
Genetic and Metabolic Determinants of Atrial Fibrillation in a General Population Sample: The CHRIS Study

 , , ,
, , ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. CHRIS Study and Phenotype Construction
2.2. Genome-Wide Association Study (GWAS)
2.3. Familial Aggregation
2.4. Sanger Sequencing
2.5. Targeted Metabolomics
2.6. Total Cholesterol, HDL and LDL
3. Results
3.1. AF Cases in CHRIS Study
3.2. Genome-Wide Association Study: Two Novel Loci Associated with AF
3.3. Familial Aggregation
3.4. PBX1 and PCCA Genes in a Family with AF
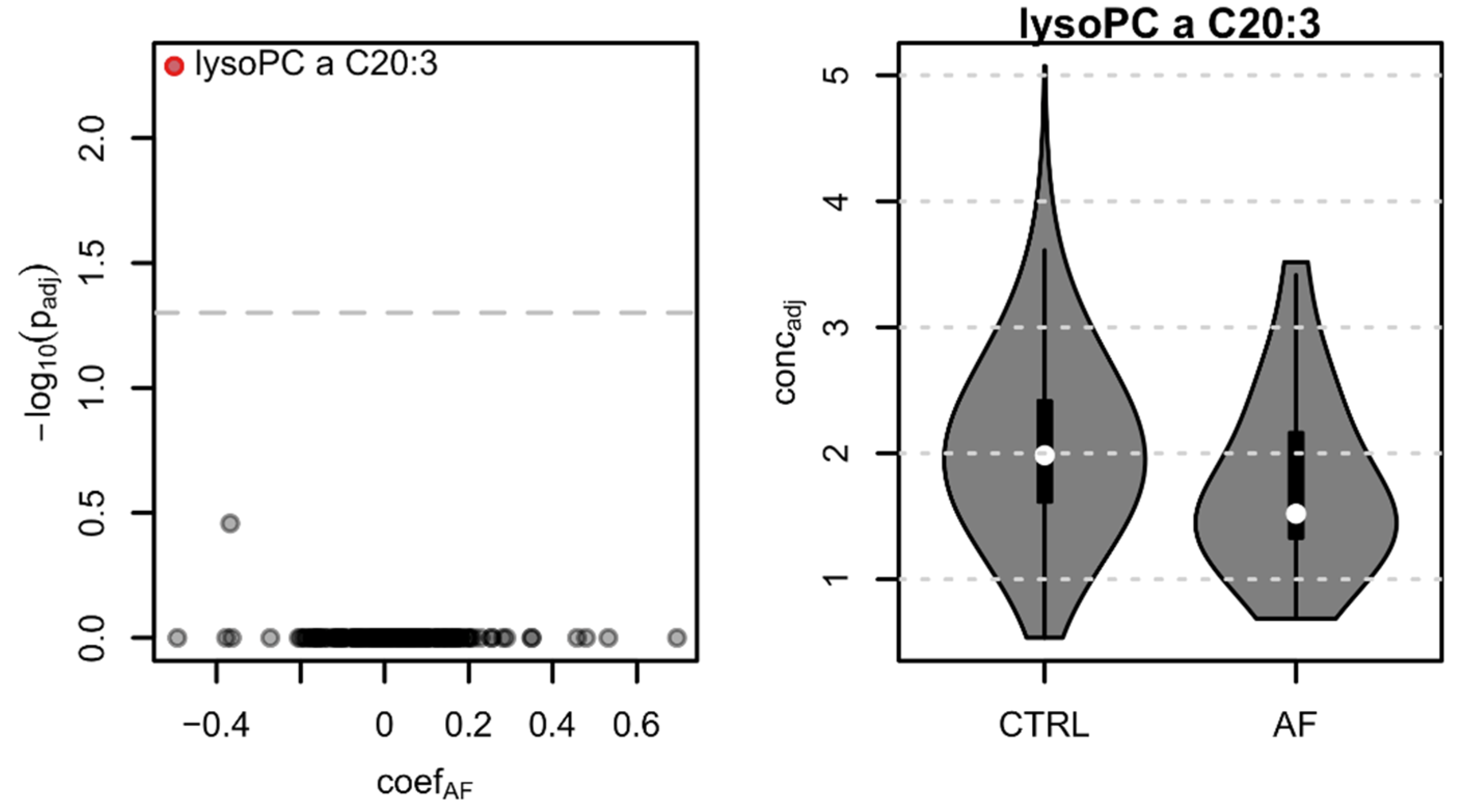
3.5. Lysophosphatidylcholine lysoPC a C20:3, HDL, LDL, and Total Cholesterol Are Reduced in AF Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867. [Google Scholar] [CrossRef]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castellá, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart J. 2016, 37, 2893–2962. [Google Scholar] [CrossRef] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Kopecky, S.; Gersh, B.; McGoon, M.; Whisnant, J.P.; Holmes, D.R.; Ilstrup, D.M.; Frye, R.L. The natural history of lone atrial fibrillation. A population-based study over three decades. N. Eng. J. Med. 1987, 317, 669–674. [Google Scholar] [CrossRef]
- Fox, C.S.; Parise, H.; D’Agostino, R.B.; Lloyd-Jones, D.M.; Vasan, R.S.; Wang, T.J.; Levy, D.; Wolf, P.A.; Benjamin, E.J. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA 2004, 291, 2851–2855. [Google Scholar] [CrossRef]
- Lubitz, S.A.; Yin, X.; Fontes, J.D.; Magnani, J.W.; Rienstra, M.; Pai, M.; Villalon, M.L.; Vasan, R.S.; Pencina, M.J.; Levy, D.; et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA 2010, 304, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.H.; Andreasen, L.; Olesen, M.S. Atrial fibrillation-a complex polygenetic disease. Eur. J. Hum. Genet. 2021, 29, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; De Souza, A.; et al. Combined metabolomics and proteomic analysis of human atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Yu, B.; Qureshi, W.T.; Grams, M.E.; Selvin, E.; Soliman, E.Z.; Loehr, L.R.; Chen, L.Y.; Agarwal, S.K.; Alexander, D.; et al. Metabolomics and incidence of atrial fibrillation in African Americans: The atherosclerosis risk in communities (ARIC) study. PLoS ONE 2015, 10, e0142610. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Tanabe, N.; Yagihara, N.; Watanabe, T.; Aizawa, Y.; Kodama, M. Association between lipid profile and risk of atrial fibrillation. Circ. J. 2011, 75, 2767–2774. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Cho, Y.; Kim, N.; Oh, I.Y.; Kang, S.W.; Choi, E.K.; Hwang, G.S. Lipidomic profiling reveals free fatty acid alterations in plasma from patients with atrial fibrillation. PLoS ONE 2018, 13, e0196709. [Google Scholar] [CrossRef] [Green Version]
- Pattaro, C.; Gögele, M.; Mascalzoni, D.; Melotti, R.; Schwienbacher, C.; De Grandi, A.; Foco, L.; D’Elia, Y.; Linder, B.; Fuchsberger, C.; et al. The Cooperative Health Research in South Tyrol (CHRIS) study: Rationale, objectives, and preliminary results. J. Transl. Med. 2015, 13, 348. [Google Scholar] [CrossRef] [PubMed]
