
Type I Interferon Receptor Subunit 1 Deletion Attenuates Experimental Abdominal Aortic Aneurysm Formation

 , , , , , and
, , , , , and 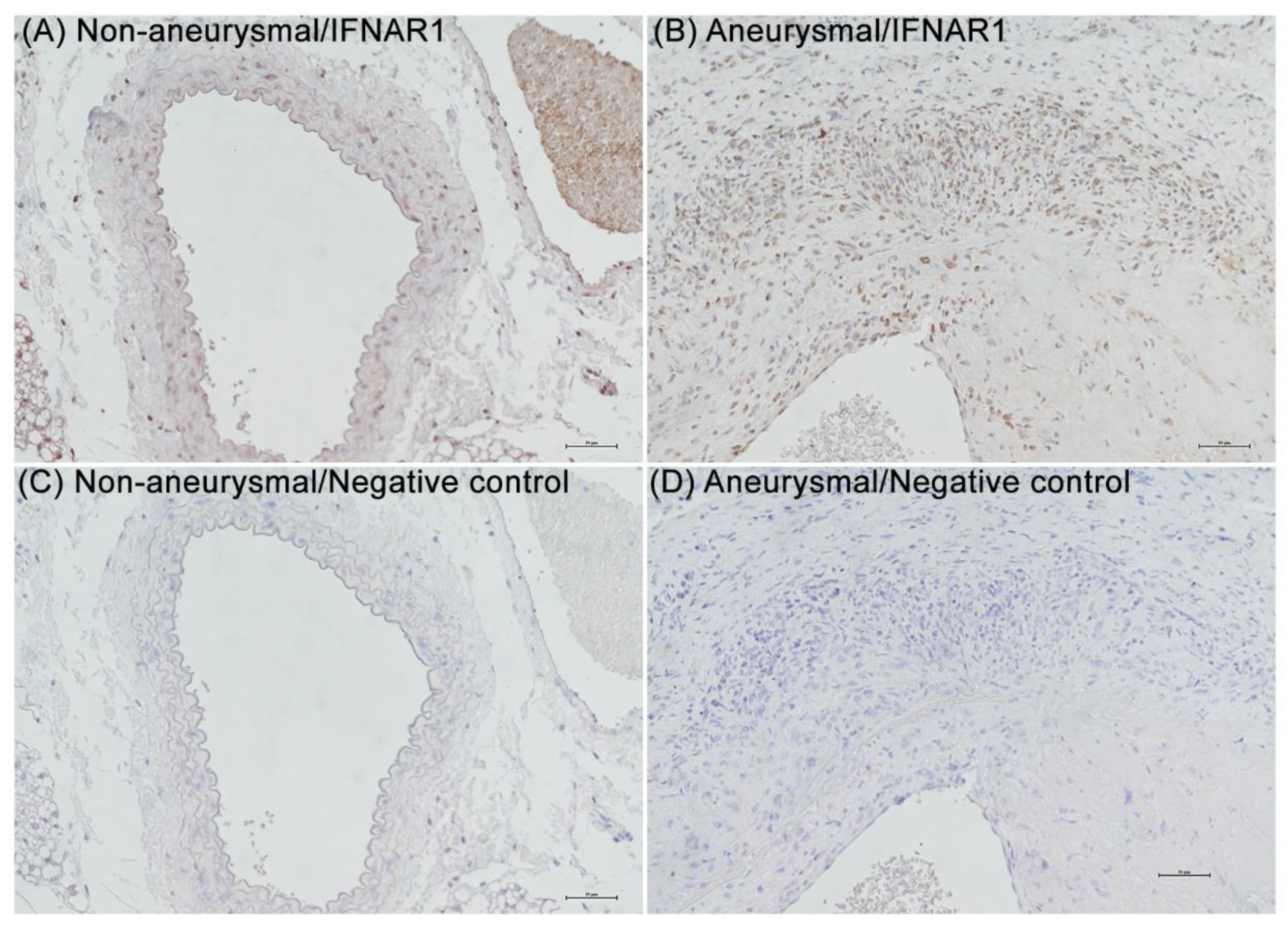{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental AAA Modeling
2.2. Immunohistochemistry for IFNAR1
2.3. In Vivo Assessment of AAA Formation and Progression
2.4. Histological Analyses
2.5. Data Analysis
3. Results
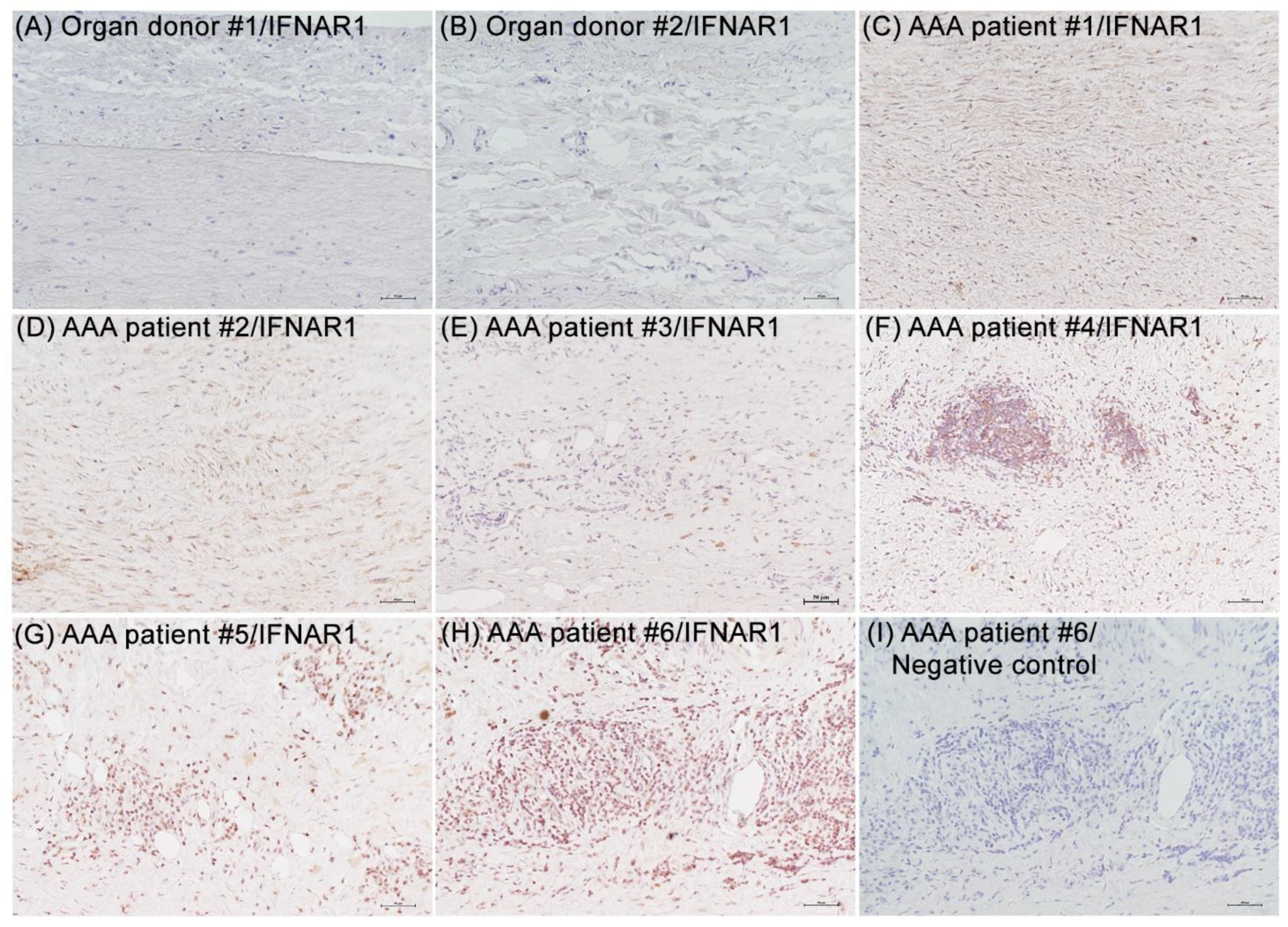
3.1. IFNAR1 Expression in Experimental and Clinical AAAs
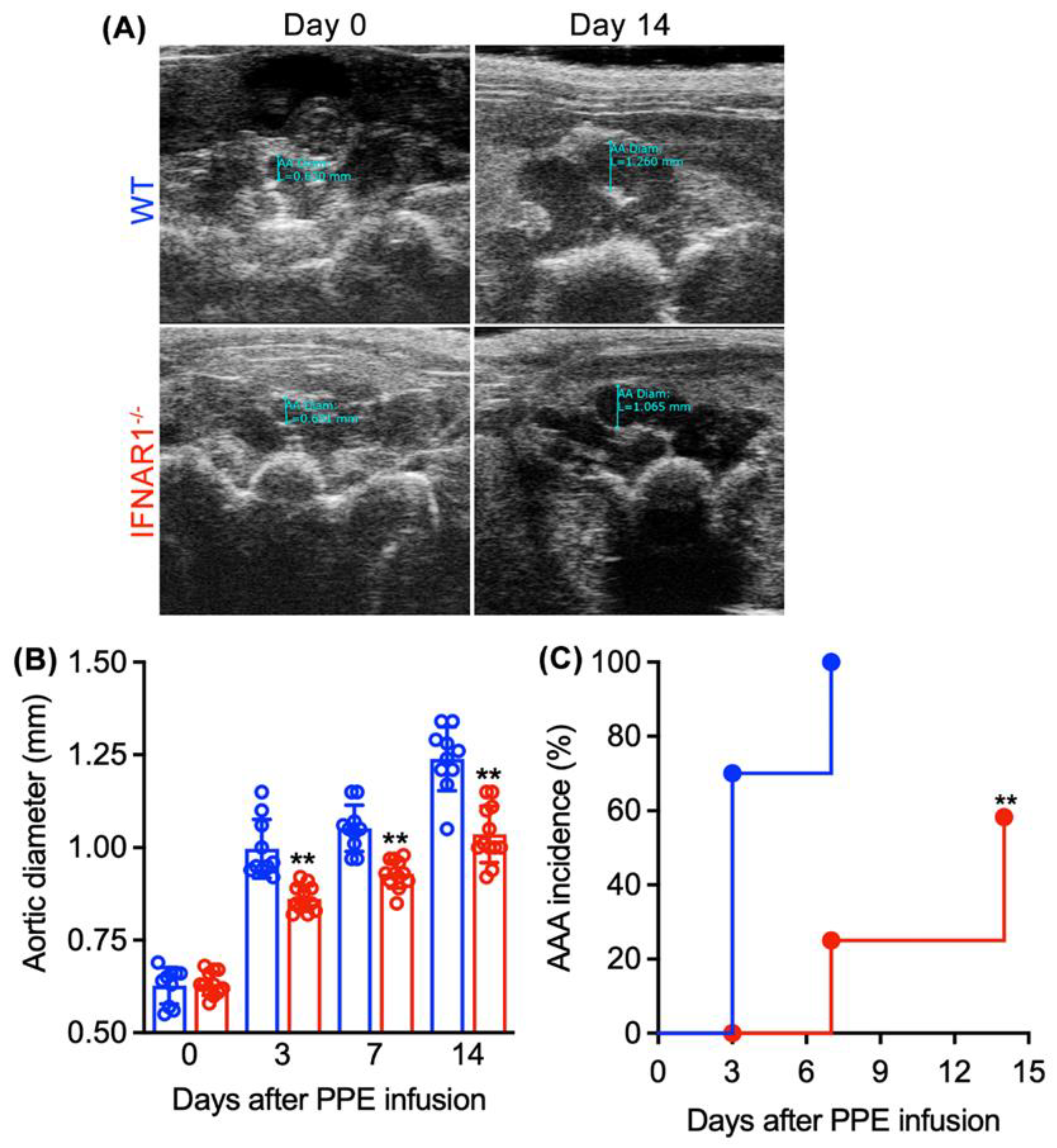
3.2. Attenuated AAA Formation and Progression in IFNAR1−/− Mice
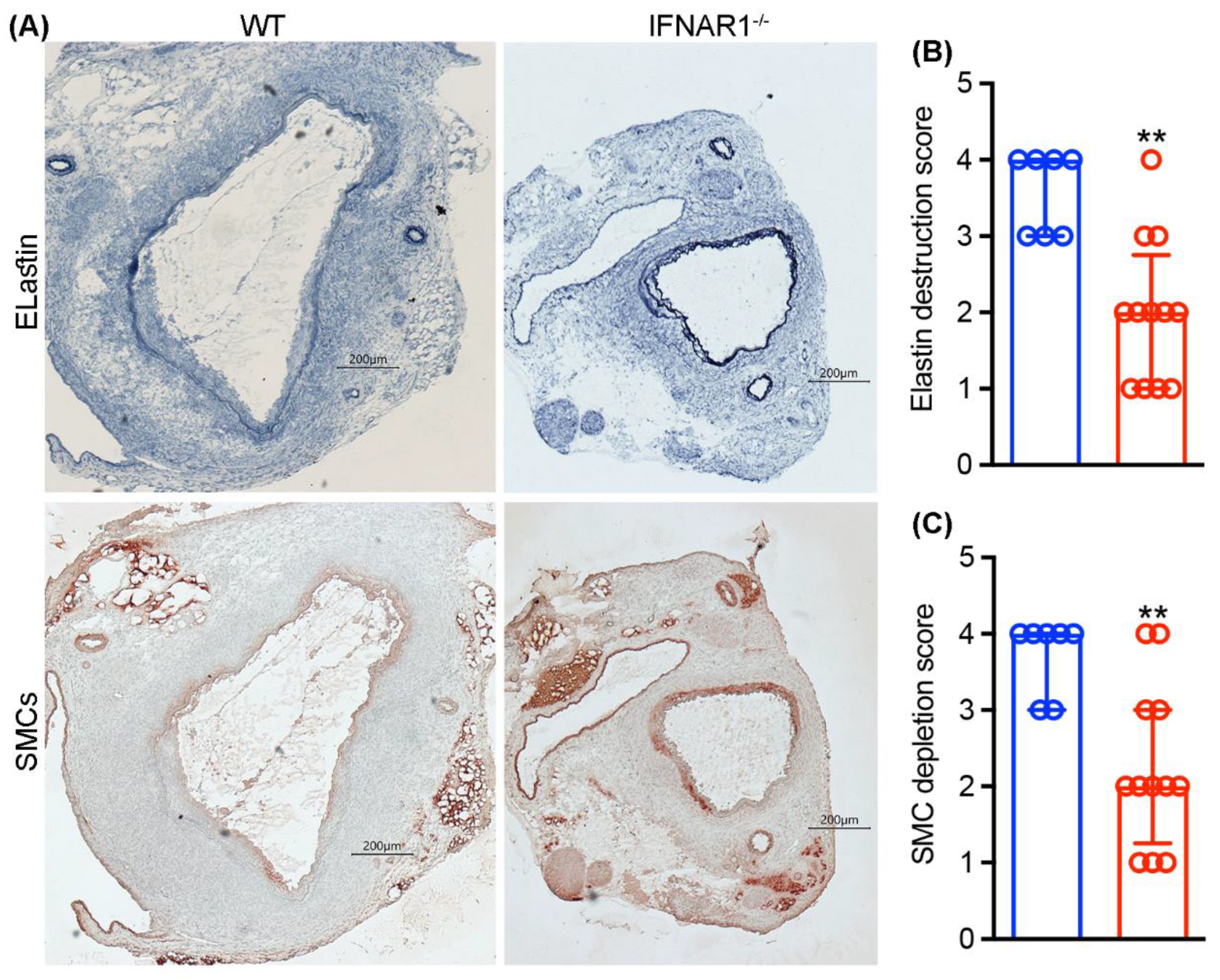
3.3. Attenuated Medial Elastin Degradation and Smooth Muscle Cell Depletion in IFNAR1−/− Mice
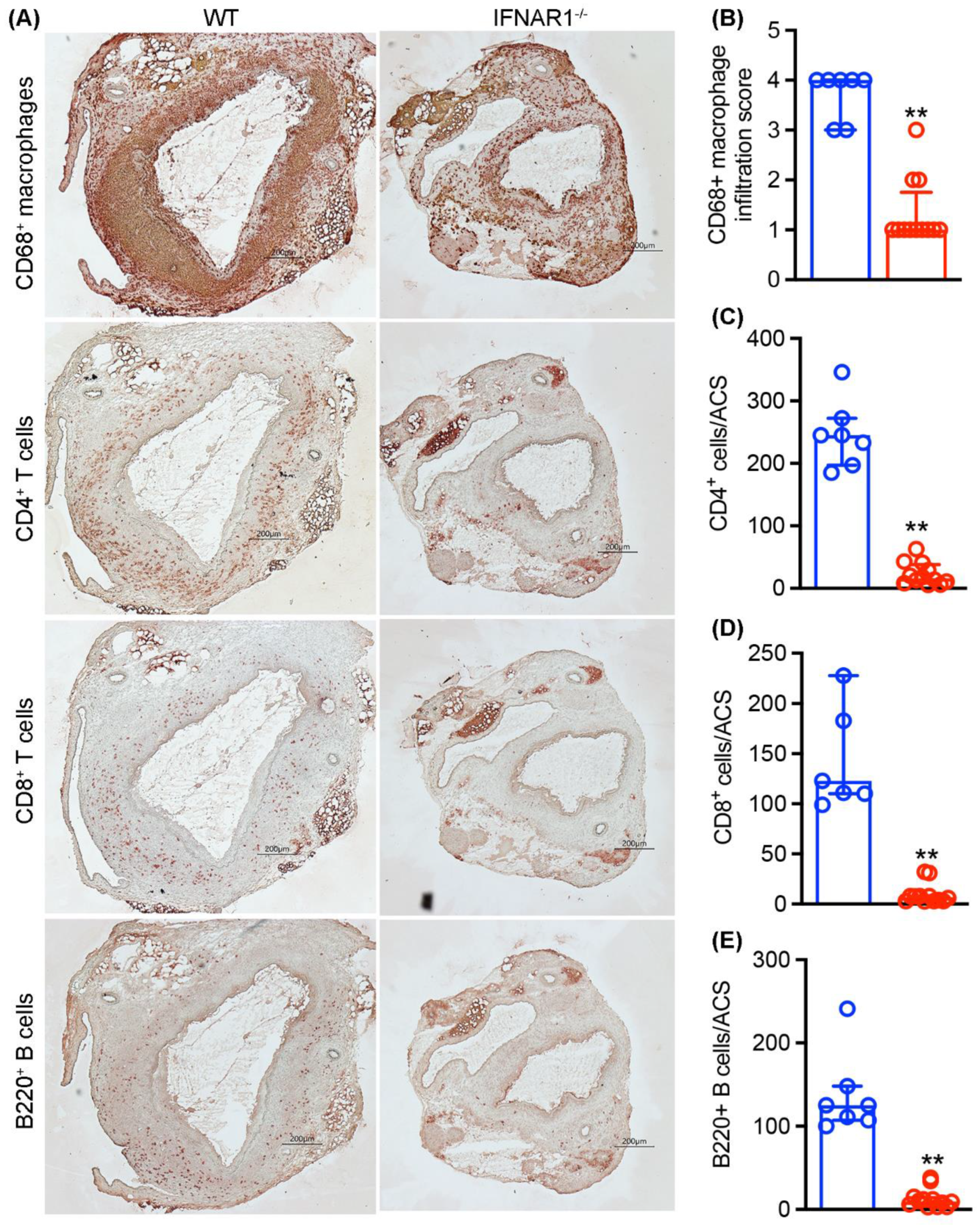
3.4. Attenuated Mural Leukocyte Accumulation in IFNAR1−/− Mice
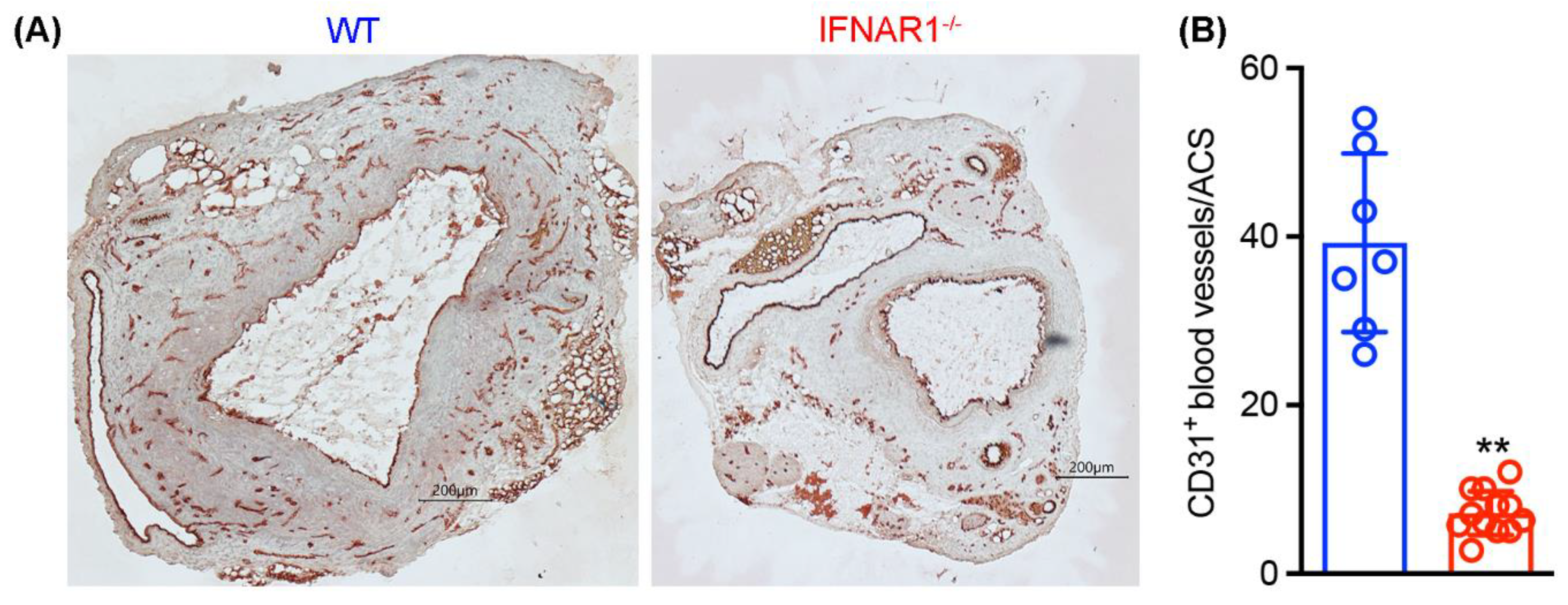
3.5. Attenuated Mural Angiogenesis in IFNAR1−/− Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, J.; Xu, J.; Xia, H.; Wang, Y.; Zhang, C.; Chen, W.; Zhang, H.; Liu, Q.; Zhu, R.; et al. Comprehensive investigations revealed consistent pathophysiological alterations after vaccination with COVID-19 vaccines. Cell Discov. 2021, 7, 99. [Google Scholar] [CrossRef]
- Krämer, B.; Knoll, R.; Bonaguro, L.; ToVinh, M.; Raabe, J.; Astaburuaga-García, R.; Schulte-Schrepping, J.; Kaiser, K.M.; Rieke, G.J.; Bischoff, J.; et al. Early IFN-α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity 2021, 54, 2650–2669.e14. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Lefeuvre, C.; Preisser, L.; Pivert, A.; Soleti, R.; Blanchard, S.; Delneste, Y.; Ducancelle, A.; Couez, D.; Jeannin, P. Age-Related Expression of IFN-λ1 Versus IFN-I and Beta-Defensins in the Nasopharynx of SARS-CoV-2-Infected Individuals. Front. Immunol. 2021, 12, 750279. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O′garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Li, Q.; Xu, B.; Michie, S.A.; Rubins, K.H.; Schreriber, R.D.; McDevitt, H.O. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12439–12444. [Google Scholar] [CrossRef] [Green Version]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Type I Interferons in Autoimmune Disease. Annu. Rev. Pathol. 2019, 14, 369–393. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, R.; Niewold, T.B. Type I Interferons in Autoimmunity. J. Investig. Dermatol. 2022, 142, 793–803. [Google Scholar] [CrossRef]
