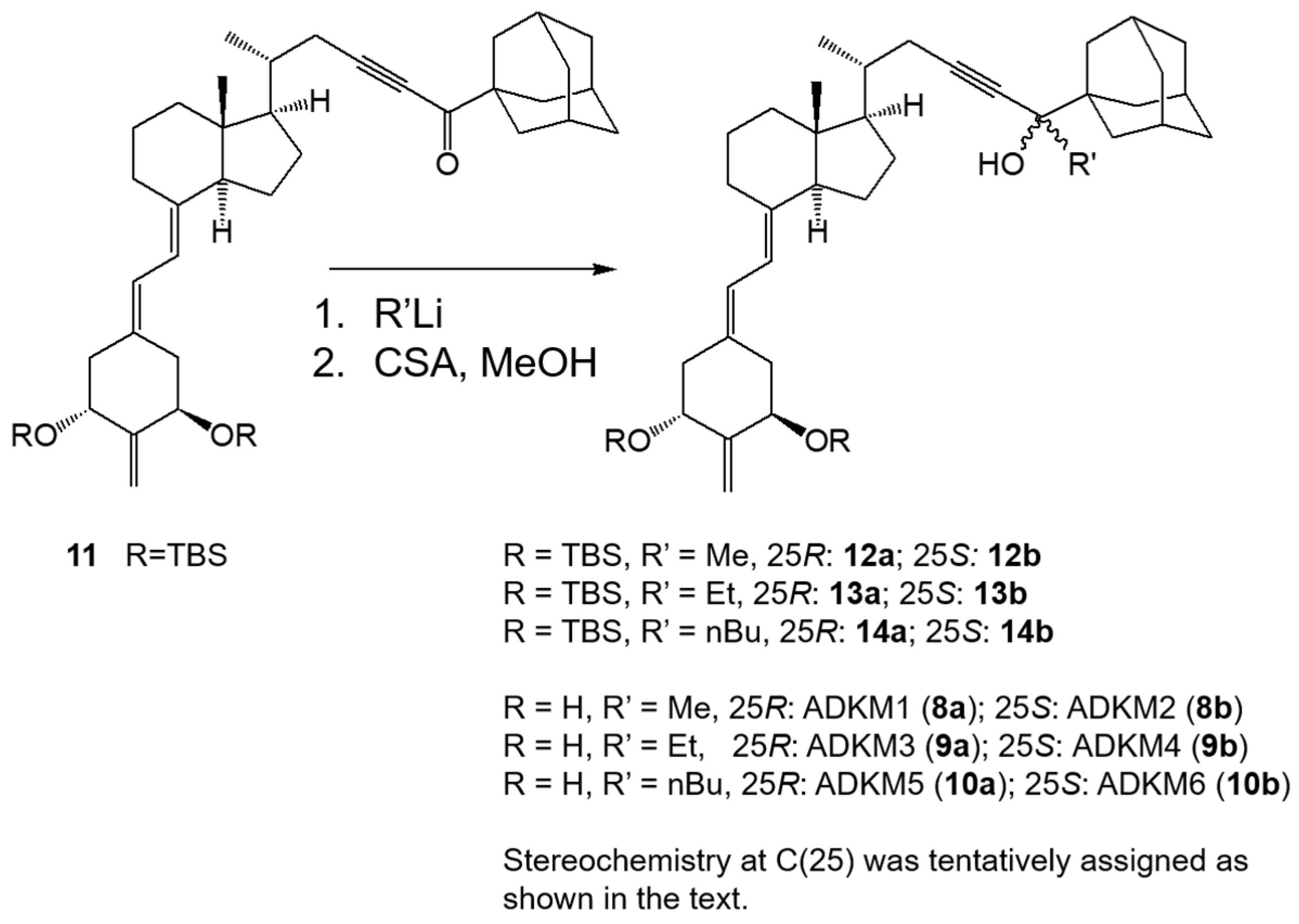
2.1. Synthesis of (25R)- and (25S)-25-adamantyl-25-alkyl-2-methylidene-23-yne-19-norvitamin D Compounds (8a, 8b, 9a, 9b, 10a and 10b)
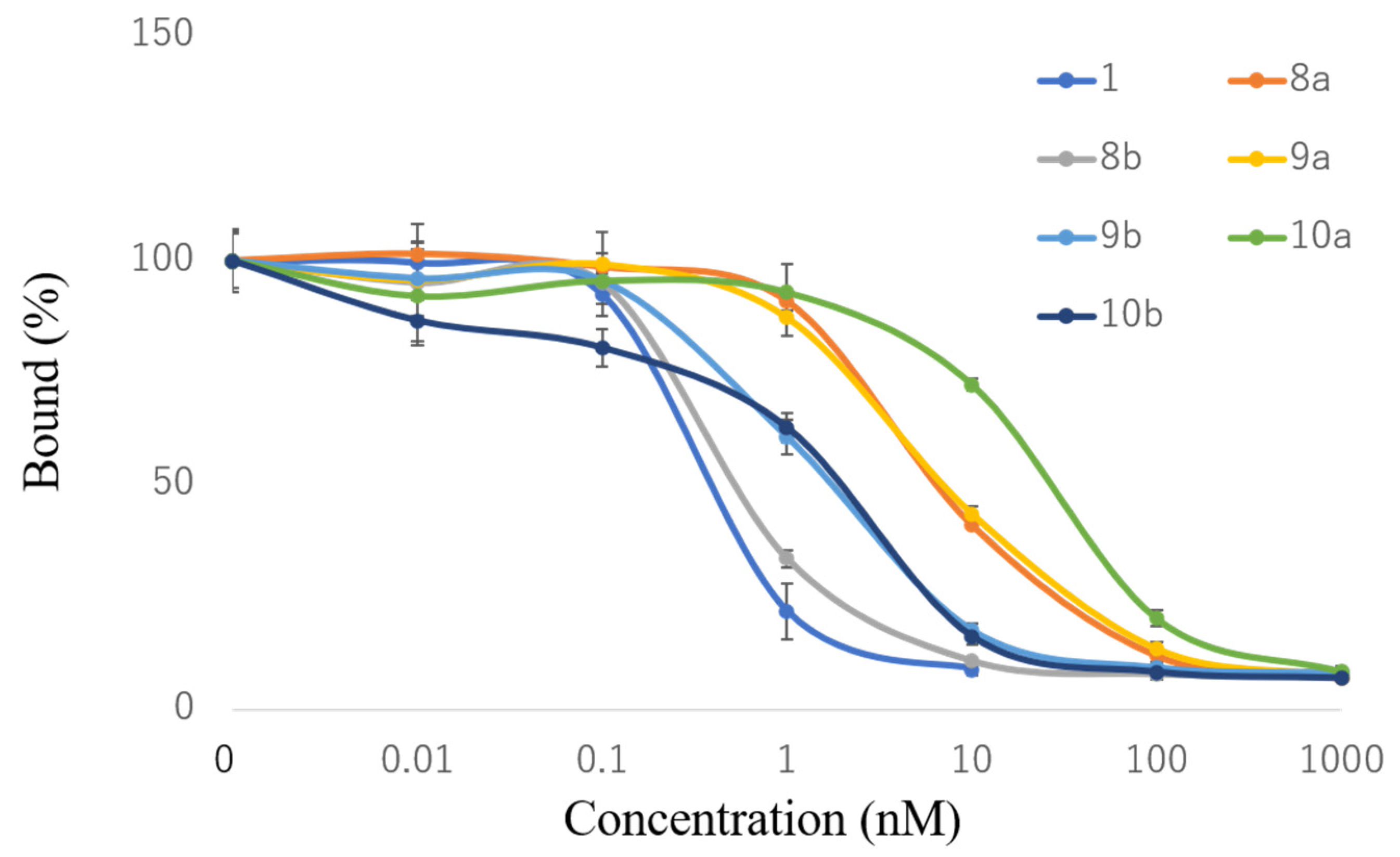
2.2. VDR-Binding Affinity
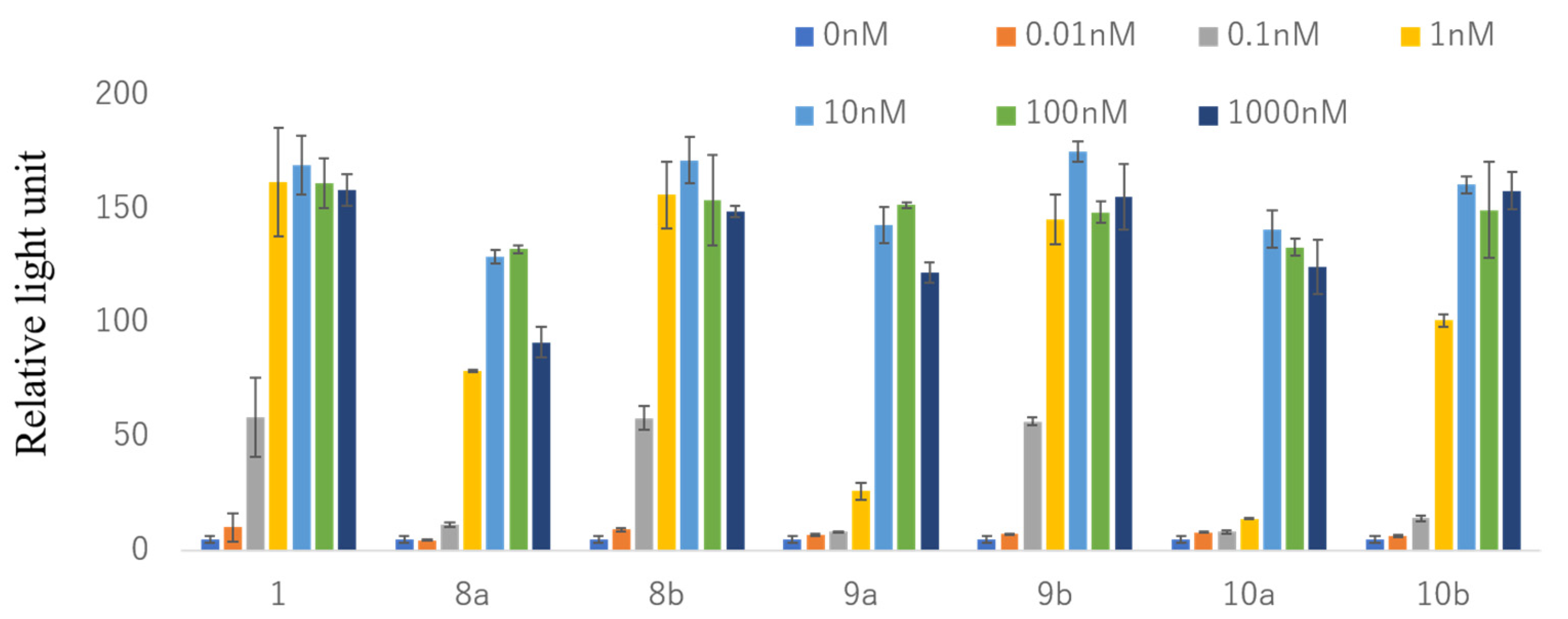
2.3. VDR Transactivation Activity
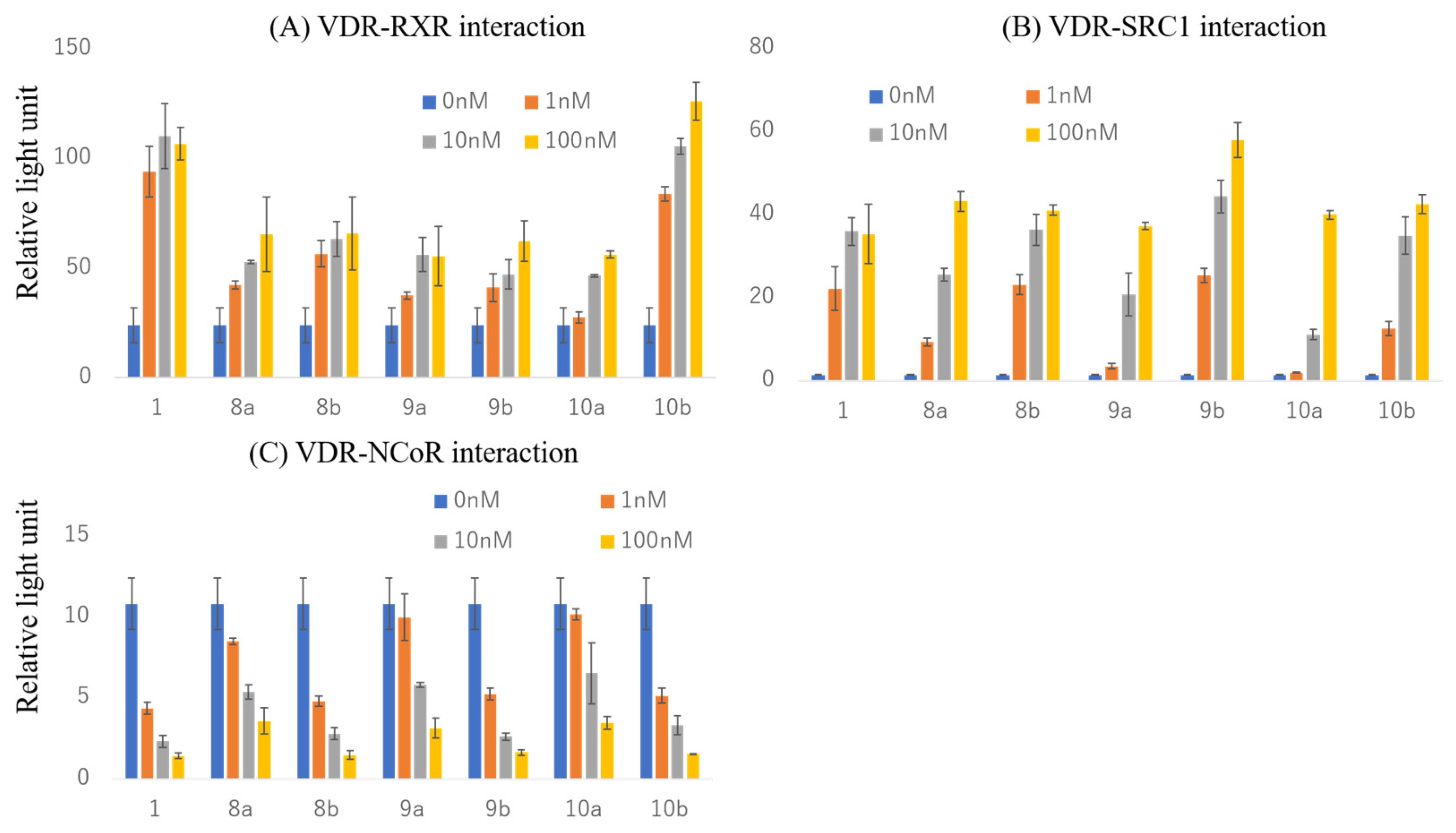
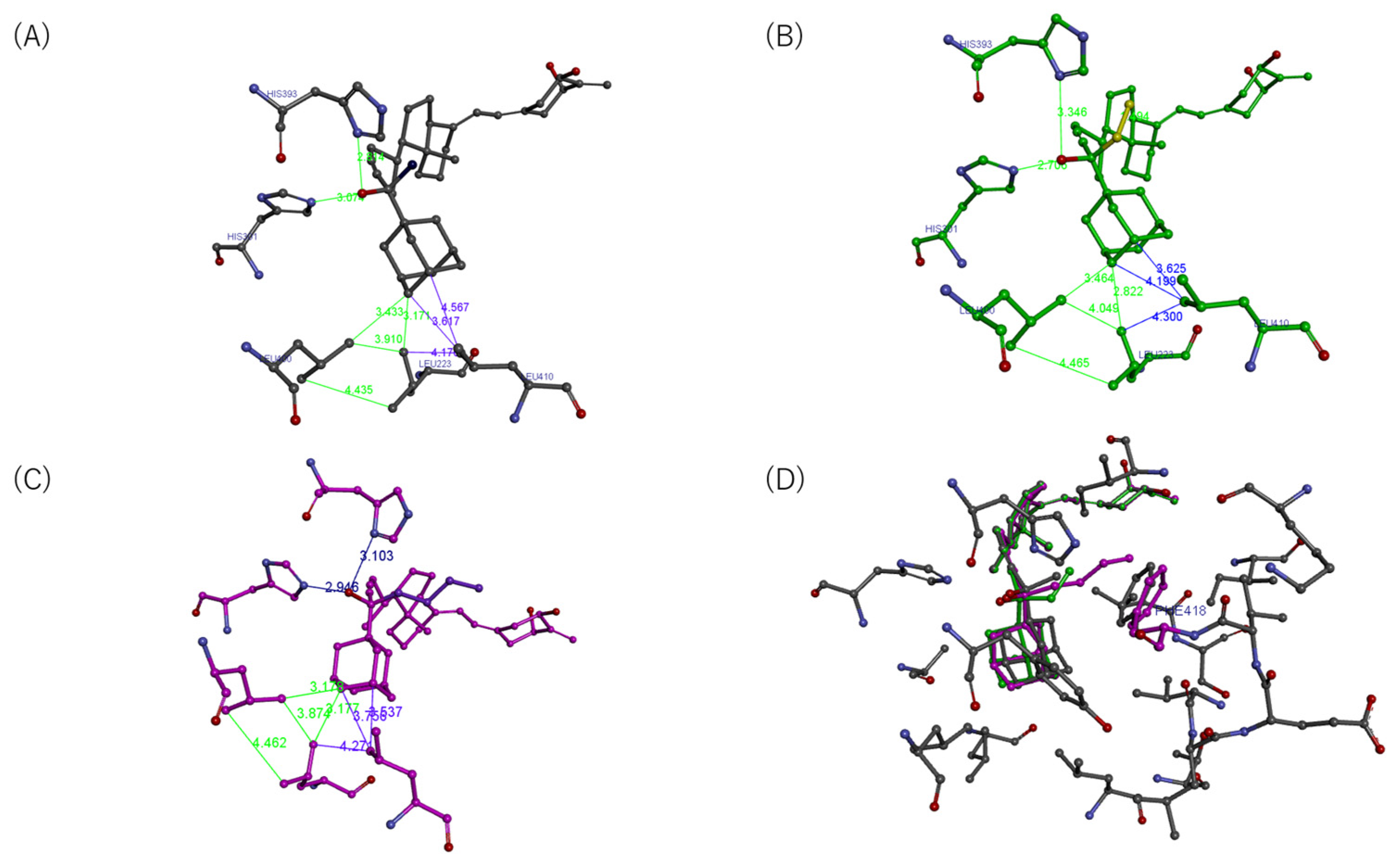
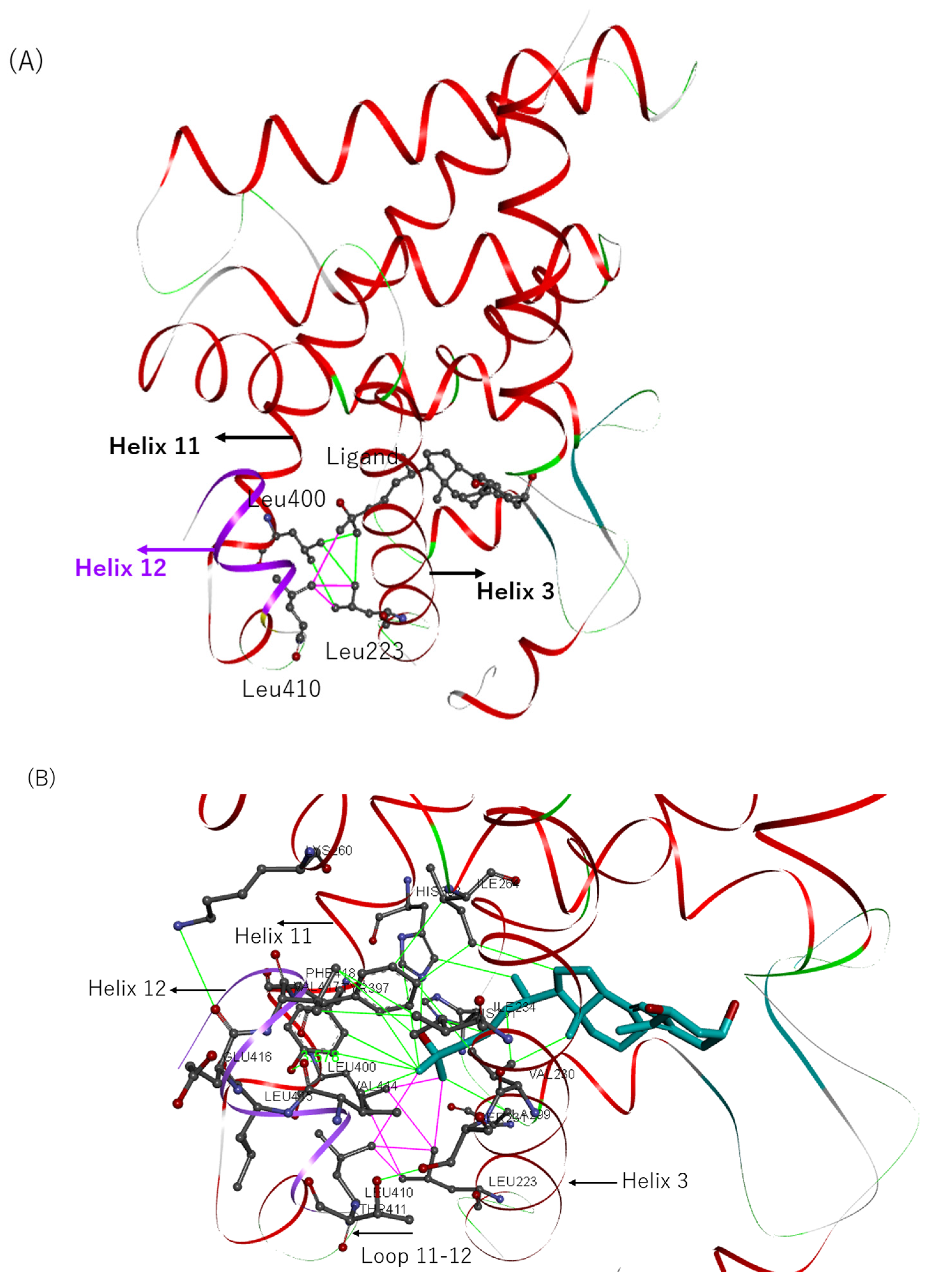
2.4. Effects on Interactions of the VDR with RXRα and Cofactors
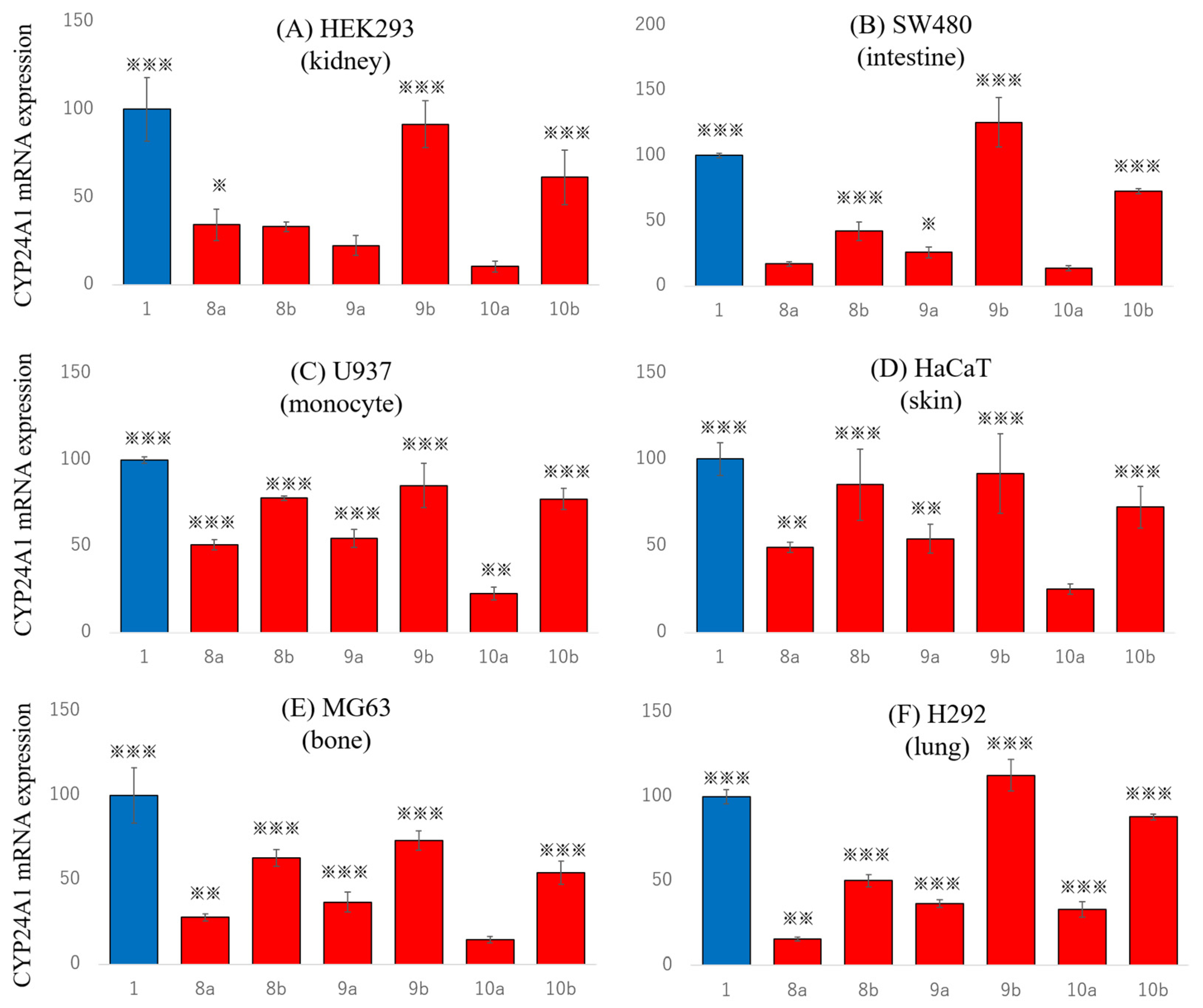
2.5. Effects on Endogenous Gene Expression in Various Tissue Cells
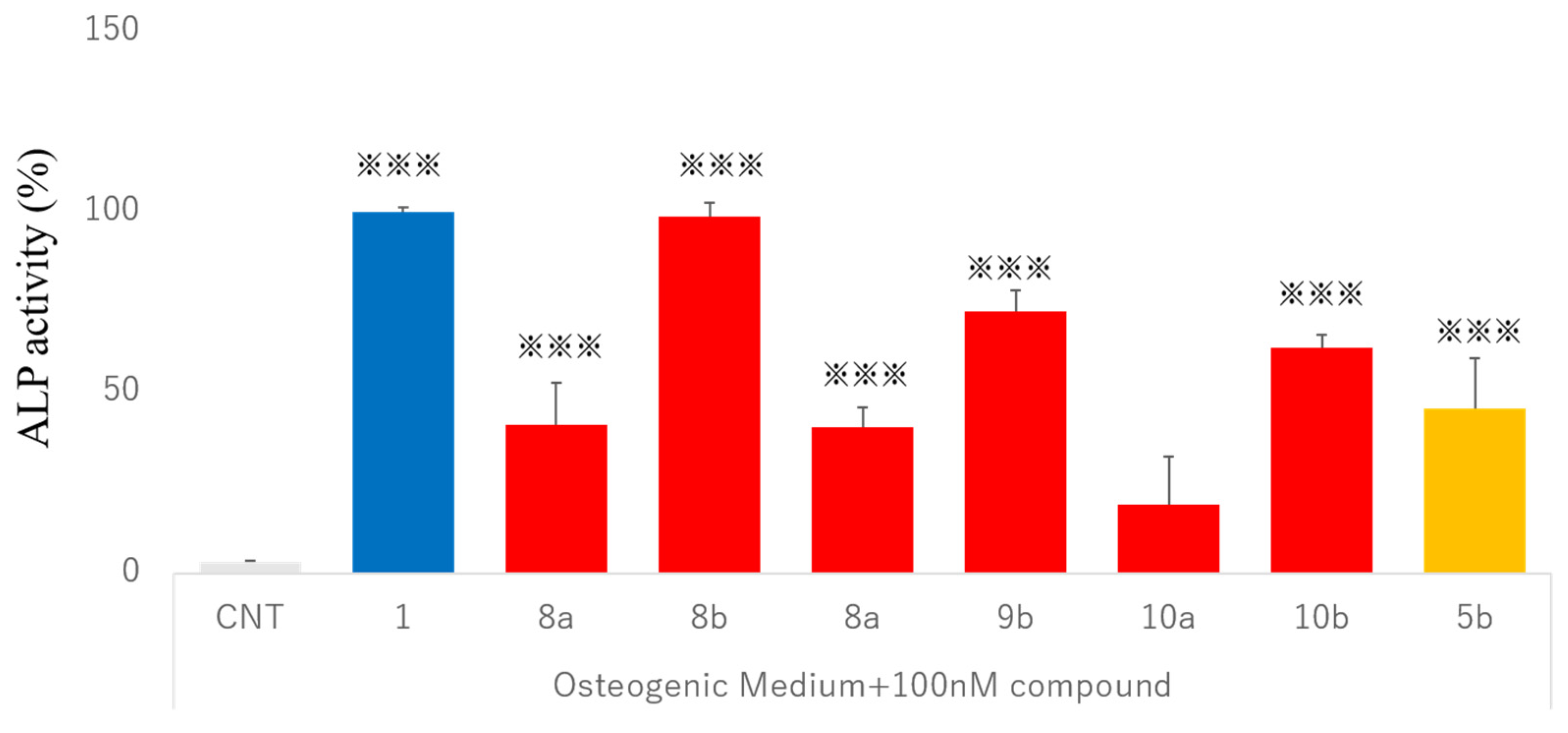
2.6. Osteogenic Differentiation Activity in Human Dedifferentiated Fat Cells
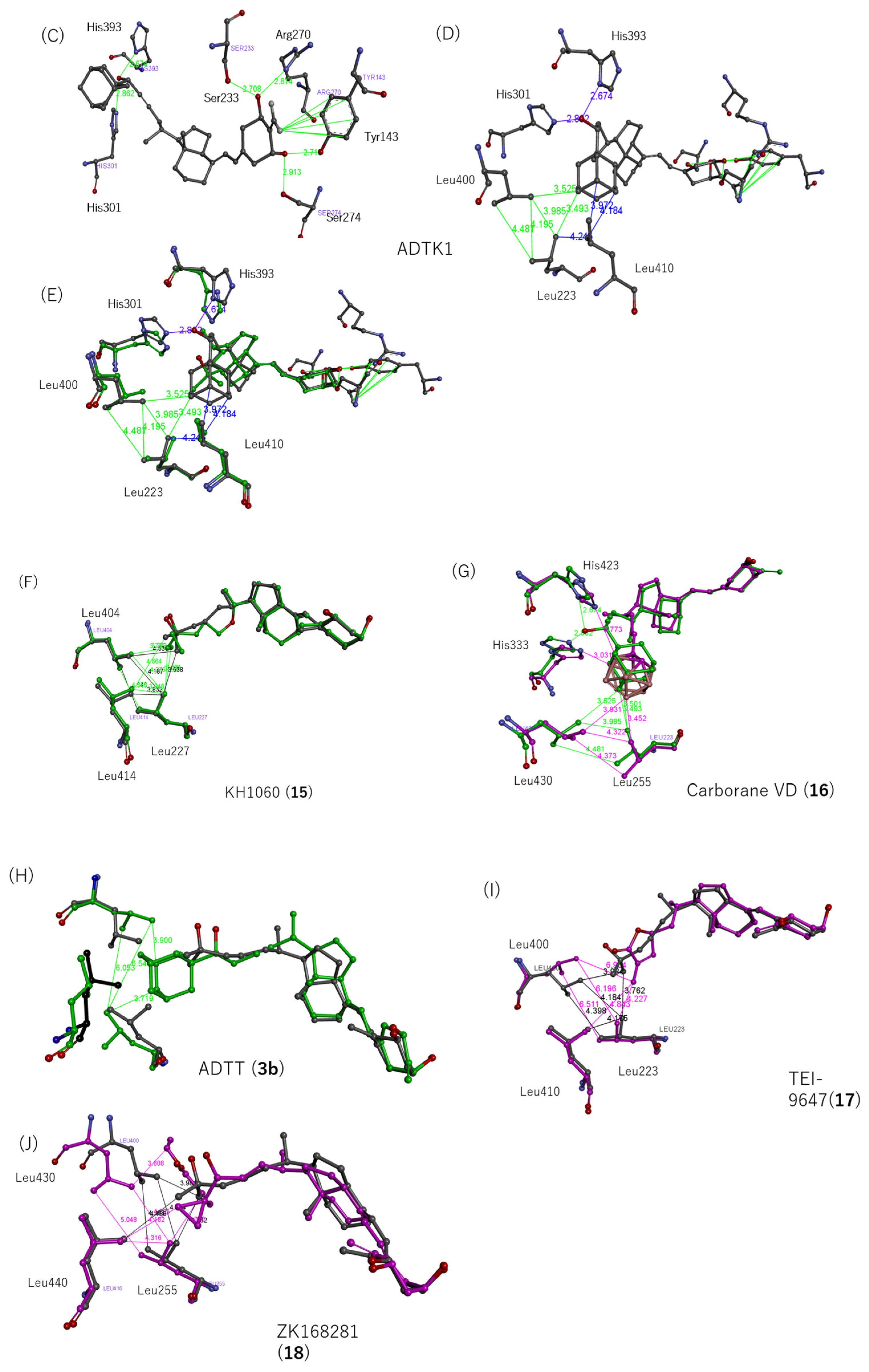
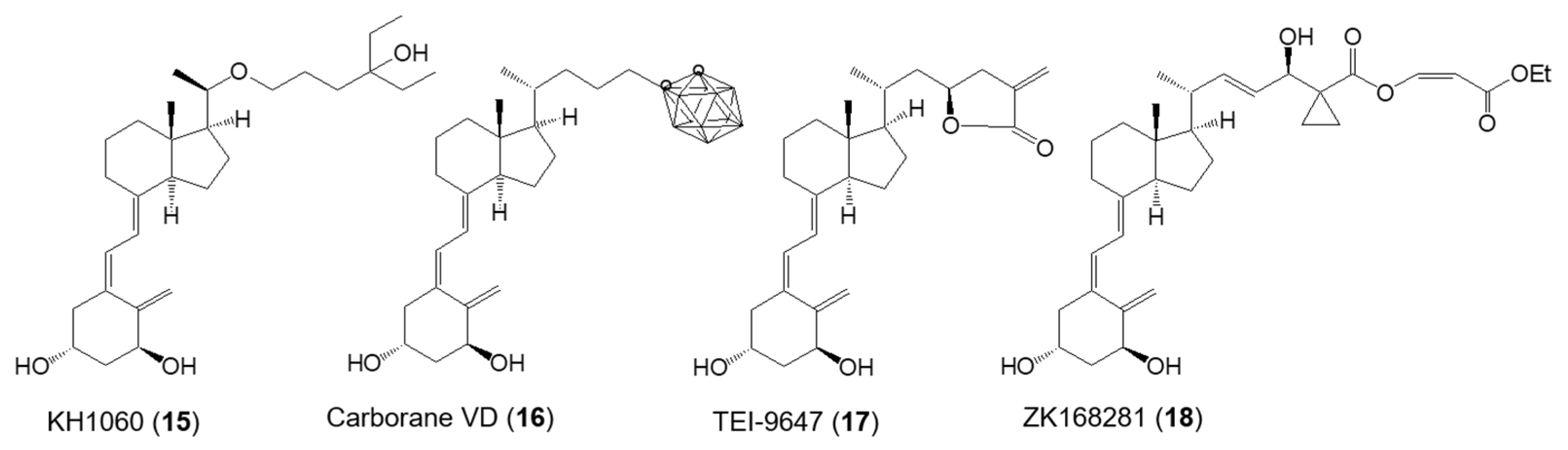
2.7. Structure–Activity Relationship of VDR Agonists and Antagonists
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.2. Plasmids
4.3. Vitamin D Receptor-Binding Assay
4.4. Cell Line Cultures
4.5. Luciferase Reporter Assays for VDR Transactivation and Mammalian Two-Hybrid Assays
4.6. Reverse Transcription and Quantitative Real-Time PCR Analysis
4.7. Human Dedifferentiated Fat Cell Isolation and Culture
4.8. Osteogenic Differentiation Assay
4.9. Statistical Analysis
4.10. Modeling and Computation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanis, J.A.; Cooper, C.; Rizzoli, R.; Reginster, J.Y. Scientific Advisory Board of the European Society for, C.; Economic Aspects of, O.; the Committees of Scientific, A.; National Societies of the International Osteoporosis, F. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos. Int. 2019, 30, 3–44. [Google Scholar] [PubMed] [Green Version]
- Williams, R.J. Calcium: Outside/inside homeostasis and signalling. Biochim. Biophys. Acta 1998, 1448, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, M. Calcium ion as a second messenger with special reference to excitation-contraction coupling. J. Pharmacol. Sci. 2006, 100, 519–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F.; Schnoes, H.K.; DeLuca, H.F.; Suda, T.; Cousins, R.J. Isolation and identification of 1,25-dihydroxycholecalciferol. A metabolite of vitamin D active in intestine. Biochemistry 1971, 10, 2799–2804. [Google Scholar]
- Jones, G.; Strugnell, S.A.; DeLuca, H.F. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar] [CrossRef] [Green Version]