- Noce, D.; Gögele, M.; Schwienbacher, C.; Caprioli, G.; De Grandi, A.; Foco, L.; Platzgummer, S.; Pramstaller, P.P.; Pattaro, C. Sequential recruitment of study participants may inflate genetic heritability estimates. Hum. Genet. 2017, 136, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Murgia, F.; Melotti, R.; Foco, L.; Gögele, M.; Meraviglia, V.; Motta, B.; Steger, A.; Toifl, M.; Sinnecker, D.; Müller, A.; et al. Effects of smoking status, history and intensity on heart rate variability in the general population: The CHRIS study. PLoS ONE 2019, 14, e0215053. [Google Scholar] [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef] [Green Version]
- Fuchsberger, C.; Abecasis, G.R.; Hinds, D.A. minimac2: Faster genotype imputation. Bioinformatics 2015, 31, 782–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Nielsen, J.B.; Fritsche, L.G.; Dey, R.; Gabrielsen, M.E.; Wolford, B.N.; LeFaive, J.; VandeHaar, P.; Gagliano, S.A.; Gifford, A.; et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018, 50, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Rainer, J.; Taliun, D.; D’Elia, Y.; Pattaro, C.; Domingues, F.S.; Weichenberger, C.X. FamAgg: An R package to evaluate familial aggregation of traits in large pedigrees. Bioinformatics 2016, 32, 1583–1585. [Google Scholar] [CrossRef]
- Weichenberger, C.X.; Rainer, J.; Pattaro, C.; Pramstaller, P.P.; Domingues, F.S. Comparative assessment of different familial aggregation methods in the context of large and unstructured pedigrees. Bioinformatics 2019, 35, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Thiele, H.; Nürnberg, P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics 2005, 21, 1730–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Fujiki, A.; Origasa, H.; Ogawa, S.; Okumura, K.; Kubota, I.; Aizawa, Y.; Yamashita, T.; Atarashi, H.; Horie, M.; et al. Prevalence of atrial fibrillation in the general population of Japan: An analysis based on periodic health examination. Int. J. Cardiol. 2009, 137, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Barrios, V.; Calderón, A.; Escobar, C.; de la Figuera, M. Patients with atrial fibrillation in a primary care setting: Val-FAAP study. Rev. Esp. Cardiol. 2012, 65, 47–53. [Google Scholar] [CrossRef]
- Heeringa, J.; van der Kuip, D.A.M.; Hofman, A.; Kors, J.A.; van Herpen, G.; Stricker, B.H.C.; Stijnen, T.; Lip, G.Y.H.; Witteman, J.C.M. Prevalence, incidence and lifetime risk of atrial fibrillation: The Rotterdam study. Eur. Heart J. 2006, 27, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Bonhorst, D.; Mendes, M.; Adragão, P.; De Sousa, J.; Primo, J.; Leiria, E.; Rocha, P. Prevalence of atrial fibrillation in the Portuguese population aged 40 and over: The FAMA study. Rev. Port. Cardiol. 2010, 29, 331–350. [Google Scholar]