- Chen, H.J.; Tas, S.W.; de Winther, M.P.J. Type-I interferons in atherosclerosis. J. Exp. Med. 2020, 217, e20190459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Downes, C.E.; Wong, C.H.; Brody, K.M.; Guio-Agulair, P.L.; Gould, J.; Ates, R.; Hertzog, P.J.; Taylor, J.M.; Crack, P.J. Type-I interferon signalling through IFNAR1 plays a deleterious role in the outcome after stroke. Neurochem. Int. 2017, 108, 472–480. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Oliver, E.; Dorfmuller, P.; Dubois, O.D.; Reed, D.M.; Kirkby, N.S.; Mohamed, N.A.; Perros, F.; Antigny, F.; Fadel, E.; et al. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ. Res. 2014, 114, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadière, C.; Rénia, L.; et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Sukhova, G.K.; Yang, M.; Wolters, P.J.; MacFarlane, L.A.; Libby, P.; Sun, C.; Zhang, Y.; Liu, J.; Ennis, T.L.; et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J. Clin. Investig. 2007, 117, 3359–3368. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.B.; Zhou, H.F.; Ennis, T.L.; Wu, X.; Lambris, J.D.; Atkinson, J.P.; Thompson, R.W.; Hourcade, D.E.; Pham, C.T. Complement-dependent neutrophil recruitment is critical for the development of elastase-induced abdominal aortic aneurysm. Circulation 2009, 119, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Schaheen, B.; Downs, E.A.; Serbulea, V.; Almenara, C.C.; Spinosa, M.; Su, G.; Zhao, Y.; Srikakulapu, P.; Butts, C.; McNamara, C.A.; et al. B-Cell Depletion Promotes Aortic Infiltration of Immunosuppressive Cells and Is Protective of Experimental Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2191–2202. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Zhao, Y.; Prall, A.; Greiner, T.C.; Baxter, B.T. Key roles of CD4+ T cells and IFN-gamma in the development of abdominal aortic aneurysms in a murine model. J. Immunol. 2004, 172, 2607–2612. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Lu, G.; Jester, A.; Johnston, W.F.; Zhao, Y.; Hajzus, V.A.; Saadatzadeh, M.R.; Su, G.; Bhamidipati, C.M.; Mehta, G.S.; et al. Experimental abdominal aortic aneurysm formation is mediated by IL-17 and attenuated by mesenchymal stem cell treatment. Circulation 2012, 126 (Suppl. 1), S38–S45. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; MacTaggart, J.; Knispel, R.; Worth, J.; Persidsky, Y.; Baxter, B.T. Blocking TNF-alpha attenuates aneurysm formation in a murine model. J. Immunol. 2009, 183, 2741–2746. [Google Scholar] [CrossRef]
