
Adenosine-Mimicking Derivatives of 3-Aminopyrazine-2-Carboxamide: Towards Inhibitors of Prolyl-tRNA Synthetase with Antimycobacterial Activity
 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
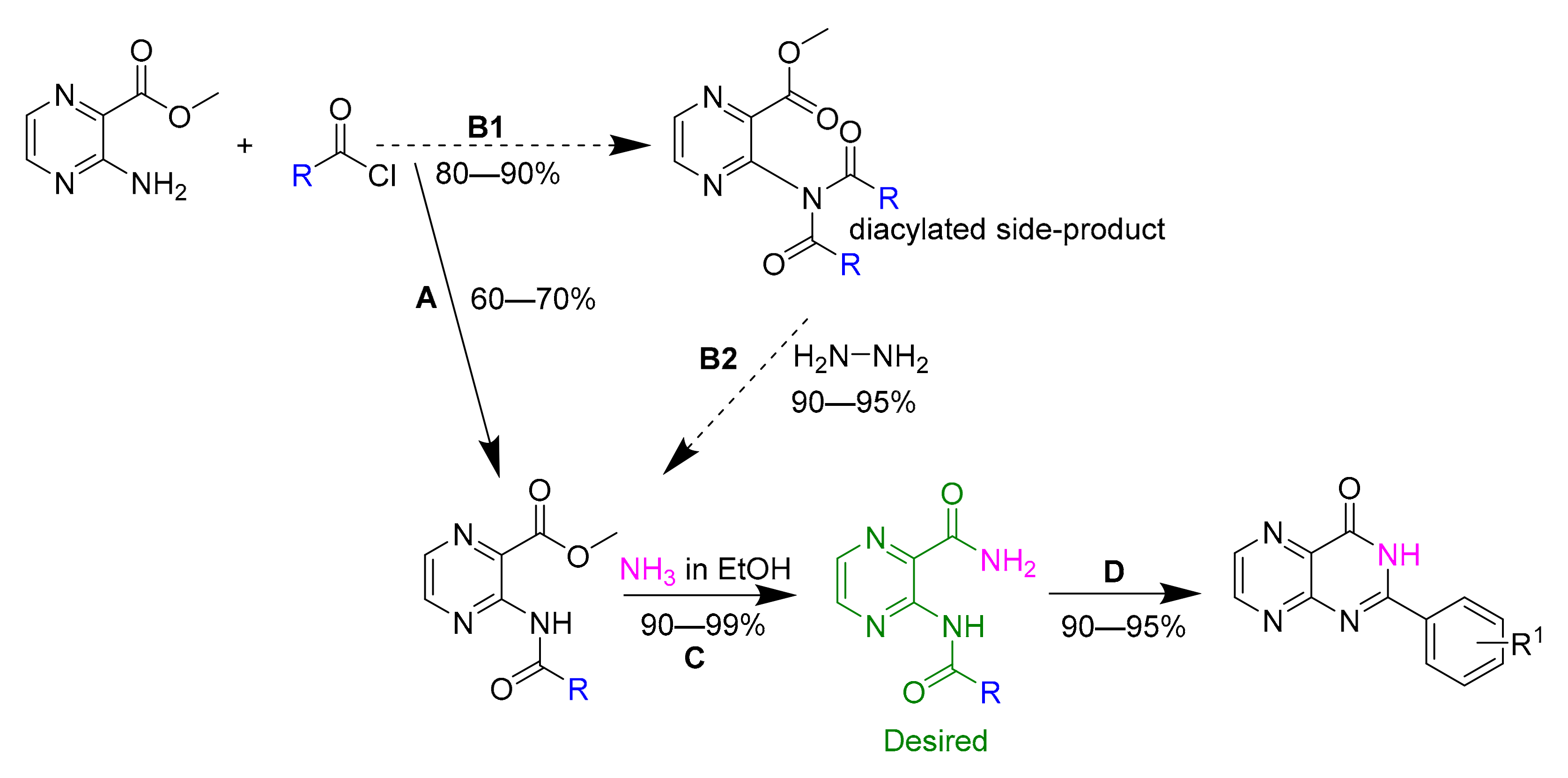
2.2. Synthesis and Purification—General Procedures
2.2.1. Acylation of Methyl 3-Aminopyrazine-2-Carboxylate
2.2.2. Ammonolysis of Methyl Ester Moiety (Compounds 1–24)
2.2.3. Synthesis of 3-(4-hydroxybenzamido)pyrazine-2-carboxamide (Compound 22) and 3-(2-hydroxybenzamido)pyrazine-2-carboxamide (Compound 23):
2.2.4. Synthesis of 3-(4-aminobenzamido)pyrazine-2-carboxamide (Compound 24):
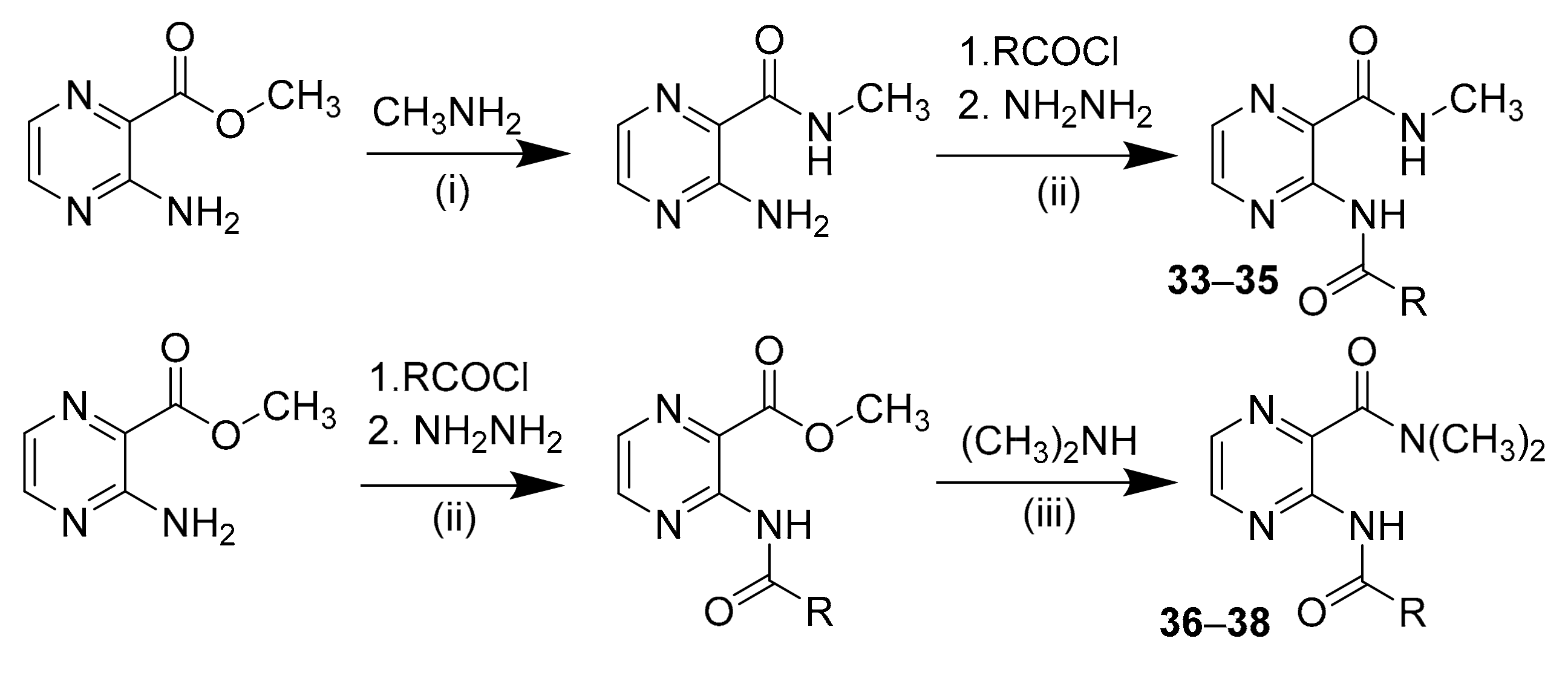
2.2.5. Synthesis of Secondary Carboxamide Derivatives (Compounds 33–35):
2.2.6. Synthesis of Tertiary Carboxamide Derivatives (Compounds 36 and 37):
2.2.7. Cyclization to Pteridine Derivatives (Compounds 39–43):
2.3. In Silico Simulations
