
Epigenetic Reprogramming of the Inflammatory Response in Obesity and Type 2 Diabetes

 , , ,
, , ,
Abstract
:1. Introduction
2. Epigenetics of Immune System and Its Role in the Development of Obesity- and T2D-Associated Inflammation
2.1. DNA Methylation
2.1.1. DNA Methylation and Regulation of Immunity
2.1.2. DNA Methylation and the Development of Inflammation in Obesity and T2D
2.2. Histone Modifications
2.2.1. Histone Modifications in Immunity
2.2.2. Histone Marks in Obesity and T2D
2.3. microRNAs
2.3.1. Role of microRNAs in the Immune Response
2.3.2. MicroRNAs and the Development of Inflammation in Metabolic Disorders
2.4. Adipocyte Hypertrophy: A Paradigm of Epigenetic Derangement in Chronic Inflammation

{kind=link}
| Study Model | Epigenetic Marks | Position | Processes | Medical Condition | Species | Ref |
|---|---|---|---|---|---|---|
| AT from Diet-induced Obesity model | DNA Hyper-methylation | Hoxa5 | Hox Gene Family, Adipogenesis, AT Macrophage Genes | Obesity, Impaired Glucose Metabolism, AT Inflammation | Mouse | [130] |
| AT from Diet-induced Obesity model | - | Hoxa5 | ER Stress Signalling pathways, M2 Macrophage Polarization | Obesity, Impaired Glucose Metabolism, AT Inflammation | Mouse | [131] |
| PMCs from Diet-induced Obesity Model | - | Zfp423 | Nf-κb Inflammatory pathway, ATM accumulation, LPS-induced Inflammation | Obesity, AT Inflammation | Mouse | [132] |
| AT from Diet-induced Obesity and from Obese subjects | DNA Hyper-methylation | Ankrd26 | Adipocyte pro-inflammatory secretion, Il-8, Mcp-1, Rantes/Pro-inflammatory profile in AT | Obesity, Impaired Glucose Metabolism, Adiposity, AT Inflammation | Mouse | [133] |
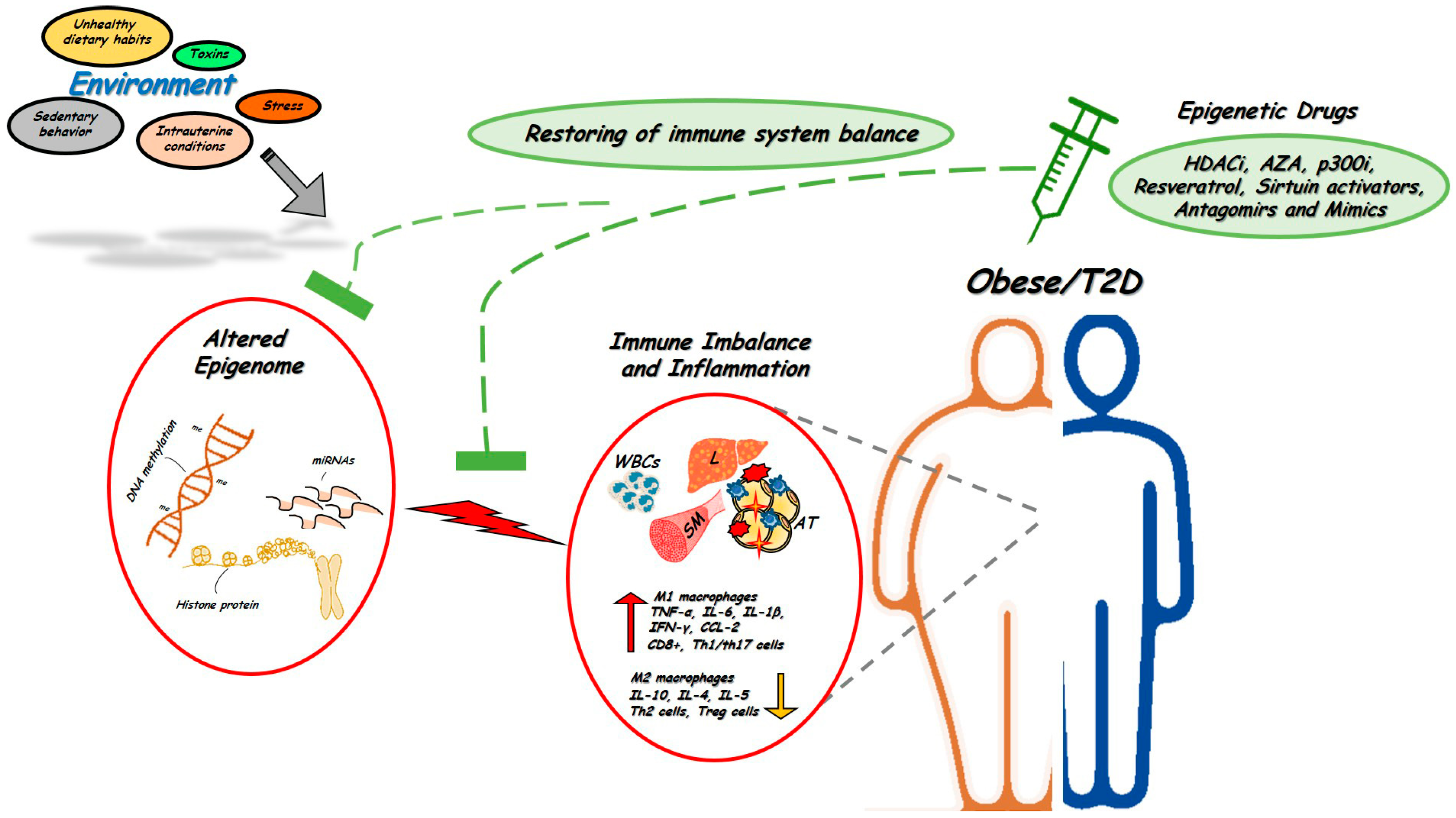
3. Epigenetic Changes as Anti-Inflammatory Targets for Treatment of Metabolic Disorders
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parrillo, L.; Spinelli, R.; Nicolo, A.; Longo, M.; Mirra, P.; Raciti, G.A.; Miele, C.; Beguinot, F. Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges. Int. J. Mol. Sci. 2019, 20, 2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, S.; Donkin, I.; Versteyhe, S.; Barres, R.; Simar, D. The Emerging Role of Epigenetics in Inflammation and Immunometabolism. Trends Endocrinol. Metab. 2016, 27, 782–795. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Bruun, J.M.; Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): Implication of macrophages resident in the AT. J. Clin. Endocrinol. Metab. 2005, 90, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni-Schnetzler, M.; Boller, S.; Debray, S.; Bouzakri, K.; Meier, D.T.; Prazak, R.; Kerr-Conte, J.; Pattou, F.; Ehses, J.A.; Schuit, F.C.; et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 2009, 150, 5218–5229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni-Schnetzler, M.; Thorne, J.; Parnaud, G.; Marselli, L.; Ehses, J.A.; Kerr-Conte, J.; Pattou, F.; Halban, P.A.; Weir, G.C.; Donath, M.Y. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 2008, 93, 4065–4074. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Akbari, M.; Hassan-Zadeh, V. The inflammatory effect of epigenetic factors and modifications in type 2 diabetes. Inflammopharmacology 2020, 28, 345–362. [Google Scholar] [CrossRef]