- Adam, M.; Kooreman, N.G.; Jagger, A.; Wagenhäuser, M.U.; Mehrkens, D.; Wang, Y.; Kayama, Y.; Toyama, K.; Raaz, U.; Schellinger, I.N.; et al. Systemic Upregulation of IL-10 (Interleukin-10) Using a Nonimmunogenic Vector Reduces Growth and Rate of Dissecting Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1796–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Li, Y.; Wang, W.; Guo, J.; Zhao, S.; Glover, K.J.; et al. Recombinant Interleukin-19 Suppresses the Formation and Progression of Experimental Abdominal Aortic Aneurysms. J. Am. Heart Assoc. 2021, 10, e022207. [Google Scholar] [CrossRef] [PubMed]
- Angelov, S.N.; Hu, J.H.; Wei, H.; Airhart, N.; Shi, M.; Dichek, D.A. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Müller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Iida, Y.; Glover, K.J.; Ge, Y.; Wang, Y.; Xuan, H.; Hu, X.; Tanaka, H.; Wang, W.; Fujimura, N.; et al. Inhibition of VEGF (Vascular Endothelial Growth Factor)-A or its Receptor Activity Suppresses Experimental Aneurysm Progression in the Aortic Elastase Infusion Model. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1652–1666. [Google Scholar] [CrossRef]
- Xuan, H.; Xu, B.; Wang, W.; Tanaka, H.; Fujimura, N.; Miyata, M.; Michie, S.A.; Dalman, R.L. Inhibition or deletion of angiotensin II type 1 receptor suppresses elastase-induced experimental abdominal aortic aneurysms. J. Vasc. Surg. 2018, 67, 573–584.e2. [Google Scholar] [CrossRef] [Green Version]
- Schultz, G.; Tedesco, M.M.; Sho, E.; Nishimura, T.; Sharif, S.; Du, X.; Myles, T.; Morser, J.; Dalman, R.L.; Leung, L.L. Enhanced abdominal aortic aneurysm formation in thrombin-activatable procarboxypeptidase B-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1363–1370. [Google Scholar] [CrossRef] [Green Version]
- Sénémaud, J.; Caligiuri, G.; Etienne, H.; Delbosc, S.; Michel, J.B.; Coscas, R. Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Ikezoe, T.; Shoji, T.; Guo, J.; Shen, F.; Lu, H.S.; Daugherty, A.; Nunokawa, M.; Kubota, H.; Miyata, M.; Xu, B.; et al. No Effect of Hypercholesterolemia on Elastase-Induced Experimental Abdominal Aortic Aneurysm Progression. Biomolecules 2021, 11, 1434. [Google Scholar] [CrossRef]