2.3.1. Software
2.3.2. Molecular Docking
2.3.3. Molecular Dynamics
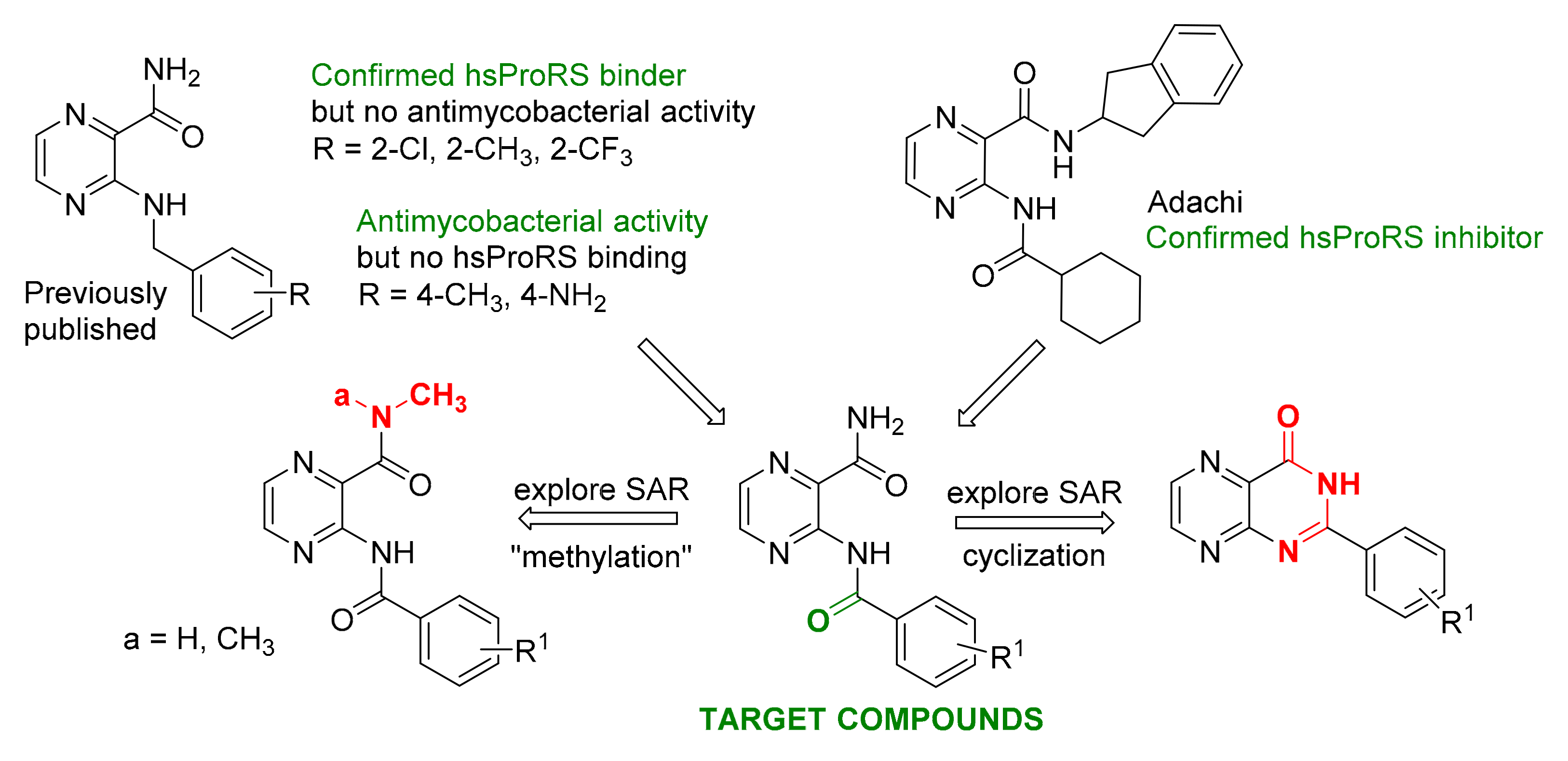
3. Results and Discussion
3.1. Chemistry
3.2. Analytical Description
3.3. Biological Evaluation
3.3.1. In Vitro Antimycobacterial Evaluation
3.3.2. In Vitro Antimycobacterial Evaluation on Drug-Sensitive and Multidrug-Resistant Strains of M. Tuberculosis
3.3.3. In Vitro Antibacterial and Antifungal Testing
3.3.4. In Vitro Cytotoxic Studies
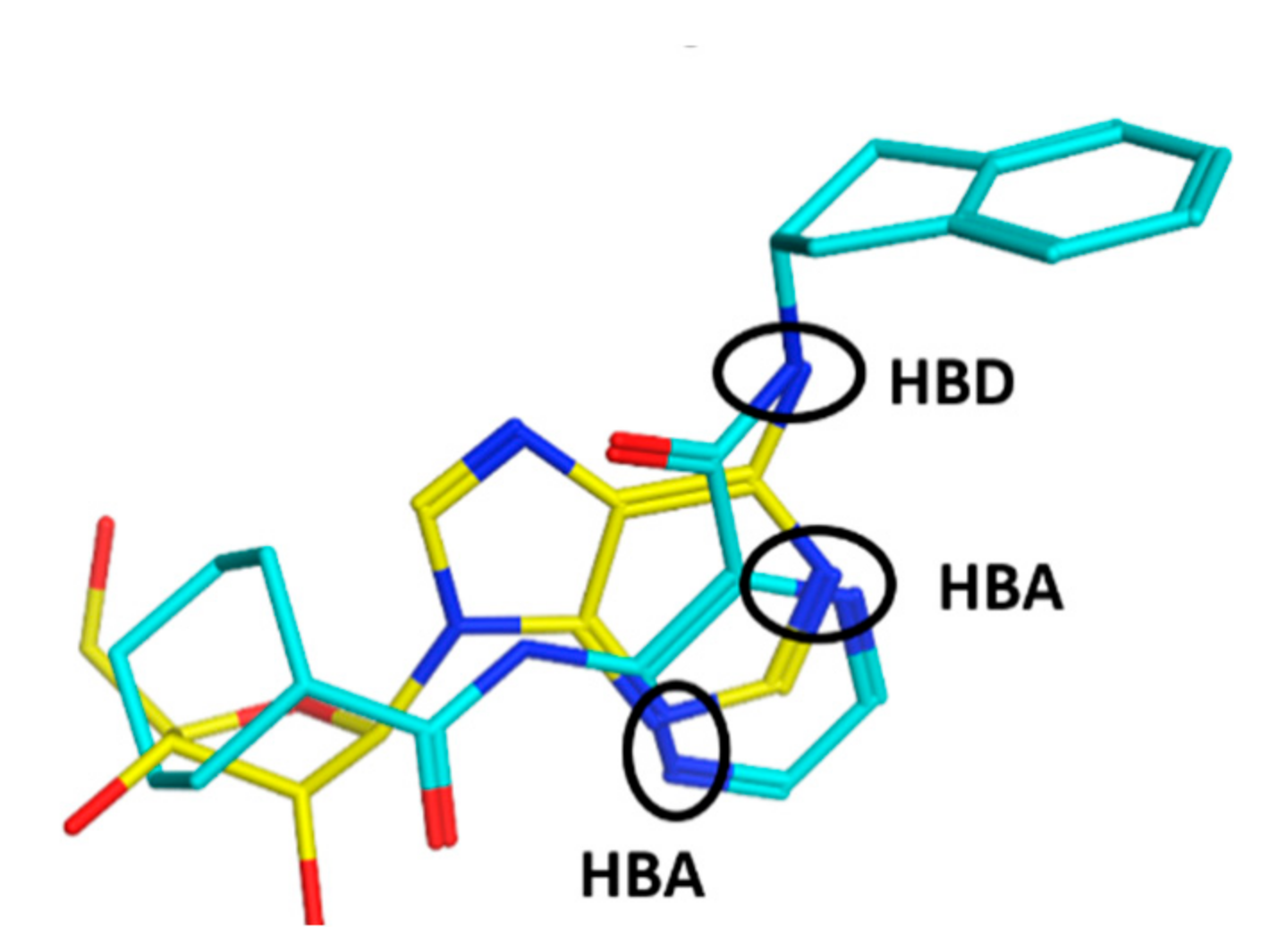
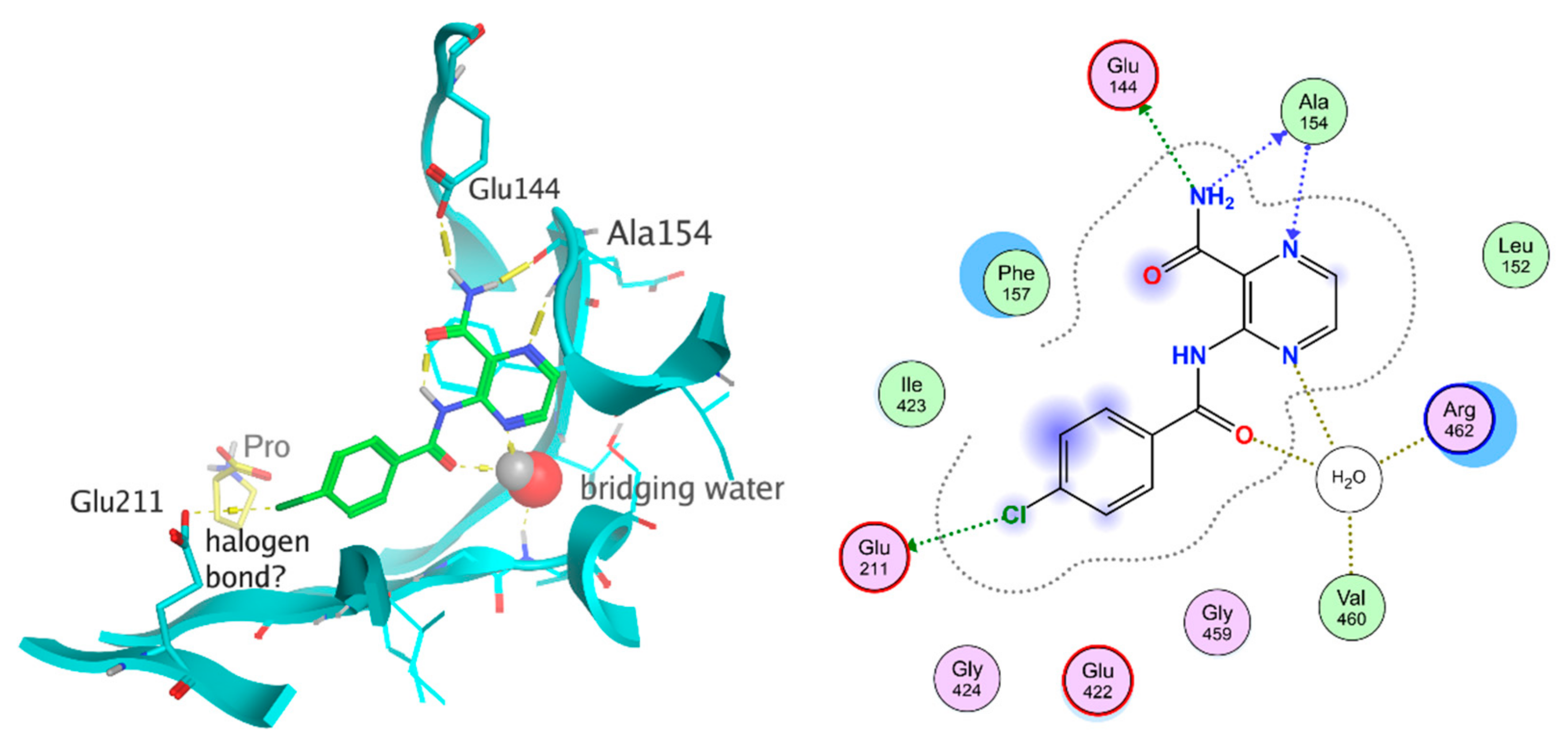
3.4. In Silico Simulations
3.4.1. Docking to Homology Model of Mycobacterial ProRS
3.4.2. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mullard, A. Tackling antimicrobial drug resistance. Nat. Rev. Drug Discov. 2016, 15, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Algammal, A.M.; Hetta, H.F.; Elkelish, A.; Alkhalifah, D.H.H.; Hozzein, W.N.; Batiha, G.E.; El