- Potts, J.T. Parathyroid hormone: Past and present. J. Endocrinol. 2005, 187, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Orimo, H.; Shiraki, M.; Hayashi, Y.; Hoshino, T.; Onaya, T.; Miyazaki, S.; Kurosawa, H.; Nakamura, T.; Ogawa, N. Effects of 1 α-hydroxyvitamin D3 on lumbar bone mineral density and vertebral fractures in patients with postmenopausal osteoporosis. Calcif. Tissue Int. 1994, 54, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Endo, I. Eldecalcitol for the treatment of osteoporosis. Drugs Today 2012, 48, 189–196. [Google Scholar] [CrossRef]
- Matsumoto, T.; Yamamoto, K.; Takeuchi, T.; Tanaka, Y.; Tanaka, S.; Nakano, T.; Ito, M.; Tomomitsu, T.; Hirakawa, A.; Soen, S. Eldecalcitol is superior to alfacalcidol in maintaining bone mineral density in glucocorticoid-induced osteoporosis patients (e-GLORIA). J. Bone Miner. Metab. 2020, 38, 522–532. [Google Scholar] [CrossRef]
- Sanford, M.; McCormack, P.L. Eldecalcitol: A review of its use in the treatment of osteoporosis. Drugs 2011, 71, 1755–1770. [Google Scholar] [CrossRef]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Kumamoto, H.; Nakamura, M.; Sugawara, S.; Takano-Yamamoto, T.; Sasaki, K.; Takahashi, T. Underlying mechanisms and therapeutic strategies for bisphosphonate-related osteonecrosis of the jaw (BRONJ). Biol. Pharm. Bull. 2017, 40, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraenzlin, M.E.; Meier, C. Parathyroid hormone analogues in the treatment of osteoporosis. Nat. Rev. Endocrinol. 2011, 7, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Shikano, K.; Kawazoe, M.; Kawai, S.; Nanki, T. Efficacy of denosumab for osteoporosis in patients with rheumatic diseases. Intern. Med. 2022, 61, 2405–2415. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Boonen, S.; Vanderschueren, D.; Haentjens, P.; Lips, P. Calcium and vitamin D in the prevention and treatment of osteoporosis—A clinical update. J. Intern. Med. 2006, 259, 539–552. [Google Scholar] [CrossRef]
- Bislev, L.S.; Langagergaard Rødbro, L.; Rolighed, L.; Sikjaer, T.; Rejnmark, L. Bone microstructure in response to vitamin D3 supplementation: A randomized placebo-controlled trial. Calcif. Tissue Int. 2019, 104, 160–170. [Google Scholar] [CrossRef]
- Holick, M.F.; MacLaughlin, J.A.; Clark, M.B.; Holick, S.A.; Potts, J.T., Jr.; Anderson, R.R.; Blank, I.H.; Parrish, J.A.; Elias, P. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science 1980, 210, 203–205. [Google Scholar] [CrossRef]