- Iida, Y.; Xu, B.; Schultz, G.M.; Chow, V.; White, J.J.; Sulaimon, S.; Hezi-Yamit, A.; Peterson, S.R.; Dalman, R.L. Efficacy and mechanism of angiotensin II receptor blocker treatment in experimental abdominal aortic aneurysms. PLoS ONE 2012, 7, e49642. [Google Scholar] [CrossRef]
- Wang, W.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Wang, L.; Huang, J.; Fu, W.; Michie, S.A.; Dalman, R.L. Hypoxia-inducible factor 1 in clinical and experimental aortic aneurysm disease. J. Vasc. Surg. 2018, 68, 1538–1550.e2. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.O.; Pinchbeck, J.; Elmore, K.; Jones, R.E.; Moxon, J.V.; Golledge, J. The reproducibility of measuring maximum abdominal aortic aneurysm diameter from ultrasound images. Ultrasound J. 2021, 13, 13. [Google Scholar] [CrossRef]
- Meecham, L.; Evans, R.; Buxton, P.; Allingham, K.; Hughes, M.; Rajagopalan, S.; Fairhead, J.; Asquith, J.R.; Pherwani, A.D. Abdominal Aortic Aneurysm Diameters: A Study on the Discrepancy between Inner to Inner and Outer to Outer Measurements. Eur. J. Vasc. Endovasc. Surg. 2015, 49, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Iida, Y.; Xu, B.; Xuan, H.; Glover, K.J.; Tanaka, H.; Hu, X.; Fujimura, N.; Wang, W.; Schultz, J.R.; Turner, C.R.; et al. Peptide inhibitor of CXCL4-CCL5 heterodimer formation, MKEY, inhibits experimental aortic aneurysm initiation and progression. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Zhou, H.F.; Akk, A.; Hu, Y.; Springer, L.E.; Ennis, T.L.; Pham, C.T. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1660–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Xuan, H.; Fujimura, N.; Michie, S.A.; Dalman, R.L. Abstract 107: Depletion of Plasmacytoid Dendritic Cells Inhibits Experimental Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2016, 36, A107. [Google Scholar] [CrossRef]
- Zhou, H.F.; Yan, H.; Cannon, J.L.; Springer, L.E.; Green, J.M.; Pham, C.T. CD43-mediated IFN-γ production by CD8+ T cells promotes abdominal aortic aneurysm in mice. J. Immunol. 2013, 190, 5078–5085. [Google Scholar] [CrossRef] [Green Version]
- Furusho, A.; Aoki, H.; Ohno-Urabe, S.; Nishihara, M.; Hirakata, S.; Nishida, N.; Ito, S.; Hayashi, M.; Imaizumi, T.; Hiromatsu, S.; et al. Involvement of B Cells, Immunoglobulins, and Syk in the Pathogenesis of Abdominal Aortic Aneurysm. J. Am. Heart Assoc. 2018, 7, e007750. [Google Scholar] [CrossRef] [Green Version]