- Xu, X.; Su, S.; Barnes, V.A.; De Miguel, C.; Pollock, J.; Ownby, D.; Shi, H.; Zhu, H.; Snieder, H.; Wang, X. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Calle-Fabregat, C.; Morante-Palacios, O.; Ballestar, E. Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease. Genes 2020, 11, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.Q.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.C.; Xi, Y.X.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, K.F.; Neele, A.E.; Jukema, J.W.; Heijmans, B.T.; de Winther, M.P.J. Human monocyte-to-macrophage differentiation involves highly localized gain and loss of DNA methylation at transcription factor binding sites. Epigenetics Chromatin 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gomez, A.; Li, T.; Kerick, M.; Catala-Moll, F.; Comet, N.R.; Rodriguez-Ubreva, J.; de la Rica, L.; Branco, M.R.; Martin, J.; Ballestar, E. TET2- and TDG-mediated changes are required for the acquisition of distinct histone modifications in divergent terminal differentiation of myeloid cells. Nucleic Acids Res. 2017, 45, 10002–10017. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Garcia-Gomez, A.; Morante-Palacios, O.; Ciudad, L.; Ozkaramehmet, S.; Van Dijck, E.; Rodriguez-Ubreva, J.; Vaquero, A.; Ballestar, E. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2020, 48, 665–681. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Merkel, A.; Heath, S.; Queiros, A.C.; Schuyler, R.P.; Castellano, G.; Beekman, R.; Raineri, E.; Esteve, A.; Clot, G.; et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015, 47, 746–756. [Google Scholar] [CrossRef]
- Shaknovich, R.; Cerchietti, L.; Tsikitas, L.; Kormaksson, M.; De, S.; Figueroa, M.E.; Ballon, G.; Yang, S.N.; Weinhold, N.; Reimers, M.; et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 2011, 118, 3559–3569. [Google Scholar] [CrossRef] [Green Version]
- Sellars, M.; Huh, J.R.; Day, K.; Issuree, P.D.; Galan, C.; Gobeil, S.; Absher, D.; Green, M.R.; Littman, D.R. Regulation of DNA methylation dictates Cd4 expression during the development of helper and cytotoxic T cell lineages. Nat. Immunol. 2015, 16, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellmeier, W.; Haust, L.; Tschismarov, R. Transcriptional control of CD4 and CD8 coreceptor expression during T cell development. Cell Mol. Life Sci. 2013, 70, 4537–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierda, R.J.; Kuipers, H.F.; van Eggermond, M.C.; Benard, A.; van Leeuwen, J.C.; Carluccio, S.; Geutskens, S.B.; Jukema, J.W.; Marquez, V.E.; Quax, P.H.; et al. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J. Cell Mol. Med. 2012, 16, 1866–1877. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Hutchins, A.S.; High, F.A.; Lee, H.W.; Sykes, K.J.; Chodosh, L.A.; Reiner, S.L. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat. Immunol. 2002, 3, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.D.; Bopp, T.; Schmitt, E.; et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef]
- Lee, D.U.; Agarwal, S.; Rao, A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity 2002, 16, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, Y.; Beier, U.H.; Han, R.; Bhatti, T.R.; Akimova, T.; Hancock, W.W. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood 2013, 121, 3631–3639. [Google Scholar] [CrossRef] [Green Version]
- Helmin, K.A.; Morales-Nebreda, L.; Torres Acosta, M.A.; Anekalla, K.R.; Chen, S.Y.; Abdala-Valencia, H.; Politanska, Y.; Cheresh, P.; Akbarpour, M.; Steinert, E.M.; et al. Maintenance DNA methylation is essential for regulatory T cell development and stability of suppressive function. J. Clin. Investig. 2020, 130, 6571–6587. [Google Scholar] [CrossRef]
- Petrus, P.; Bialesova, L.; Checa, A.; Kerr, A.; Naz, S.; Backdahl, J.; Gracia, A.; Toft, S.; Dahlman-Wright, K.; Heden, P.; et al. Adipocyte Expression of SLC19A1 Links DNA Hypermethylation to Adipose Tissue Inflammation and Insulin Resistance. J. Clin. Endocrinol. Metab. 2018, 103, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.H.; Chen, L.L.; Deng, X.L.; Song, H.J.; Liao, Y.F.; Zeng, T.S.; Zheng, J.; Li, H.Q. Methylation status of CpG sites in the MCP-1 promoter is correlated to serum MCP-1 in Type 2 diabetes. J. Endocrinol. Investig. 2012, 35, 585–589. [Google Scholar] [CrossRef]