- Friberg, L.; Bergfeldt, L. Atrial fibrillation prevalence revisited. J. Intern. Med. 2013, 274, 461–468. [Google Scholar] [CrossRef]
- Wilke, T.; Groth, A.; Mueller, S.; Pfannkuche, M.; Verheyen, F.; Linder, R.; Maywald, U.; Bauersachs, R.; Breithardt, G. Incidence and prevalence of atrial fibrillation: An analysis based on 8.3 million patients. Europace 2013, 15, 486–493. [Google Scholar] [CrossRef]
- Pistoia, F.; Sacco, S.; Tiseo, C.; Degan, D.; Ornello, R.; Carolei, A. The epidemiology of atrial fibrillation and stroke. Cardiol. Clin. 2016, 34, 255–268. [Google Scholar] [CrossRef]
- Pérez-Villacastín, J.; Castellano, N.P.; Planas, J.M. Epidemiology of atrial fibrillation in Spain in the past 20 years. Rev. Esp. Cardiol. 2013, 66, 561–565. [Google Scholar] [CrossRef]
- Mitt, M.; Kals, M.; Pärn, K.; Gabriel, S.B.; Lander, E.S.; Palotie, A.; Ripatti, S.; Morris, A.P.; Metspalu, A.; Esko, T.; et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 2017, 25, 869–876. [Google Scholar] [CrossRef]
- Kreiner-Møller, E.; Medina-Gomez, C.; Uitterlinden, A.G.; Rivadeneira, F.; Estrada, K. Improving accuracy of rare variant imputation with a two-step imputation approach. Eur. J. Hum. Genet. 2015, 23, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Christophersen, I.E.; Rienstra, M.; Roselli, C.; Yin, X.; Geelhoed, B.; Barnard, J.; Lin, H.; Arking, D.E.; Smith, A.V.; Albert, C.M.; et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat. Genet. 2017, 49, 946–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roselli, C.; Chaffin, M.D.; Wenig, L.C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 2018, 50, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Stankunas, K.; Shang, C.; Kao, S.C.; Twu, K.Y.; Cleary, M.L. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development 2008, 135, 3577–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alankarage, D.; Szot, J.O.; Pachter, N.; Slavotinek, A.; Selleri, L.; Shieh, J.T.; Winlaw, D.; Giannoulatou, E.; Chapman, G.; Dunwoodie, S.L. Functional characterization of a novel PBX1 de novo missense variant identified in a patient with syndromic congenital heart disease. Hum. Mol. Genet. 2020, 29, 1068–1082. [Google Scholar] [CrossRef]
- Lin, H.; Yin, X.; Lunetta, K.L.; Dupuis, J.; McManus, D.D.; Lubitz, S.A.; Magnani, J.W.; Joehanes, R.; Munson, P.J.; Larson, M.G.; et al. Whole blood gene expression and atrial fibrillation: The Framingham Heart Study. PLoS ONE 2014, 9, e96794. [Google Scholar] [CrossRef] [PubMed]
- Bodi, I.; Grünert, S.C.; Becker, N.; Stoelzle-Feix, S.; Spiekerkoetter, U.; Zehender, M.; Bugger, H.; Bode, C.; Odening, K.E. Mechanisms of acquired long QT syndrome in patients with propionic acidemia. Heart Rhythm 2016, 13, 1335–1345. [Google Scholar] [CrossRef]
- Benito, B.; Brugada, R.; Perich, R.M.; Lizotte, E.; Cinca, J.; Mont, L.; Berruezo, A.; Tolosana, J.M.; Freixa, X.; Brugada, P.; et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 2008, 5, 1434–1440. [Google Scholar] [CrossRef]