- Tedesco, M.M.; Terashima, M.; Blankenberg, F.G.; Levashova, Z.; Spin, J.M.; Backer, M.V.; Backer, J.M.; Sho, M.; Sho, E.; McConnell, M.V.; et al. Analysis of in situ and ex vivo vascular endothelial growth factor receptor expression during experimental aortic aneurysm progression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1452–1457. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, H.; Anzai, T.; Takahashi, T.; Kohno, T.; Shimoda, M.; Sasaki, A.; Shimizu, H.; Nagai, T.; Maekawa, Y.; Yoshimura, K.; et al. Role of vascular endothelial growth factor-A in development of abdominal aortic aneurysm. Cardiovasc. Res. 2011, 91, 358–367. [Google Scholar] [CrossRef]
- Yang, L.; Shen, L.; Li, G.; Yuan, H.; Jin, X.; Wu, X. Silencing of hypoxia inducible factor-1α gene attenuated angiotensin Ⅱ-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice. Atherosclerosis 2016, 252, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Shoji, T.; Ge, Y.; Zheng, X.; Li, Y.; Zhao, S.; Ikezoe, T.; Liu, S.; Huang, J.; Wang, W.; et al. Treatment with the Prolyl Hydroxylase Inhibitor JNJ Promotes Abdominal Aortic Aneurysm Progression in Diabetic Mice. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Miyama, N.; Dua, M.M.; Yeung, J.J.; Schultz, G.M.; Asagami, T.; Sho, E.; Sho, M.; Dalman, R.L. Hyperglycemia limits experimental aortic aneurysm progression. J. Vasc. Surg. 2010, 52, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.H.; Chang, C.H.; Wang, J.L.; Wu, L.C.; Lin, J.W.; Toh, S. Association of Infections and Use of Fluoroquinolones With the Risk of Aortic Aneurysm or Aortic Dissection. JAMA Intern. Med. 2020, 180, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Høgh, J.; Pham, M.H.C.; Knudsen, A.D.; Thudium, R.F.; Gelpi, M.; Sigvardsen, P.E.; Fuchs, A.; Kühl, J.T.; Afzal, S.; Nordestgaard, B.G.; et al. HIV infection is associated with thoracic and abdominal aortic aneurysms: A prospective matched cohort study. Eur. Heart J. 2021, 42, 2924–2931. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska, A.; Zagrapan, B.; Paradowska, E.; Neumayer, C.; Eilenberg, W.; Brostjan, C.; Klinger, M.; Nanobachvili, J.; Huk, I. Abdominal aortic aneurysm and virus infection: A potential causative role for cytomegalovirus infection? J. Med. Virol. 2021, 93, 5017–5024. [Google Scholar] [CrossRef] [PubMed]