- Roshanzamir, N.; Hassan-Zadeh, V. Methylation of Specific CpG Sites in IL-1beta and IL1R1 Genes is Affected by Hyperglycaemia in Type 2 Diabetic Patients. Immunol. Investig. 2020, 49, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Hassan-Zadeh, V. Hyperglycemia Affects the Expression of Inflammatory Genes in Peripheral Blood Mononuclear Cells of Patients with Type 2 Diabetes. Immunol. Investig. 2018, 47, 654–665. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef] [Green Version]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [Green Version]
- Dayeh, T.; Tuomi, T.; Almgren, P.; Perfilyev, A.; Jansson, P.A.; de Mello, V.D.; Pihlajamaki, J.; Vaag, A.; Groop, L.; Nilsson, E.; et al. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016, 11, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Hermsdorff, H.H.; Mansego, M.L.; Campion, J.; Milagro, F.I.; Zulet, M.A.; Martinez, J.A. TNF-alpha promoter methylation in peripheral white blood cells: Relationship with circulating TNFalpha, truncal fat and n-6 PUFA intake in young women. Cytokine 2013, 64, 265–271. [Google Scholar] [CrossRef]
- Na, Y.K.; Hong, H.S.; Lee, W.K.; Kim, Y.H.; Kim, D.S. Increased methylation of interleukin 6 gene is associated with obesity in Korean women. Mol. Cells 2015, 38, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Arpon, A.; Milagro, F.I.; Ramos-Lopez, O.; Mansego, M.L.; Riezu-Boj, J.I.; Martinez, J.A.; Project, M. Methylome-Wide Association Study in Peripheral White Blood Cells Focusing on Central Obesity and Inflammation. Genes 2019, 10, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simar, D.; Versteyhe, S.; Donkin, I.; Liu, J.; Hesson, L.; Nylander, V.; Fossum, A.; Barres, R. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism 2014, 63, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Dispirito, J.R.; Shen, H. Histone acetylation at the single-cell level: A marker of memory CD8+ T cell differentiation and functionality. J. Immunol. 2010, 184, 4631–4636. [Google Scholar] [CrossRef] [Green Version]
- Allan, R.S.; Zueva, E.; Cammas, F.; Schreiber, H.A.; Masson, V.; Belz, G.T.; Roche, D.; Maison, C.; Quivy, J.P.; Almouzni, G.; et al. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 2012, 487, 249–253. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Dorschner, M.O.; Sekimata, M.; Santer, D.M.; Shnyreva, M.; Fitzpatrick, D.R.; Stamatoyannopoulos, J.A.; Wilson, C.B. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 2007, 8, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Good-Jacobson, K.L. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front. Immunol. 2014, 5, 596. [Google Scholar] [CrossRef] [Green Version]
- Baxter, J.; Sauer, S.; Peters, A.; John, R.; Williams, R.; Caparros, M.L.; Arney, K.; Otte, A.; Jenuwein, T.; Merkenschlager, M.; et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 2004, 23, 4462–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waibel, M.; Christiansen, A.J.; Hibbs, M.L.; Shortt, J.; Jones, S.A.; Simpson, I.; Light, A.; O’Donnell, K.; Morand, E.F.; Tarlinton, D.M.; et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat. Commun. 2015, 6, 6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F.t.; Mukherjee, S.; Grembecka, J.; Cierpicki, T.; et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Rong, S.; Repa, J.J.; St Clair, R.; Parks, J.S.; Mishra, N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Kiguchi, N.; Kobayashi, Y.; Saika, F.; Kishioka, S. Epigenetic upregulation of CCL2 and CCL3 via histone modifications in infiltrating macrophages after peripheral nerve injury. Cytokine 2013, 64, 666–672. [Google Scholar] [CrossRef]