- Tam, S.P.; Strugnell, S.; Deeley, R.G.; Jones, G. 25-Hydroxylation of vitamin D3 in the human hepatoma cell lines Hep G2 and Hep 3B. J. Lipid Res. 1988, 29, 1637–1642. [Google Scholar] [CrossRef]
- Takeyama, K.; Kitanaka, S.; Sato, T.; Kobori, M.; Yanagisawa, J.; Kato, S. 25-Hydroxyvitamin D3 1α-hydroxylase and vitamin D synthesis. Science 1997, 277, 1827–1830. [Google Scholar] [CrossRef]
- MacLaughlin, J.; Holick, M.F. Aging decreases the capacity of human skin to produce vitamin D3. J. Clin. Investig. 1985, 76, 1536–1538. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, J.C. Vitamin D and aging. Endocrinol. Metab. Clin. N. Am. 2013, 42, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Vitamin D deficiency accelerates ageing and age-related diseases: A novel hypothesis. J. Physiol. 2017, 595, 6825–6836. [Google Scholar] [CrossRef]
- Haussler, M.R. Vitamin D receptors: Nature and function. Annu. Rev. Nutr. 1986, 6, 527–562. [Google Scholar] [CrossRef]
- Lee, S.M.; Riley, E.M.; Meyer, M.B.; Benkusky, N.A.; Plum, L.A.; DeLuca, H.F.; Pike, J.W. 1,25-dihydroxyvitamin D3 controls a cohort of vitamin D receptor target genes in the proximal intestine that is enriched for calcium-regulating components. J. Biol. Chem. 2015, 290, 18199–18215. [Google Scholar] [CrossRef] [Green Version]
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1α,25-dihydroxyvitamin D3 regulates mitochondrial oxygen consumption and dynamics in human skeletal muscle cells. J. Biol. Chem. 2016, 291, 1514–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Schnoes, H.K.; DeLuca, H.F. Synthesis of 25-hydroxy [23,24-3H]vitamin D3. Anal. Biochem. 1978, 85, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Kream, B.E.; Jose, M.; Yamada, S.; DeLuca, H.F. A specific high-affinity binding macromolecule for 1,25-dihydroxyvitamin D3 in fetal bone. Science 1977, 197, 1086–1088. [Google Scholar] [CrossRef]
- Kream, B.E.; Yamada, S.; Schnoes, H.K.; DeLuca, H.F. Specific cytosol-binding protein for 1,25-dihydroxyvitamin D3 in rat intestine. J. Biol. Chem. 1977, 252, 4501–4505. [Google Scholar] [CrossRef]
- Yamada, S.; Ohmori, M.; Takayama, H. Synthesis of 24, 24-difluoro-1α, 25-dihydroxyvitamin D3. Chem. Pharm. Bull. 1979, 27, 3196–3198. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Ohmori, M.; Takayama, H. Synthesis of 24, 24-difluoro-25-hydroxyvitamin D3. Tetrahedron Lett. 1979, 20, 1859–1862. [Google Scholar] [CrossRef]