- Vammen, S.; Lindholt, J.S.; Østergaard, L.; Fasting, H.; Henneberg, E.W. Randomized double-blind controlled trial of roxithromycin for prevention of abdominal aortic aneurysm expansion. Br. J. Surg. 2001, 88, 1066–1072. [Google Scholar] [CrossRef]
- Høgh, A.; Vammen, S.; Ostergaard, L.; Joensen, J.B.; Henneberg, E.W.; Lindholt, J.S. Intermittent roxithromycin for preventing progression of small abdominal aortic aneurysms: Long-term results of a small clinical trial. Vasc. Endovasc. Surg. 2009, 43, 452–456. [Google Scholar] [CrossRef]
- Karlsson, L.; Gnarpe, J.; Bergqvist, D.; Lindbäck, J.; Pärsson, H. The effect of azithromycin and Chlamydophilia pneumonia infection on expansion of small abdominal aortic aneurysms—A prospective randomized double-blind trial. J. Vasc. Surg. 2009, 50, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.E.; Jones, J.M.; Deng, Y.; Nycz, E.; Lee, A.; Iachan, R.; Gundlapalli, A.V.; Hall, A.J.; MacNeil, A. Seroprevalence of Infection-Induced SARS-CoV-2 Antibodies-United States, September 2021–February 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 606–608. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z.; Bowe, B.; Xie, Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat. Med. 2022, 28, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Morrow, A.J.; Sykes, R.; McIntosh, A.; Kamdar, A.; Bagot, C.; Bayes, H.K.; Blyth, K.G.; Briscoe, M.; Bulluck, H.; Carrick, D.; et al. A multisystem, cardio-renal investigation of post-COVID-19 illness. Nat. Med. 2022, 28, 1303–1313. [Google Scholar] [CrossRef]
- Severa, M.; Diotti, R.A.; Etna, M.P.; Rizzo, F.; Fiore, S.; Ricci, D.; Iannetta, M.; Sinigaglia, A.; Lodi, A.; Mancini, N.; et al. Differential plasmacytoid dendritic cell phenotype and type I Interferon response in asymptomatic and severe COVID-19 infection. PLoS Pathog. 2021, 17, e1009878. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, G.; Guo, J.; Ikezoe, T.; Kasirajan, K.; Zhao, S.; Dalman, R.L. Angiotensin-converting enzyme 2, coronavirus disease 2019, and abdominal aortic aneurysms. J. Vasc. Surg. 2021, 74, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xuan, H.; Iida, Y.; Miyata, M.; Dalman, R.L. Pathogenic and Therapeutic Significance of Angiotensin II Type I Receptor in Abdominal Aortic Aneurysms. Curr. Drug Targets 2018, 19, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Sweeting, M.J.; Thompson, S.G.; Brown, L.C.; Powell, J.T. Meta-analysis of individual patient data to examine factors affecting growth and rupture of small abdominal aortic aneurysms. Br. J. Surg. 2012, 99, 655–665. [Google Scholar] [CrossRef]