- Olesen, M.S.; Yuan, L.; Liang, B.; Holst, A.G.; Nielsen, N.; Nielsen, J.B.; Hedley, P.L.; Christiansen, M.; Olesen, S.P.; Haunso, S.; et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 2012, 5, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, M.D.; Duverger, J.E.; Naud, P.; Chartier, D.; Qi, X.Y.; Comtois, P.; Fabritz, L.; Kirchhof, P.; Nattel, S. Arrhythmogenic left atrial cellular electrophysiology in a murine genetic long QT syndrome model. Cardiovasc. Res. 2011, 92, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamayo, M.; Fulgencio-Covián, A.; Navarro-García, J.A.; Val-Blasco, A.; Ruiz-Hurtado, G.; Gil-Fernández, M.; Martín-Nunes, L.; Lopez, J.A.; Desviat, L.-R.; Delgado, C.; et al. Intracellular calcium mishandling leads to cardiac dysfunction and ventricular arrhythmias in a mouse model of propionic acidemia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165586. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.M.; Franz, M.R.; Clopton, P.; Pruvot, E.J.; Krummen, D.E. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 2011, 123, 2922–2930. [Google Scholar] [CrossRef] [Green Version]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Schondube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.E.; Abu-Taha, I.H.; Wang, Q.; Rosello-Diez, E.; Kamler, M.; Nattel, S.; Ravens, U.; Wehrens, X.H.T.; Hove-Madsen, L.; Heijman, J.; et al. Profibrotic, electrical, and calcium-handling remodeling of the atria in heart failure patients with and without atrial fibrillation. Front. Physiol. 2018, 9, 1383. [Google Scholar] [CrossRef] [Green Version]
- Del Greco, M.F.; Foco, L.; Teumer, A.; Verweij, N.; Paglia, G.; Meraviglia, V.; Melotti, R.; Vukovic, V.; Rauhe, W.; Joshi, P.K.; et al. Lipidomics, atrial conduction, and body mass index. Circ. Genom. Precis. Med. 2019, 12, e002384. [Google Scholar] [CrossRef] [Green Version]
- Magnani, J.W.; Williamson, M.A.; Ellinor, P.T.; Monahan, K.M.; Benjamin, E.J. P wave indices: Current status and future directions in epidemiology, clinical, and research applications. Circ. Arrhythm. Electrophysiol. 2009, 2, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Trieb, M.; Kornej, J.; Knuplez, E.; Hindricks, G.; Thiele, H.; Sommer, P.; Scharnagl, H.; Dagres, N.; Dinov, B.; Bollmann, A.; et al. Atrial fibrillation is associated with alterations in HDL function, metabolism, and particle number. Basic Res. Cardiol. 2019, 114, 27. [Google Scholar] [CrossRef]
- Gruppen, E.G.; Connelly, M.A.; Otvos, J.D.; Bakker, S.J.L.; Dullaart, R.P.F. A novel protein glycan biomarker and LCAT activity in metabolic syndrome. Eur. J. Clin. Investig. 2015, 45, 850–859. [Google Scholar] [CrossRef]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Fender, A.C.; Kleeschulte, S.; Stolte, S.; Leineweber, K.; Kamler, M.; Bode, J.; Li, N.; Dobrev, D. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res. Cardiol. 2020, 115, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Tu, D.; Liu, X.; Niu, S.; Suo, Y.; Liu, T.; Li, G.; Liu, C. Role of NLRP3-inflammasome/Caspase-1/Galectin-3 pathway on atrial remodeling in diabetic rabbits. J. Cardiovasc. Transl. Res. 2020, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular basis of atrial fibrillation pathophysiology and therapy: A translational perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef]