- Shiina, Y.; Abe, E.; Miyaura, C.; Tanaka, H.; Yamada, S.; Ohmori, M.; Nakayama, K.; Takayama, H.; Matsunaga, I.; Nishii, Y.; et al. Biological activity of 24,24-difluoro-1 α, 25-dihydroxyvitamin D3 and 1 α, 25-dihydroxyvitamin D3-26,23-lactone in inducing differentiation of human myeloid leukemia cells. Arch. Biochem. Biophys. 1983, 220, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Takahashi, T.; Uratsuka, S.; Yamada, S. Synthesis of highly fluorescent dienophiles for detecting conjugated dienes in biological fluid. J. Chem. Soc. Chem. Commun. 1990, 20, 1416–1417. [Google Scholar] [CrossRef]
- Shimizu, M.; Kamachi, S.; Nishii, Y.; Yamada, S. Synthesis of a reagent for fluorescence-labeling of vitamin D and its use in assaying vitamin D metabolites. Anal. Biochem. 1991, 194, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Gao, Y.; Aso, T.; Nakatsu, K.; Yamada, S. Fluorometric assay of 25-hydroxyvitamin D3 and 24R,25-dihydroxyvitamin D3 in plasma. Anal. Biochem. 1992, 204, 258–264. [Google Scholar] [CrossRef]
- Choi, M.; Yamamoto, K.; Itoh, T.; Makishima, M.; Mangelsdorf, D.J.; Moras, D.; DeLuca, H.F.; Yamada, S. Interaction between vitamin D receptor and vitamin D ligands: Two-dimensional alanine scanning mutational analysis. Chem. Biol. 2003, 10, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Abe, D.; Yoshimoto, N.; Choi, M.; Yamagishi, K.; Tokiwa, H.; Shimizu, M.; Makishima, M.; Yamada, S. Vitamin D receptor: Ligand recognition and allosteric network. J. Med. Chem. 2006, 49, 1313–1324. [Google Scholar] [CrossRef]
- Igarashi, M.; Yoshimoto, N.; Yamamoto, K.; Shimizu, M.; Ishizawa, M.; Makishima, M.; DeLuca, H.F.; Yamada, S. Identification of a highly potent vitamin D receptor antagonist: (25S)-26-adamantyl-25-hydroxy-2-methylene-22,23-didehydro-19,27-dinor-20-epi-vitamin D3 (ADMI3). Arch. Biochem. Biophys. 2007, 460, 240–253. [Google Scholar] [CrossRef]
- Inaba, Y.; Yamamoto, K.; Yoshimoto, N.; Matsunawa, M.; Uno, S.; Yamada, S.; Makishima, M. Vitamin D3 derivatives with adamantane or lactone ring side chains are cell type-selective vitamin D receptor modulators. Mol. Pharmacol. 2007, 71, 1298–1311. [Google Scholar] [CrossRef]
- Yoshimoto, N. Vitamin D Receptor Antagonists: Design, Synthesis, Biological Activities and Molecular Basis of Antagonism. Ph.D. Thesis, Tokyo Medical and Dental University, Tokyo, Japan, 2007. [Google Scholar]