- Schlösser, F.J.; Tangelder, M.J.; Verhagen, H.J.; van der Heijden, G.J.; Muhs, B.E.; van der Graaf, Y.; Moll, F.L. and Smart Study Group. Growth predictors and prognosis of small abdominal aortic aneurysms. J. Vasc. Surg. 2008, 47, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Powell, J.T.; Sweeting, M.J.; Brown, L.C.; Gotensparre, S.M.; Fowkes, F.G.; Thompson, S.G. Systematic review and meta-analysis of growth rates of small abdominal aortic aneurysms. Br. J. Surg. 2011, 98, 609–618. [Google Scholar] [CrossRef]
- Bozzani, A.; Arici, V.; Ticozzelli, G.; Franciscone, M.M.; Sterpetti, A.V.; Ragni, F. Sudden Rupture of Abdominal Aortic Aneurysm in COVID19 Patients. J. Endovasc. Ther. 2022. [Google Scholar] [CrossRef]
- Bozzani, A.; Arici, V.; Franciscone, M.; Ticozzelli, G.; Sterpetti, A.V.; Ragni, F. COVID-19 patients with abdominal aortic aneurysm may be at higher risk for sudden enlargement and rupture. J. Vasc. Surg. 2022, 75, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Bozzani, A.; Arici, V.; Ticozzelli, G.; Franciscone, M.M.; Sterpetti, A.V.; Ragni, F. Increased rates of ruptured abdominal aortic aneurysm during the COVID-19 pandemic. J. Vasc. Surg. 2021, 74, 2119–2120. [Google Scholar] [CrossRef] [PubMed]
- Guy, A.; Tiosano, S.; Comaneshter, D.; Tekes-Manova, D.; Shovman, O.; Cohen, A.D.; Amital, H. Aortic aneurysm association with SLE-a case-control study. Lupus 2016, 25, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Khalid, U.; Egeberg, A.; Ahlehoff, O.; Smedegaard, L.; Gislason, G.H.; Hansen, P.R. Nationwide Study on the Risk of Abdominal Aortic Aneurysms in Patients With Psoriasis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1043–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, J.C.; Kiran, A.; Maskell, J.; Hutchings, A.; Arden, N.; Dasgupta, B.; Hamilton, W.; Emin, A.; Culliford, D.; Luqmani, R.A. The relative risk of aortic aneurysm in patients with giant cell arteritis compared with the general population of the UK. Ann. Rheum. Dis. 2015, 74, 129–135. [Google Scholar] [CrossRef]