Nahhas, N.; Mabrok, M.A. Methicillin-Resistant Staphylococcus aureus (MRSA): One Health Perspective Approach to the Bacterium Epidemiology, Virulence Factors, Antibiotic-Resistance, and Zoonotic Impact. Infect. Drug Resist. 2020, 13, 3255–3265. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.S.; Gigante, V.; Sati, H.; Paulin, S.; Al-Sulaiman, L.; Rex, J.H.; Fernandes, P.; Arias, C.A.; Paul, M.; Thwaites, G.E.; et al. Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed. Antimicrob. Agents Chemother. 2022, 66, e0199121. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Ho, J.M.; Bakkalbasi, E.; Söll, D.; Miller, C.A. Drugging tRNA aminoacylation. RNA Biol. 2018, 15, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, L.P.; Weeks, S.D.; Van Aerschot, A. Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Int. J. Mol. Sci. 2021, 22, 34. [Google Scholar] [CrossRef]
- Bouz, G.; Zitko, J. Inhibitors of aminoacyl-tRNA synthetases as antimycobacterial compounds: An up-to-date review. Bioorgan. Chem. 2021, 110, 104806. [Google Scholar] [CrossRef] [PubMed]
- Adachi, R.; Okada, K.; Skene, R.; Ogawa, K.; Miwa, M.; Tsuchinaga, K.; Ohkubo, S.; Henta, T.; Kawamoto, T. Discovery of a novel prolyl-tRNA synthetase inhibitor and elucidation of its binding mode to the ATP site in complex with l-proline. Biochem. Biophys. Res. Commun. 2017, 488, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Weeks, S.D.; Juhás, M.; Strelkov, S.V.; Zitko, J.; Van Aerschot, A. Towards Novel 3-Aminopyrazinamide-Based Prolyl-tRNA Synthetase Inhibitors: In Silico Modelling, Thermal Shift Assay and Structural Studies. Int. J. Mol. Sci. 2021, 22, 7793. [Google Scholar] [CrossRef] [PubMed]