- Nakabayashi, M.; Yamada, S.; Yoshimoto, N.; Tanaka, T.; Igarashi, M.; Ikura, T.; Ito, N.; Makishima, M.; Tokiwa, H.; DeLuca, H.F.; et al. Crystal structures of rat vitamin D receptor bound to adamantyl vitamin D analogs: Structural basis for vitamin D receptor antagonism and partial agonism. J. Med. Chem. 2008, 51, 5320–5329. [Google Scholar] [CrossRef]
- Kudo, T.; Ishizawa, M.; Maekawa, K.; Nakabayashi, M.; Watarai, Y.; Uchida, H.; Tokiwa, H.; Ikura, T.; Ito, N.; Makishima, M.; et al. Combination of triple bond and adamantane ring on the vitamin D side chain produced partial agonists for vitamin D receptor. J. Med. Chem. 2014, 57, 4073–4087. [Google Scholar] [CrossRef]
- Otero, R.; Ishizawa, M.; Numoto, N.; Ikura, T.; Ito, N.; Tokiwa, H.; Mouriño, A.; Makishima, M.; Yamada, S. 25 S-sdamantyl-23-yne-26,27-dinor-1α,25-dihydroxyvitamin D(3): Synthesis, tissue selective biological activities, and X-ray crystal structural analysis of its vitamin D receptor complex. J. Med. Chem. 2018, 61, 6658–6673. [Google Scholar] [CrossRef]
- Watarai, Y.; Ishizawa, M.; Ikura, T.; Zacconi, F.C.; Uno, S.; Ito, N.; Mouriño, A.; Tokiwa, H.; Makishima, M.; Yamada, S. Synthesis, biological activities, and X-ray crystal structural analysis of 25-hydroxy-25(or 26)-adamantyl-17-[20,23-diynyl]-21-norvitamin D compounds. J. Med. Chem. 2015, 58, 9510–9521. [Google Scholar] [CrossRef]
- Miura, D.; Manabe, K.; Ozono, K.; Saito, M.; Gao, Q.; Norman, A.W.; Ishizuka, S. Antagonistic action of novel 1α,25-dihydroxyvitamin D3-26, 23-lactone analogs on differentiation of human leukemia cells (HL-60) induced by 1α,25-dihydroxyvitamin D3. J. Biol. Chem. 1999, 274, 16392–16399. [Google Scholar] [CrossRef] [Green Version]
- Belorusova, A.Y.; Chalhoub, S.; Rovito, D.; Rochel, N. Structural analysis of VDR complex with ZK168281 antagonist. J. Med. Chem. 2020, 63, 9457–9463. [Google Scholar] [CrossRef] [PubMed]
- Väisänen, S.; Peräkylä, M.; Kärkkäinen, J.I.; Steinmeyer, A.; Carlberg, C. Critical role of helix 12 of the vitamin D(3) receptor for the partial agonism of carboxylic ester antagonists. J. Mol. Biol. 2002, 315, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Otero, R.; Seoane, S.; Sigüeiro, R.; Belorusova, A.Y.; Maestro, M.A.; Pérez-Fernández, R.; Rochel, N.; Mouriño, A. Carborane-based design of a potent vitamin D receptor agonist. Chem. Sci. 2016, 7, 1033–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Brown, M. Molecular determinants for the tissue specificity of SERMs. Science 2002, 295, 2465–2468. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kano, K.; Kondo, D.; Fukuda, N.; Iribe, Y.; Tanaka, N.; Matsubara, Y.; Sakuma, T.; Satomi, A.; Otaki, M.; et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J. Cell. Physiol. 2008, 215, 210–222. [Google Scholar] [CrossRef]