- Wang, S.H.; Chang, Y.S.; Liu, C.J.; Lai, C.C.; Chen, T.J.; Chen, W.S. Incidence and risk analysis of aortic aneurysm and aortic dissection among patients with systemic lupus erythematosus: A nationwide population-based study in Taiwan. Lupus 2014, 23, 665–671. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, J.; Kong, R.; Cheng, C.; Chen, Q.; Xia, Y.; Li, X.; Zhang, T.; Cai, Q. Prevalence, clinical features, risk factors, and outcomes of SLE patients with aortic aneurysm: A cross-sectional retrospective study in a Chinese single center. Clin. Rheumatol. 2022, 41, 377–386. [Google Scholar] [CrossRef]
- Thompson, A.; Cooper, J.A.; Fabricius, M.; Humphries, S.E.; Ashton, H.A.; Hafez, H. An analysis of drug modulation of abdominal aortic aneurysm growth through 25 years of surveillance. J. Vasc. Surg. 2010, 52, 55–61.e2. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.W.; Gracon, A.S.; Murphy, M.P.; Wilkes, D.S. Exploring autoimmunity in the pathogenesis of abdominal aortic aneurysms. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H719–H727. [Google Scholar] [CrossRef] [Green Version]
- Tilson, M.D. Autoimmunity in the Abdominal Aortic Aneurysm and its Association with Smoking. AORTA 2017, 5, 159–167. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O′Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.M.; Hanna, R.N.; Rajan, B.; Zerrouki, K.; Karnell, J.L.; Sagar, D.; Vainshtein, I.; Farmer, E.; Rosenthal, K.; Morehouse, C.; et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci. Med. 2018, 5, e000261. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoji, T.; Guo, J.; Ge, Y.; Li, Y.; Li, G.; Ikezoe, T.; Wang, W.; Zheng, X.; Zhao, S.; Fujimura, N.; et al. Type I Interferon Receptor Subunit 1 Deletion Attenuates Experimental Abdominal Aortic Aneurysm Formation. Biomolecules 2022, 12, 1541. https://doi.org/10.3390/biom12101541
Shoji T, Guo J, Ge Y, Li Y, Li G, Ikezoe T, Wang W, Zheng X, Zhao S, Fujimura N, et al. Type I Interferon Receptor Subunit 1 Deletion Attenuates Experimental Abdominal Aortic Aneurysm Formation. Biomolecules. 2022; 12(10):1541. https://doi.org/10.3390/biom12101541
Chicago/Turabian StyleShoji, Takahiro, Jia Guo, Yingbin Ge, Yankui Li, Gang Li, Toru Ikezoe, Wei Wang, Xiaoya Zheng, Sihai Zhao, Naoki Fujimura, and et al. 2022. "Type I Interferon Receptor Subunit 1 Deletion Attenuates Experimental Abdominal Aortic Aneurysm Formation" Biomolecules 12, no. 10: 1541. https://doi.org/10.3390/biom12101541