- Jandourek, O.; Tauchman, M.; Paterova, P.; Konecna, K.; Navratilova, L.; Kubicek, V.; Holas, O.; Zitko, J.; Dolezal, M. Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation. Molecules 2017, 22, 223. [Google Scholar] [CrossRef] [PubMed]
- Wamhoff, H.; Kroth, E. Dihalogentriphenylphosphorane in der Heterocyclensynthese, 29. Eine einfache Synthese von Pteridin-4-onen aus 3-Amino-2-pyrazincarbonsäuremethylester und Pyrazino[3,1]oxazin-4-onen. Synthesis 1994, 1994, 405–410. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhás, M.; Pallabothula, V.S.K.; Grabrijan, K.; Šimovičová, M.; Jan’ourek, O.; Konečná, K.; Bárta, P.; Paterová, P.; Gobec, S.; Sosič, I.; et al. Design, synthesis and biological evaluation of substituted 3-amino-N-(thiazol-2-yl)pyrazine-2-carboxamides as inhibitors of mycobacterial methionine aminopeptidase 1. Bioorgan. Chem. 2022, 118, 105489. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, M.C.; Blinov, N.; Wishart, D.; Kovalenko, A.; Pande, V.S. Calculation of Local Water Densities in Biological Systems: A Comparison of Molecular Dynamics Simulations and the 3D-RISM-KH Molecular Theory of Solvation. J. Phys. Chem. B 2011, 115, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society for Clinical Microbiology and Infectious Diseases (ESCMID). EUCAST Discussion Document E. Dis 5.1: Determination of Minimum Inhibitory Concentrations (MICs) of Antibacterial Agents by Broth Dilution. Clin. Microbiol. Infec. 2003, 9, 1–7. Available online: http://www.eucast.org/documents/publications_in_journals/ (accessed on 11 December 2019).
- EUCAST DEFINITIVE DOCUMENT E.DEF 7.3.1. Method for the Determination of Broth Dilution Minimum Inhibitory Concentrations of Antifungal Agents for Yeasts. 2017. Available online: http://www.eucast.org/astoffungi/methodsinantifungalsusceptibilitytesting/susceptibility_testing_of_yeasts/ (accessed on 11 December 2019).