- De Kok, I.J.; Hicok, K.C.; Padilla, R.J.; Young, R.G.; Cooper, L.F. Effect of vitamin D pretreatment of human mesenchymal stem cells on ectopic bone formation. J. Oral Implantol. 2006, 32, 103–109. [Google Scholar] [CrossRef]
- Fuggle, N.R.; Cooper, C.; Oreffo, R.O.C.; Price, A.J.; Kaux, J.F.; Maheu, E.; Cutolo, M.; Honvo, G.; Conaghan, P.G.; Berenbaum, F.; et al. Alternative and complementary therapies in osteoarthritis and cartilage repair. Aging Clin. Exp. Res. 2020, 32, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, B.; Hyun, J.S.; Montoro, D.T.; Lo, D.D.; Chan, C.K.; Hu, S.; Sun, N.; Lee, M.; Grova, M.; Connolly, A.J.; et al. In vivo directed differentiation of pluripotent stem cells for skeletal regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 20379–20384. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, S.; Ishizuka, S.; Eguchi, H.; Mizwicki, M.T.; Norman, A.W.; Takimoto-Kamimura, M. Structural basis of the histidine-mediated vitamin D receptor agonistic and antagonistic mechanisms of (23S)-25-dehydro-1α-hydroxyvitamin D3-26,23-lactone. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 918–926. [Google Scholar] [CrossRef]
- Tocchini-Valentini, G.; Rochel, N.; Wurtz, J.M.; Moras, D. Crystal structures of the vitamin D nuclear receptor liganded with the vitamin D side chain analogues calcipotriol and seocalcitol, receptor agonists of clinical importance. Insights into a structural basis for the switching of calcipotriol to a receptor antagonist by further side chain modification. J. Med. Chem. 2004, 47, 1956–1961. [Google Scholar]
- Kaneko, E.; Matsuda, M.; Yamada, Y.; Tachibana, Y.; Shimomura, I.; Makishima, M. Induction of intestinal ATP-binding cassette transporters by a phytosterol-derived liver X receptor agonist. J. Biol. Chem. 2003, 278, 36091–36098. [Google Scholar] [CrossRef] [Green Version]
- Tavangar, K.; Hoffman, A.R.; Kraemer, F.B. A micromethod for the isolation of total RNA from adipose tissue. Anal. Biochem. 1990, 186, 60–63. [Google Scholar] [CrossRef]