- Ding, Q.; Gao, Z.; Chen, K.; Zhang, Q.; Hu, S.; Zhao, L. Inflammation-Related Epigenetic Modification: The Bridge Between Immune and Metabolism in Type 2 Diabetes. Front. Immunol. 2022, 13, 883410. [Google Scholar] [CrossRef]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Farha, M.; Tiss, A.; Abubaker, J.; Khadir, A.; Al-Ghimlas, F.; Al-Khairi, I.; Baturcam, E.; Cherian, P.; Elkum, N.; Hammad, M.; et al. Proteomics analysis of human obesity reveals the epigenetic factor HDAC4 as a potential target for obesity. PLoS ONE 2013, 8, e75342. [Google Scholar] [CrossRef]
- Shanaki, M.; Omidifar, A.; Shabani, P.; Toolabi, K. Association between HDACs and pro-inflammatory cytokine gene expressions in obesity. Arch. Physiol. Biochem. 2020, 1–7. [Google Scholar] [CrossRef]
- Hanzu, F.A.; Musri, M.M.; Sanchez-Herrero, A.; Claret, M.; Esteban, Y.; Kaliman, P.; Gomis, R.; Parrizas, M. Histone demethylase KDM1A represses inflammatory gene expression in preadipocytes. Obesity 2013, 21, E616–E625. [Google Scholar] [CrossRef]
- Tian, W.; Xu, H.; Fang, F.; Chen, Q.; Xu, Y.; Shen, A. Brahma-related gene 1 bridges epigenetic regulation of proinflammatory cytokine production to steatohepatitis in mice. Hepatology 2013, 58, 576–588. [Google Scholar] [CrossRef]
- Mikula, M.; Majewska, A.; Ledwon, J.K.; Dzwonek, A.; Ostrowski, J. Obesity increases histone H3 lysine 9 and 18 acetylation at Tnfa and Ccl2 genes in mouse liver. Int. J. Mol. Med. 2014, 34, 1647–1654. [Google Scholar] [CrossRef] [Green Version]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [Green Version]
- Arab Sadeghabadi, Z.; Nourbakhsh, M.; Pasalar, P.; Emamgholipour, S.; Golestani, A.; Larijani, B.; Razzaghy-Azar, M. Reduced gene expression of sirtuins and active AMPK levels in children and adolescents with obesity and insulin resistance. Obes. Res. Clin. Pract. 2018, 12, 167–173. [Google Scholar] [CrossRef]
- Zhou, S.; Tang, X.; Chen, H.Z. Sirtuins and Insulin Resistance. Front. Endocrinol. 2018, 9, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crujeiras, A.B.; Parra, D.; Goyenechea, E.; Martinez, J.A. Sirtuin gene expression in human mononuclear cells is modulated by caloric restriction. Eur. J. Clin. Investig. 2008, 38, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Z.; Schaffert, S.; Fragoso, R.; Loh, C. Regulation of immune responses and tolerance: The microRNA perspective. Immunol. Rev. 2013, 253, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Kurkewich, J.L.; Boucher, A.; Klopfenstein, N.; Baskar, R.; Kapur, R.; Dahl, R. The mirn23a and mirn23b microrna clusters are necessary for proper hematopoietic progenitor cell production and differentiation. Exp. Hematol. 2018, 59, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Lu, J.; Schlanger, R.; Zhang, H.; Wang, J.Y.; Fox, M.C.; Purton, L.E.; Fleming, H.H.; Cobb, B.; Merkenschlager, M.; et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proc. Natl. Acad. Sci. USA 2010, 107, 14229–14234. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Liu, Y.; Liang, G.; Zhao, M.; Wu, H.; Liang, Y.; Qiu, X.; Tan, Y.; Dai, Y.; Yung, S.; et al. The role of microRNA-1246 in the regulation of B cell activation and the pathogenesis of systemic lupus erythematosus. Clin. Epigenet. 2015, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Marcais, A.; Blevins, R.; Graumann, J.; Feytout, A.; Dharmalingam, G.; Carroll, T.; Amado, I.F.; Bruno, L.; Lee, K.; Walzer, T.; et al. microRNA-mediated regulation of mTOR complex components facilitates discrimination between activation and anergy in CD4 T cells. J. Exp. Med. 2014, 211, 2281–2295. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F.; Thomas, M.F.; Hu, J.K.; Yang, Z.; Babiarz, J.E.; Allen, C.D.; Matloubian, M.; Blelloch, R.; Ansel, K.M. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity 2011, 35, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtale, G.; Rubino, M.; Locati, M. MicroRNAs as Molecular Switches in Macrophage Activation. Front. Immunol. 2019, 10, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; McCurdy, S.; Huang, S.; Zhu, X.; Peplowska, K.; Tiirikainen, M.; Boisvert, W.A.; Garmire, L.X. Time Series miRNA-mRNA integrated analysis reveals critical miRNAs and targets in macrophage polarization. Sci. Rep. 2016, 6, 37446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kang, Y.; Zhang, H.; Zhao, D.; Xia, J.; Lu, Z.; Wang, H.; Xu, F.; Shi, L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015, 194, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.A.; So, A.Y.; Sinha, N.; Gibson, W.S.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Liu, L.; Li, X. Downregulation of miR-320 Alleviates Endoplasmic Reticulum Stress and Inflammatory Response in 3T3-L1 Adipocytes. Exp. Clin. Endocrinol. Diabetes 2021, 129, 131–137. [Google Scholar] [CrossRef]
- Arner, E.; Mejhert, N.; Kulyte, A.; Balwierz, P.J.; Pachkov, M.; Cormont, M.; Lorente-Cebrian, S.; Ehrlund, A.; Laurencikiene, J.; Heden, P.; et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 2012, 61, 1986–1993. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.M.; Guo, L.; Chi, M.H.; Sun, H.M.; Chen, X.W. Identification of active miRNA and transcription factor regulatory pathways in human obesity-related inflammation. BMC Bioinform. 2015, 16, 76. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wei, Z.; Wu, X.; Yang, H. Screening of exosomal miRNAs derived from subcutaneous and visceral adipose tissues: Determination of targets for the treatment of obesity and associated metabolic disorders. Mol. Med. Rep. 2018, 18, 3314–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijmans, J.G.; Diehl, K.J.; Bammert, T.D.; Kavlich, P.J.; Lincenberg, G.M.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Influence of Overweight and Obesity on Circulating Inflammation-Related microRNA. Microrna 2018, 7, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Q.; Xiao, X.; Wu, C.; Gao, R.; Peng, C.; Li, D.; Zhang, W.; Du, T.; Wang, Y.; et al. miR-1934, downregulated in obesity, protects against low-grade inflammation in adipocytes. Mol. Cell. Endocrinol. 2016, 428, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Ge, Q.; Gerard, J.; Noel, L.; Scroyen, I.; Brichard, S.M. MicroRNAs regulated by adiponectin as novel targets for controlling adipose tissue inflammation. Endocrinology 2012, 153, 5285–5296. [Google Scholar] [CrossRef] [Green Version]
- Runtsch, M.C.; Nelson, M.C.; Lee, S.H.; Voth, W.; Alexander, M.; Hu, R.; Wallace, J.; Petersen, C.; Panic, V.; Villanueva, C.J.; et al. Anti-inflammatory microRNA-146a protects mice from diet-induced metabolic disease. PLoS Genet. 2019, 15, e1007970. [Google Scholar] [CrossRef]
- Balasubramanyam, M.; Aravind, S.; Gokulakrishnan, K.; Prabu, P.; Sathishkumar, C.; Ranjani, H.; Mohan, V. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Mol. Cell. Biochem. 2011, 351, 197–205. [Google Scholar] [CrossRef]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef]
- Ye, E.A.; Liu, L.; Jiang, Y.; Jan, J.; Gaddipati, S.; Suvas, S.; Steinle, J.J. miR-15a/16 reduces retinal leukostasis through decreased pro-inflammatory signaling. J. Neuroinflamm. 2016, 13, 305. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.M.; Guerau-de-Arellano, M.; Costinean, S.; Williams, J.L.; Bottoni, A.; Mavrikis Cox, G.; Satoskar, A.R.; Croce, C.M.; Racke, M.K.; Lovett-Racke, A.E.; et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J. Immunol. 2012, 189, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Zhu, E.; Wang, X.; Zheng, B.; Wang, Q.; Hao, J.; Chen, S.; Zhao, Q.; Zhao, L.; Wu, Z.; Yin, Z. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORgammat and STAT3. J. Immunol. 2014, 192, 5599–5609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatandoost, N.; Amini, M.; Iraj, B.; Momenzadeh, S.; Salehi, R. Dysregulated miR-103 and miR-143 expression in peripheral blood mononuclear cells from induced prediabetes and type 2 diabetes rats. Gene 2015, 572, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.A.; Jin, W.; Villeneuve, L.; Wang, M.; Lanting, L.; Todorov, I.; Kato, M.; Natarajan, R. Pro-inflammatory role of microrna-200 in vascular smooth muscle cells from diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.A.; Das, S.; Zhuo, C.; Jin, W.; Wang, M.; Lanting, L.; Natarajan, R. Regulation of Vascular Smooth Muscle Cell Dysfunction Under Diabetic Conditions by miR-504. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 864–873. [Google Scholar] [CrossRef] [Green Version]
- InterAct, C.; Scott, R.A.; Langenberg, C.; Sharp, S.J.; Franks, P.W.; Rolandsson, O.; Drogan, D.; van der Schouw, Y.T.; Ekelund, U.; Kerrison, N.D.; et al. The link between family history and risk of type 2 diabetes is not explained by anthropometric, lifestyle or genetic risk factors: The EPIC-InterAct study. Diabetologia 2013, 56, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Parrillo, L.; Spinelli, R.; Longo, M.; Desiderio, A.; Mirra, P.; Nigro, C.; Fiory, F.; Hedjazifar, S.; Mutarelli, M.; Carissimo, A.; et al. Altered PTPRD DNA methylation associates with restricted adipogenesis in healthy first-degree relatives of Type 2 diabetes subjects. Epigenomics 2020, 12, 873–888. [Google Scholar] [CrossRef]
- Henninger, A.M.; Eliasson, B.; Jenndahl, L.E.; Hammarstedt, A. Adipocyte hypertrophy, inflammation and fibrosis characterize subcutaneous adipose tissue of healthy, non-obese subjects predisposed to type 2 diabetes. PLoS ONE 2014, 9, e105262. [Google Scholar] [CrossRef]
- Weyer, C.; Foley, J.E.; Bogardus, C.; Tataranni, P.A.; Pratley, R.E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar] [CrossRef] [Green Version]
- Lonn, M.; Mehlig, K.; Bengtsson, C.; Lissner, L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010, 24, 326–331. [Google Scholar] [CrossRef]
- Spinelli, R.; Florese, P.; Parrillo, L.; Zatterale, F.; Longo, M.; D’Esposito, V.; Desiderio, A.; Nerstedt, A.; Gustafson, B.; Formisano, P.; et al. ZMAT3 hypomethylation contributes to early senescence of preadipocytes from healthy first-degree relatives of type 2 diabetics. Aging Cell 2022. [Google Scholar] [CrossRef] [PubMed]
- Parrillo, L.; Spinelli, R.; Costanzo, M.; Florese, P.; Cabaro, S.; Desiderio, A.; Prevenzano, I.; Raciti, G.A.; Smith, U.; Miele, C.; et al. Epigenetic Dysregulation of the Homeobox A5 (HOXA5) Gene Associates with Subcutaneous Adipocyte Hypertrophy in Human Obesity. Cells 2022, 11, 728. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Raciti, G.A.; Zatterale, F.; Parrillo, L.; Desiderio, A.; Spinelli, R.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Nigro, C.; et al. Epigenetic modifications of the Zfp/ZNF423 gene control murine adipogenic commitment and are dysregulated in human hypertrophic obesity. Diabetologia 2018, 61, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiderio, A.; Longo, M.; Parrillo, L.; Campitelli, M.; Cacace, G.; De Simone, S.; Spinelli, R.; Zatterale, F.; Cabaro, S.; Dolce, P.; et al. Epigenetic silencing of the ANKRD26 gene correlates to the pro-inflammatory profile and increased cardio-metabolic risk factors in human obesity. Clin. Epigenetics 2019, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Desiderio, A.; Spinelli, R.; Nigro, C.; Longo, M.; Parrillo, L.; D’Esposito, V.; Carissimo, A.; Hedjazifar, S.; Smith, U.; et al. Adipocyte precursor cells from first degree relatives of type 2 diabetic patients feature changes in hsa-mir-23a-5p, -193a-5p, and -193b-5p and insulin-like growth factor 2 expression. FASEB J. 2021, 35, e21357. [Google Scholar] [CrossRef]