- EUCAST DEFINITIVE DOCUMENT E.DEF 9.3.1. Method for the Determination of Broth Dilution Minimum Inhibitory Concentrations of Antifungal Agents for Conidia Forming Moulds. 2017. Available online: http://www.eucast.org/astoffungi/methodsinantifungalsusceptibilitytesting/susceptibility_testing_of_moulds/ (accessed on 11 December 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Code | R1 | Mtb H37Ra MIC [µg/mL] | M. smegmatis MIC [µg/mL] | M. aurum MIC [µg/mL] | HepG2 IC50 [µM] | Log P |
| 1 | H | 250 | ≥500 | ≥500 | >250 * | 0.955 |
| 2 | 2-Me | ≥500 | ≥500 | ≥500 | 422 | 1.114 |
| 3 | 3-Me | 62.5 | ≥250 | ≥250 | >100 * | 1.454 |
| 4 | 4-Me | 7.81 | ≥250 | ≥250 | >100 * | 1.454 |
| 5 | 4-Et | ≥500 | ≥500 | ≥500 | >100 * | 1.983 |
| 6 | 4-tBu | 15.625 | 15.625 | ≥500 | 113.7 | 2.781 |
| 7 | 2-OMe | 125 | 62.5 | ≥500 | >500 * | 0.969 |
| 8 | 3-OMe | ≥250 | ≥250 | ≥250 | >100 * | 1.039 |
| 9 | 4-OMe | ≥125 | ≥125 | ≥125 | >250 * | 1.039 |
| 10 | 2-F | 250 | ≥500 | ≥500 | 556.1 | 0.697 |
| 11 | 3-F | ≥125 | ≥125 | ≥125 | >50 * | 1.107 |
| 12 | 4-F | 31.25 | 250 | ≥500 | >250 * | 1.107 |
| 13 | 2-Cl | ≥500 | ≥500 | ≥500 | >250 * | 0.847 |
| 14 | 3-Cl | ≥125 | ≥125 | ≥125 | >25 * | 1.677 |
| 15 | 4-Cl | 3.91 | 3.91 | ≥250 | >100 * | 1.677 |
| 16 | 2-Br | ≥250 | ≥250 | ≥250 | >500 * | 0.907 |
| 17 | 3-Br | ≥125 | ≥125 | ≥125 | >5 * | 1.827 |
| 18 | 4-Br | 1.95 | 125 | ≥250 | >25 * | 1.827 |
| 19 | 3,5-diCl | ≥62.5 | ≥62.5 | ≥62.5 | >250 * | 2.393 |
| 20 | 2-CF3 | 500 | ≥500 | ≥500 | >250 * | 0.648 |
| 21 | 4-NO2 | 250 | 250 | ≥500 | >100 * | 0.727 |
| 22 | 2-OH | ≥500 | ≥ 500 | ≥500 | >250 * | 1.577 |
| 23 | 4-OH | ≥500 | ≥ 500 | ≥500 | >500 * | 0.627 |
| 24 | 4-NH2 | 125 | ≥ 250 | ≥250 | 929.0 | 0.047 |
| CIP | - | 0.25 | 0.063 | 0.016 | >500 * | −0.725 |
| RIF | - | 0.003 | 12.5 | 0.39 | >500 * | −0.640 |
| INH | - | 0.25 | 15.625 | 3.91 | >1000 ** | −0.668 |
 | ||||||
|---|---|---|---|---|---|---|
| Code | n | Mtb H37Ra MIC [µg/mL] | M. smegmatis MIC [µg/mL] | M. aurum MIC [µg/mL] | HepG2 IC50 [µM] | Log P |
| 25 | - | ≥250 | ≥250 | ≥250 | >100 * | 1.146 |
| 26 | - | 125 | ≥500 | ≥500 | >500 * | 1.562 |
| 27 | - | 125 | 250 | ≥500 | >1000 ** | 1.497 |
| 28 | - | 31.25 | 125 | 125 | 366.6 | 2.125 |
| 29 | 2 | ≥500 | ≥500 | ≥500 | >1000 ** | 0.524 |
| 30 | 4 | ≥500 | ≥500 | ≥250 | 560.3 | 1.582 |
| 31 | 5 | ≥500 | ≥500 | ≥500 | >250 * | 2.111 |
| 32 | 6 | 250 | 250 | 125 | >1000 ** | 2.640 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Code | R1 | a1 | a2 | Mtb H37Ra MIC [µg/mL] | M. smegmatis MIC [µg/mL] | M. aurum MIC [µg/mL] | HepG2 IC50 [µM] | Log P |
| 33 | 4-Me | Me | H | 250 | ≥500 | 125 | 764.1 | 1.806 |
| 34 | 4-Cl | Me | H | ≥500 | ≥500 | ≥500 | >100 * | 2.028 |
| 35 | 4-Br | Me | H | ≥500 | ≥500 | ≥500 | >50 * | 2.178 |