- Li, W.L.; Hua, L.G.; Qu, P.; Yan, W.H.; Ming, C.; Jun, Y.D.; Yuan, L.D.; Nan, N. NLRP3 inflammasome: A novel link between lipoproteins and atherosclerosis. Arch. Med. Sci. 2016, 12, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence 5′–3′ | Used for Sequencing |
|---|---|---|
| rs528903211-F | GCCCAGACCGTGAAAACATT | |
| rs528903211-R | GGCATTTCGTCTCTGCCCTAG | Yes |
| rs573729400-F | ACAATCTGATTTGCCCAGCG | |
| rs573729400-R | CATGGGATGGGTCAAATTCTATGC | Yes |
| rs745582874-F | CCACTGATGATGGCCTTGG | Yes |
| rs745582874-R | GACATGGTCCTAGAGCTGCT | |
| rs768476991-F | TCTGTTCACACGGCTTAGATGA | Yes |
| rs768476991-R | GGTAATCCAACACTGTCCAAACA |
| Cases (n = 110) | Controls (n = 10,399) | ||||||
|---|---|---|---|---|---|---|---|
| Missings, n (%) | Mean | Min/Max | Missings, n (%) | Mean | Min/Max | p-Value | |
| (SD) | (SD) | ||||||
| Age at participation—years | 0 | 66.51 | 18/93 | 0 | 45.78 | 18/94 | <0.001 |
| (19.66) | (16.1) | ||||||
| Sex, n (%) | |||||||
| male | 67 (60.9%) | 4642 (45%) | 0.001 | ||||
| female | 43 (39.1%) | 5757 (55%) | |||||
| Do you have atrial fibrillation diagnosed by doctor? n (%) | |||||||
| yes | 50 (45.5%) | 51 (0.5%) | <0.001 | ||||
| no | 60 (54.5%) | 10,334 (99.4%) | |||||
| missing | 0 | 14 (0.1%) | |||||
| P wave (ms) | 0 | 13.42 | 0/150 | 71 (0.7%) | 105.93 | 46/208 | <0.001 |
| (37.46) | (12.49) | ||||||
| P wave = 0, n (%) | |||||||
| yes | 97 (88.2%) | 0 | <0.001 | ||||
| no | 13 (11.8%) | 10,328 (99.3%) | |||||
| missing | 0 | 71 (0.7%) | |||||
| Medication corresponding to the ATC classification group “C”, n (%) | |||||||
| yes | 76 (69.1%) | 1590 (15.3%) | <0.001 | ||||
| no | 34 (30.9%) | 8420 (81%) | |||||
| missing | 0 | 389 (3.7%) | |||||
| Body Mass Index—kg/m2 | 5 (4.5%) | 27.83 | 18.3/40.2 | 65 (0.6%) | 25.82 | 15.5/55.1 | <0.001 |
| (4.69) | (4.6) | ||||||
| Systolic BP, mean of 3 measurements—mm/Hg | 5 (4.5%) | 131.45 | 92/175 | 273 (2.6%) | 122.16 | 81/226 | <0.001 |
| (18.5) | (16.37) | ||||||
| Diastolic BP, mean of 3 measurements—mm/Hg | 5 (4.5%) | 81.13 | 59/122 | 273 (2.6%) | 78.26 | 51/145 | 0.002 |
| (11.43) | (9.32) | ||||||
| Cholesterol total—mg/dL | 0 | 196.98 | 95/287 | 6 (0.06%) | 211.26 | 87/586 | 0.0004 |
| (38.79) | (41.72) | ||||||
| Cholesterol High Density Lipoprotein (HDL)—mg/dL | 0 | 56.62 | 27/92 | 7 (0.07%) | 61.13 | 20/147 | 0.0028 |
| (15.35) | (15.78) | ||||||
| Cholesterol Low Density Lipoprotein (LDL)—mg/dL | 0 | 120.14 | 31/220 | 8 (0.08%) | 131.42 | 24/332 | 0.0016 |
| (35.71) | (37.27) | ||||||
| C-Reactive Protein (CRP)—mg/dL | 0 | 0.42 | 0.02/2.85 | 5 (0.05%) | 0.27 | 0/17.24 | 0.002 |