- Tsai, F.J.; Yang, C.F.; Chen, C.C.; Chuang, L.M.; Lu, C.H.; Chang, C.T.; Wang, T.Y.; Chen, R.H.; Shiu, C.F.; Liu, Y.M.; et al. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet. 2010, 6, e1000847. [Google Scholar] [CrossRef] [Green Version]
- Pike, K.A.; Tremblay, M.L. Protein Tyrosine Phosphatases: Regulators of CD4 T Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2504. [Google Scholar] [CrossRef]
- Hale, A.J.; Ter Steege, E.; den Hertog, J. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev. Biol. 2017, 428, 283–292. [Google Scholar] [CrossRef]
- Vilborg, A.; Glahder, J.A.; Wilhelm, M.T.; Bersani, C.; Corcoran, M.; Mahmoudi, S.; Rosenstierne, M.; Grander, D.; Farnebo, M.; Norrild, B.; et al. The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc. Natl. Acad. Sci. USA 2009, 106, 15756–15761. [Google Scholar] [CrossRef] [Green Version]
- Parrillo, L.; Costa, V.; Raciti, G.A.; Longo, M.; Spinelli, R.; Esposito, R.; Nigro, C.; Vastolo, V.; Desiderio, A.; Zatterale, F.; et al. Hoxa5 undergoes dynamic DNA methylation and transcriptional repression in the adipose tissue of mice exposed to high-fat diet. Int. J. Obes. 2016, 40, 929–937. [Google Scholar] [CrossRef]
- Cao, W.; Zhang, T.; Feng, R.; Xia, T.; Huang, H.; Liu, C.; Sun, C. Hoxa5 alleviates obesity-induced chronic inflammation by reducing ER stress and promoting M2 macrophage polarization in mouse adipose tissue. J. Cell. Mol. Med. 2019, 23, 7029–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, B.; Shao, M.; Zhang, Q.; Hepler, C.; Paschoal, V.A.; Barnes, S.D.; Vishvanath, L.; An, Y.A.; Jia, L.; Malladi, V.S.; et al. Perivascular mesenchymal cells control adipose-tissue macrophage accrual in obesity. Nat. Metab. 2020, 2, 1332–1349. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Spinelli, R.; Desiderio, A.; Longo, M.; Parrillo, L.; Nigro, C.; D’Esposito, V.; Mirra, P.; Fiory, F.; Pilone, V.; et al. Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity. Sci. Rep. 2017, 7, 43526. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lin, L.; Li, Q.; Liu, K.; Huang, Y.; Wang, X.; Cao, K.; Chen, X.; Cao, W.; Li, F.; et al. IGF-2 Preprograms Maturing Macrophages to Acquire Oxidative Phosphorylation-Dependent Anti-inflammatory Properties. Cell Metab. 2019, 29, 1363–1375. [Google Scholar] [CrossRef]
- Woo, C.Y.; Jang, J.E.; Lee, S.E.; Koh, E.H.; Lee, K.U. Mitochondrial Dysfunction in Adipocytes as a Primary Cause of Adipose Tissue Inflammation. Diabetes Metab. J. 2019, 43, 247–256. [Google Scholar] [CrossRef]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef]
- Arguelles, A.O.; Meruvu, S.; Bowman, J.D.; Choudhury, M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discov. Today 2016, 21, 499–509. [Google Scholar] [CrossRef]
- Stathis, A.; Hotte, S.J.; Chen, E.X.; Hirte, H.W.; Oza, A.M.; Moretto, P.; Webster, S.; Laughlin, A.; Stayner, L.A.; McGill, S.; et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin. Cancer Res. 2009, 27, 3528. [Google Scholar] [CrossRef]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Lundh, M.; Galbo, T.; Poulsen, S.S.; Mandrup-Poulsen, T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metab. 2015, 17, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhou, R.; Wang, B.; Mi, M.T. Effect of resveratrol on glucose control and insulin sensitivity: A meta-analysis of 11 randomized controlled trials. Am. J. Clin. Nutr. 2014, 99, 1510–1519. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, D.; Ghosh, S. Regulation of the NF-kappaB-Mediated Transcription of Inflammatory Genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Pivari, F.; Mingione, A.; Brasacchio, C.; Soldati, L. Curcumin and Type 2 Diabetes Mellitus: Prevention and Treatment. Nutrients 2019, 11, 1837. [Google Scholar] [CrossRef] [Green Version]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; He, Y.; Robinson, V.; Yang, Z.; Hessler, P.; Lasko, L.M.; Lu, X.; Bhathena, A.; Lai, A.; Uziel, T.; et al. Targeting Lineage-specific MITF Pathway in Human Melanoma Cell Lines by A-485, the Selective Small-molecule Inhibitor of p300/CBP. Mol. Cancer Ther. 2018, 17, 2543–2550. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.L.; Wang, Y.M.; Zhou, X.X.; Yao, H.P.; Zheng, S.; Xiang, Z.; Ji, F. An inhibitor of the acetyltransferases CBP/p300 exerts antineoplastic effects on gastrointestinal stromal tumor cells. Oncol. Rep. 2016, 36, 2763–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landman, S.; van der Horst, C.; van Erp, P.E.J.; Joosten, I.; de Vries, R.; Koenen, H. Immune responses to azacytidine in animal models of inflammatory disorders: A systematic review. J. Transl. Med. 2021, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Nam, K.H.; Kim, J.; Baek, M.W.; Park, J.E.; Park, H.Y.; Kwon, H.J.; Kwon, O.S.; Kim, D.Y.; Oh, G.T. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2404–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, T.; Lugrin, J.; Le Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, F.U.; Rehman, M.S.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [Green Version]

| Study Model | Epigenetic Marks | Position | Processes | Medical Condition | Species | Ref |
|---|---|---|---|---|---|---|
| SAT APC and PBL from FDR, PBL from Obese subjects | DNA Hypo-methylation | Global/PTPRD | Inflammation by chemokine and cytokine pathway and Adipogenesis/PTPs | SAT Hypertrophy, Familiarity for T2D, Obesity | Human | [117] |
| SAT APC from FDR | DNA Hypo-methylation | ZMAT3 | Pro-Inflammatory markers: IL-6, MCP-1, RANTES, IL-8, MIP1b/Senescence and Aging | SAT Hypertrophy, Familiarity for T2D | Human | [121] |
| SAT APC and PBL from FDR, PBL from Obese subjects | DNA Hyper-methylation | HOXA5 | WNT-Signaling Pathway, Adipogenesis | SAT Hypertrophy, Familiarity for T2D, Obesity | Human | [122] |
| SAT APC from Human Hypertrophic Obesity | DNA Hyper-methylation | ZNF423 | Adipogenesis | SAT Hypertrophy | Human | [123] |
| PBL from Obese Subjects | DNA Hyper-methylation | ANKRD26 | Adipocyte Pro-Inflammatory Markers: IL-1β, IL-6, IL-12, IL-8, IP-10, MIP-1α, MIP-1β, RANTES | Obesity, Cardio- metabolic Risk | Human | [124] |
| SAT APC and PBL from FDR | Deregulation of miRNA expression | Mir-23a-5p, mir193a-5p, mir-193b-5p | Pro-inflammatory pathway, adipogenesis, IGF2 signaling | SAT Hypertrophy, Familiarity for T2D | Human | [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zatterale, F.; Raciti, G.A.; Prevenzano, I.; Leone, A.; Campitelli, M.; De Rosa, V.; Beguinot, F.; Parrillo, L. Epigenetic Reprogramming of the Inflammatory Response in Obesity and Type 2 Diabetes. Biomolecules 2022, 12, 982. https://doi.org/10.3390/biom12070982
Zatterale F, Raciti GA, Prevenzano I, Leone A, Campitelli M, De Rosa V, Beguinot F, Parrillo L. Epigenetic Reprogramming of the Inflammatory Response in Obesity and Type 2 Diabetes. Biomolecules. 2022; 12(7):982. https://doi.org/10.3390/biom12070982
Chicago/Turabian StyleZatterale, Federica, Gregory Alexander Raciti, Immacolata Prevenzano, Alessia Leone, Michele Campitelli, Veronica De Rosa, Francesco Beguinot, and Luca Parrillo. 2022. "Epigenetic Reprogramming of the Inflammatory Response in Obesity and Type 2 Diabetes" Biomolecules 12, no. 7: 982. https://doi.org/10.3390/biom12070982