| 36 | 4-Me | Me | Me | ≥500 | ≥500 | ≥500 | >1000 ** | 0.684 |
| 37 | 4-Cl | Me | Me | ≥500 | ≥500 | ≥500 | >1000 ** | 0.907 |
| 38 | - | - | - | 62.5 | ≥500 | ≥500 | 19.6 | 3.380 |
 | ||||||
|---|---|---|---|---|---|---|
| Code | R1 | Mtb H37Ra MIC [µg/mL] | M. smegmatis MIC [µg/mL] | M. aurum MIC [µg/mL] | HepG2 IC50 [µM] | Log P |
| 39 | H | 62.5 | ≥500 | 125 | >1000 ** | 0.483 |
| 40 | 4-Me | 31.25 | ≥250 | 250 | >1000 ** | 0.982 |
| 41 | 4-F | ≥500 | ≥500 | ≥500 | >1000 ** | 0.628 |
| 42 | 4-Cl | ≥500 | ≥500 | ≥500 | >250 * | 1.198 |
| 43 | 4-Br | 31.25 | ≥500 | ≥500 | >250 * | 1.347 |
| Code | R1 | Mtb H37Ra MIC [µg/mL] | Mtb H37Rv MIC [µg/mL] | Mtb IZAK MIC [µg/mL] | Mtb MATI MIC [µg/mL] |
|---|---|---|---|---|---|
| 4 | 4-Me | 7.81 | 6.25 | 12.5 | 12.5 |
| 12 | 4-F | 31.25 | 50 | 100 | 100 |
| 15 | 4-Cl | 3.91 | 6.25 | 12.5 | 12.5 |
| 18 | 4-Br | 1.95 | 6.25 | 12.5 | 12.5 |
| INH | - | 0.25–0.5 | 0.2 | 4 R | >8 R |
| RFM | - | 0.002 | n.d. | >8 * R | >8 * R |
| EMB | - | 0.5 | 0.39 | 0.39/0.5 * S | 0.39/0.5 * S |
| PZA | - | n.d. | n.d. | >16 * R | >128 * R |
| STM | - | 0.5 | n.d. | 4 * R | >16 * R |
| CIP | - | 0.25 | 0.2 | 0.2 S | 0.2 S |
| Compound 15 | Compound 34 | ||||||
|---|---|---|---|---|---|---|---|
| acceptor | repl_1 | repl_2 | repl_3 | repl_1 | repl_2 | repl_3 | |
| CONH | Ala154-bb | 77.36% | 77.48% | 74.66% | 16.32% | 23.72% | 35.30% |
| Ala154-bb | N-1 | 76.38% | 72.44% | 74.22% | 24.88% | 30.38% | 56.58% |
| HOH586 * | N-4 or 3-NHCO | 63.82% | 56.06% | 63.32% | 67.04% | 70.20% | 61.94% |
| CONH | Glu144-Side | 44.12% | 10.12% | 45.66% | n.d. | n.d. | n.d. |
| Arg462-sc | N-4 | 11.68% | 2.56% | 14.38% | <1% | <1% | <1% |
| Arg142-sc | CONH | 10.66% | 0.02% | 1.02% | <1% | <1% | <1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallabothula, V.S.K.; Kerda, M.; Juhás, M.; Janďourek, O.; Konečná, K.; Bárta, P.; Paterová, P.; Zitko, J. Adenosine-Mimicking Derivatives of 3-Aminopyrazine-2-Carboxamide: Towards Inhibitors of Prolyl-tRNA Synthetase with Antimycobacterial Activity. Biomolecules 2022, 12, 1561. https://doi.org/10.3390/biom12111561
Pallabothula VSK, Kerda M, Juhás M, Janďourek O, Konečná K, Bárta P, Paterová P, Zitko J. Adenosine-Mimicking Derivatives of 3-Aminopyrazine-2-Carboxamide: Towards Inhibitors of Prolyl-tRNA Synthetase with Antimycobacterial Activity. Biomolecules. 2022; 12(11):1561. https://doi.org/10.3390/biom12111561
Chicago/Turabian StylePallabothula, Vinod Sukanth Kumar, Marek Kerda, Martin Juhás, Ondřej Janďourek, Klára Konečná, Pavel Bárta, Pavla Paterová, and Jan Zitko. 2022. "Adenosine-Mimicking Derivatives of 3-Aminopyrazine-2-Carboxamide: Towards Inhibitors of Prolyl-tRNA Synthetase with Antimycobacterial Activity" Biomolecules 12, no. 11: 1561. https://doi.org/10.3390/biom12111561