| (0.49) | (0.48) | ||||||
| Creatinine—mg/dL | 0 | 1.02 | 0.68/2.29 | 6 (0.06%) | 0.88 | 0.36/8.69 | <0.001 |
| (0.26) | (0.17) | ||||||
| eGFR—mL/min/1.73 m2 | 0 | 73.24 | 26.06/134.25 | 7 (0.07%) | 91.9 | 6.19/137.17 | <0.001 |
| (21.97) | (16.12) | ||||||
| Smoking habit, n (%) | |||||||
| never | 60 (54.5%) | 5540 (53.3%) | 0.042 | ||||
| past | 39 (35.5%) | 2975 (28.6%) | |||||
| current | 10 (9.1%) | 1836 (17.6%) | |||||
| missing | 1 (0.9%) | 48 (0.5%) | |||||
| Has a doctor ever said that you have high blood pressure or hypertension? n (%) | |||||||
| yes | 56 (51%) | 2337 (22.5%) | <0.001 | ||||
| no | 53 (48%) | 8006 (77.0%) | |||||
| missing | 1 (0.9%) | 56 (0.5%) | |||||
| Do you have diabetes mellitus? n (%) | |||||||
| yes | 11 (10.0%) | 269 (2.6%) | <0.001 | ||||
| no | 98 (89.1%) | 10,111 (97.2%) | |||||
| missing | 1 (0.9%) | 19 (0.2%) | |||||
| Has a doctor ever told you that you have a heart failure? n (%) | |||||||
| yes | 17 (15.4%) | 50 (0.4%) | <0.001 | ||||
| no | 86 (78.2%) | 10,286 (99.0%) | |||||
| missing | 7 (6.4%) | 63 (0.6%) | |||||
| Have you ever been told by a doctor that you had a stroke? n (%) | |||||||
| yes | 13 (11.8%) | 88 (0.7%) | <0.001 | ||||
| no | 96 (87.3%) | 10,278 (99.0%) | |||||
| missing | 1 (0.9%) | 33 (0.3%) | |||||
| Have you ever been told by a doctor that you had a myocardial infarction? n (%) | |||||||
| yes | 9 (8.2%) | 118 (0.1%) | <0.001 | ||||
| no | 98 (89.1%) | 10,257 (98.6%) | |||||
| missing | 3 (2.7%) | 24 (0.3%) | |||||
| Chr | POS (GRCh37) | REF/ALT | rsID | MAF | MAC | p-value | R2 |
|---|---|---|---|---|---|---|---|
| 1 | 164062528 | C/G | rs528903211 | 0.00050 | 10 | 1.1 × 10−8 | 0.47 |
| 1 | 164110550 | G/A | rs573729400 | 0.00056 | 9 | 2.4 × 10−8 | 0.43 |
| 1 | 165331300 | T/G | rs745582874 | 0.00066 | 13 | 4.3 × 10−8 | 0.81 |
| 13 | 101142909 | C/T | rs768476991 | 0.00028 | 5 | 4.1 × 10−8 | 0.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emmert, D.B.; Vukovic, V.; Dordevic, N.; Weichenberger, C.X.; Losi, C.; D’Elia, Y.; Volpato, C.; Hernandes, V.V.; Gögele, M.; Foco, L.; et al. Genetic and Metabolic Determinants of Atrial Fibrillation in a General Population Sample: The CHRIS Study. Biomolecules 2021, 11, 1663. https://doi.org/10.3390/biom11111663
Emmert DB, Vukovic V, Dordevic N, Weichenberger CX, Losi C, D’Elia Y, Volpato C, Hernandes VV, Gögele M, Foco L, et al. Genetic and Metabolic Determinants of Atrial Fibrillation in a General Population Sample: The CHRIS Study. Biomolecules. 2021; 11(11):1663. https://doi.org/10.3390/biom11111663
Chicago/Turabian StyleEmmert, David B., Vladimir Vukovic, Nikola Dordevic, Christian X. Weichenberger, Chiara Losi, Yuri D’Elia, Claudia Volpato, Vinicius V. Hernandes, Martin Gögele, Luisa Foco, and et al. 2021. "Genetic and Metabolic Determinants of Atrial Fibrillation in a General Population Sample: The CHRIS Study" Biomolecules 11, no. 11: 1663. https://doi.org/10.3390/biom11111663