- Kitaura, K.; Sawai, T.; Asada, T.; Nakano, T.; Uebayasi, M. Pair interaction molecular orbital method: An approximate computational method for molecular interactions. Chem. Phys. Lett. 1999, 312, 319–324. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Tanaka, S.; Fukuzawa, K. Recent Advances of the Fragment Molecular Orbital Method; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Arulmozhiraja, S.; Matsuo, N.; Ishitsubo, E.; Okazaki, S.; Shimano, H.; Tokiwa, H. Comparative binding analysis of dipeptidyl peptidase IV (DPP-4) with antidiabetic drugs—An ab initio fragment molecular orbital study. PLoS ONE 2016, 11, e0166275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group. Molecular Operating Environment (MOE), version 2013.08; Chemical Computing Group: Montreal, QC, Canada, 2016. [Google Scholar]
- Yamagishi, K.; Yamamoto, K.; Yamada, S.; Tokiwa, H. Functions of key residues in the ligand-binding pocket of vitamin D receptor: Fragment molecular orbital–interfragment interaction energy analysis. Chem. Phys. Lett. 2006, 420, 465–468. [Google Scholar] [CrossRef]
- Yamagishi, K.; Tokiwa, H.; Makishima, M.; Yamada, S. Interactions between 1α,25(OH)2D3 and residues in the ligand-binding pocket of the vitamin D receptor: A correlated fragment molecular orbital study. J. Steroid Biochem. Mol. Biol. 2010, 121, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Caricato, M.X.; Marenich, A.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, J.L.; et al. Gaussian 09, Revision A. 02.; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Feyereisen, M.; Fitzgerald, G.; Komornicki, A. Use of approximate integrals in ab initio theory. An application in MP2 energy calculations. Chem. Phys. Lett. 1993, 208, 359–363. [Google Scholar] [CrossRef]
- Bernholdt, D.E.; Harrison, R.J. Large-scale correlated electronic structure calculations: The RI-MP2 method on parallel computers. Chem. Phys. Lett. 1996, 250, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Ishikura, T.; Kuwata, K. Theoretical study of the prion protein based on the fragment molecular orbital method. J. Comput. Chem. 2009, 30, 2594–2601. [Google Scholar] [CrossRef]
- RmsdByResidue. Available online: https://pymolwiki.org/index.php/RmsdByResidue (accessed on 12 May 2023).
- DeLano, W.L. The PyMOL Molecular Graphics System; Delano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | VDR Affinity IC50 | Transcriptional Activity EC50 (% Efficacy of That of 1) | Antagonistic Activity IC50 |
|---|---|---|---|
| 1,25(OH)2D3 (1) | 1 × 10−10 M | 0.2 × 10−9 M | |
| ADTT (3b) [40,41] | 1.3 × 10−10 M | 1 × 10−9 M (15%) | 3 × 10−9 M |
| ADNY93 (3a) [40] | 1 × 10−10 M | 1 × 10−9 M (17%) | 1.2 × 10−9 M |
| AD47 (2) [39] | 5 × 10−9 M | 3 × 10−7 M (17%) | 4 × 10−7 M |
| ADMI3 (4c) [38] | 6 × 10−10 M (17%) | 3 × 10−9 M | 3 × 10−9 M |
| ADYW2 [44] (7a, n = 0, 25S) | 5 × 10−9 M | 1.5 × 10−9 M (86%) | |
| ADYW1 [44] (7b, n = 0, 25R) | 1.25 × 10−8 M | 7 × 10−9 M (41%) | |
| ADTK2 (25R) (5a) [42] | 12 × 10−9 M | 6.6 × 10−9 M (>38%) | |
| ADTK1 (25S) (5b) [42] | 0.5 × 10−9 M | 0.1 × 10−9 M (81%) | |
| ADOR2 (25R) (6a) [43] | 0.6 × 10−9 M | 5.7 × 10−9 M (>52%) | |
| ADOR1 (25S) (6b) [43] | 2.4 × 10−10 M | 0.3 × 10−10 M (87%) | |
| ADKM1 (25R) * (8a) | 6.2 × 10−9 M | 0.5 × 10−9 M (78%) | |
| ADKM2 (25S) * (8b) | 0.6 × 10−9 M | 0.2 × 10−9 M (101%) | |
| ADKM3 (25R) * (9a) | 6.7 × 10−9 M | 2.3 × 10−9 M (90%) | |
| ADKM4 (25S) * (9b) | 1.3 × 10−9 M | 0.2 × 10−9 M (104%) | |
| ADKM5 (25R) * (10a) | 23.4 × 10−9 M | 2.6 × 10−9 M (83%) | |
| ADKM6 (25S) * (10a) | 1.6 × 10−9 M | 0.6 × 10−9 M (95%) | |
| TEI-9647 (17) [45] | 10−9 M | 5 × 10−8 M | 10−7 M |
| ZK168281 (18) [46,47] | 5 × 10−8 M | ||
| 1α-OH,24-carboranyl VD (16) [48] | 2.9 × 10−9 M | 1.4 × 10−10 M |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maekawa, K.; Ishizawa, M.; Ikawa, T.; Sajiki, H.; Matsumoto, T.; Tokiwa, H.; Makishima, M.; Yamada, S. Syntheses of 25-Adamantyl-25-alkyl-2-methylidene-1α,25-dihydroxyvitamin D3 Derivatives with Structure–Function Studies of Antagonistic and Agonistic Active Vitamin D Analogs. Biomolecules 2023, 13, 1082. https://doi.org/10.3390/biom13071082
Maekawa K, Ishizawa M, Ikawa T, Sajiki H, Matsumoto T, Tokiwa H, Makishima M, Yamada S. Syntheses of 25-Adamantyl-25-alkyl-2-methylidene-1α,25-dihydroxyvitamin D3 Derivatives with Structure–Function Studies of Antagonistic and Agonistic Active Vitamin D Analogs. Biomolecules. 2023; 13(7):1082. https://doi.org/10.3390/biom13071082
Chicago/Turabian StyleMaekawa, Kazuki, Michiyasu Ishizawa, Takashi Ikawa, Hironao Sajiki, Taro Matsumoto, Hiroaki Tokiwa, Makoto Makishima, and Sachiko Yamada. 2023. "Syntheses of 25-Adamantyl-25-alkyl-2-methylidene-1α,25-dihydroxyvitamin D3 Derivatives with Structure–Function Studies of Antagonistic and Agonistic Active Vitamin D Analogs" Biomolecules 13, no. 7: 1082. https://doi.org/10.3390/biom13071082