
Mitochondrial Neurodegenerative Diseases: Three Mitochondrial Ribosomal Proteins as Intermediate Stage in the Pathway That Associates Damaged Genes with Alzheimer’s and Parkinson’s


Abstract
:Simple Summary
Abstract
1. Introduction
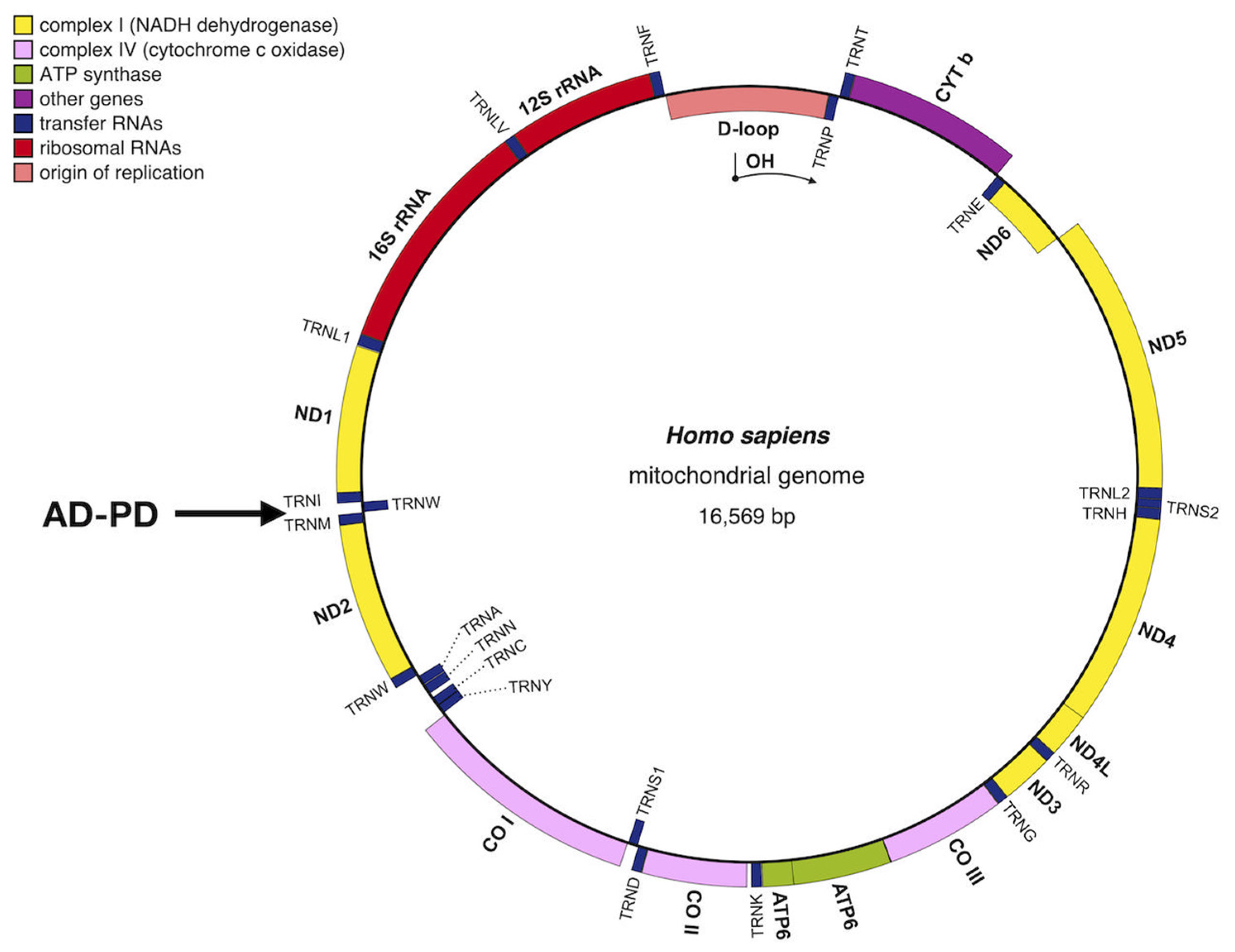
2. Mitochondrial Neurodegenerative Diseases

{kind=link}
{kind=link}
| Mitochondrial Pathology | mtDNA Mutations | Clinical Syndromes | References |
|---|---|---|---|
| CPEO | single deletion | Loss of the muscle functions involved in eye and eyelid movement | [51,52] |
| KSS | single deletion | Neuromuscular disorder | [53,54] |
| PS | single deletion | It affects various parts of the body, especially bone marrow and the pancreas | [55,56] |
| Diabetes and deafness | single deletion | Hyperglycemia and reduction or absence of hearing ability | [57,58] |
| Encephalomyopathy | multiple deletions | Muscle weakness and pain, recurrent headaches, loss of appetite, vomiting and seizures | [59,60] |
| Recurrent myoglobinuria | multiple deletions | Metabolic disturbances that include hypokalemia, hypophosphatemia, hyponatremia, hypocalcemia and hypernatremia | [61,62] |
| SANDO | multiple deletions | Impaired coordination (ataxia), slurred speech (dysarthria) and weakness of the eye muscles (ophthalmoparesis) | [63,64] |
| LHON | point mutation | Progressive visual loss due to optic neuropathy | [65,66] |
| MELAS | point mutation | Disease primarily affecting the nervous system and muscles | [67,68] |
| NARP | point mutation | Neurogenic muscle weakness, sensory-motor neuropathy, ataxia and pigmentary retinopathy | [69,70] |
| MERRF | point mutation | Progressive myoclonus and seizures, cerebellar ataxia, myopathy, cardiac arrhythmia, sensorineural hearing loss, optic atrophy and dementia | [71,72] |
| CPEO | point mutation | Loss of the muscle functions involved in eye and eyelid movement | [19,73] |
| Leigh syndrome | point mutation | Neurological disorder involving elevated blood and/or cerebrospinal fluid levels of lactate, developmental retardation, hypotonia, followed by respiratory dysfunction, epileptic seizures, poor feeding and weakness | [74,75] |
| AD | nuclear gene mutation | Brain disorder that slowly destroys memory and thinking skills and, eventually, the ability to carry out the simplest tasks | [41,76] |
| PD | nuclear gene mutation | Combinations of motor problems—namely, bradykinesia, resting tremor, rigidity, flexed posture, “freezing,” and loss of postural reflexes | [76,77] |
| FRDA | nuclear gene mutation | Progressive ataxia, absent lower limb reflexes, upgoing plantar responses and peripheral sensory neuropathy. | [76,78] |
| HD | nuclear gene mutation | Disorder that causes nerve cells (neurons) in parts of the brain to gradually break down and die | [76,79] |
| ALS | nuclear gene mutation | Progressive nervous system disease that affects nerve cells in the brain and spinal cord, causing loss of muscle control | [76,80] |
| HSP | nuclear gene mutation | Disorder that causes the small blood vessels in skin, joints, intestines and kidneys to become inflamed and bleed | [76,81] |
| Aging | nuclear gene mutation | Accumulation of biological changes leading to functional decrease in the organism | [76,82] |

2.1. Alzheimer’s Disease in Brief
2.2. Parkinson’s Disease in Brief
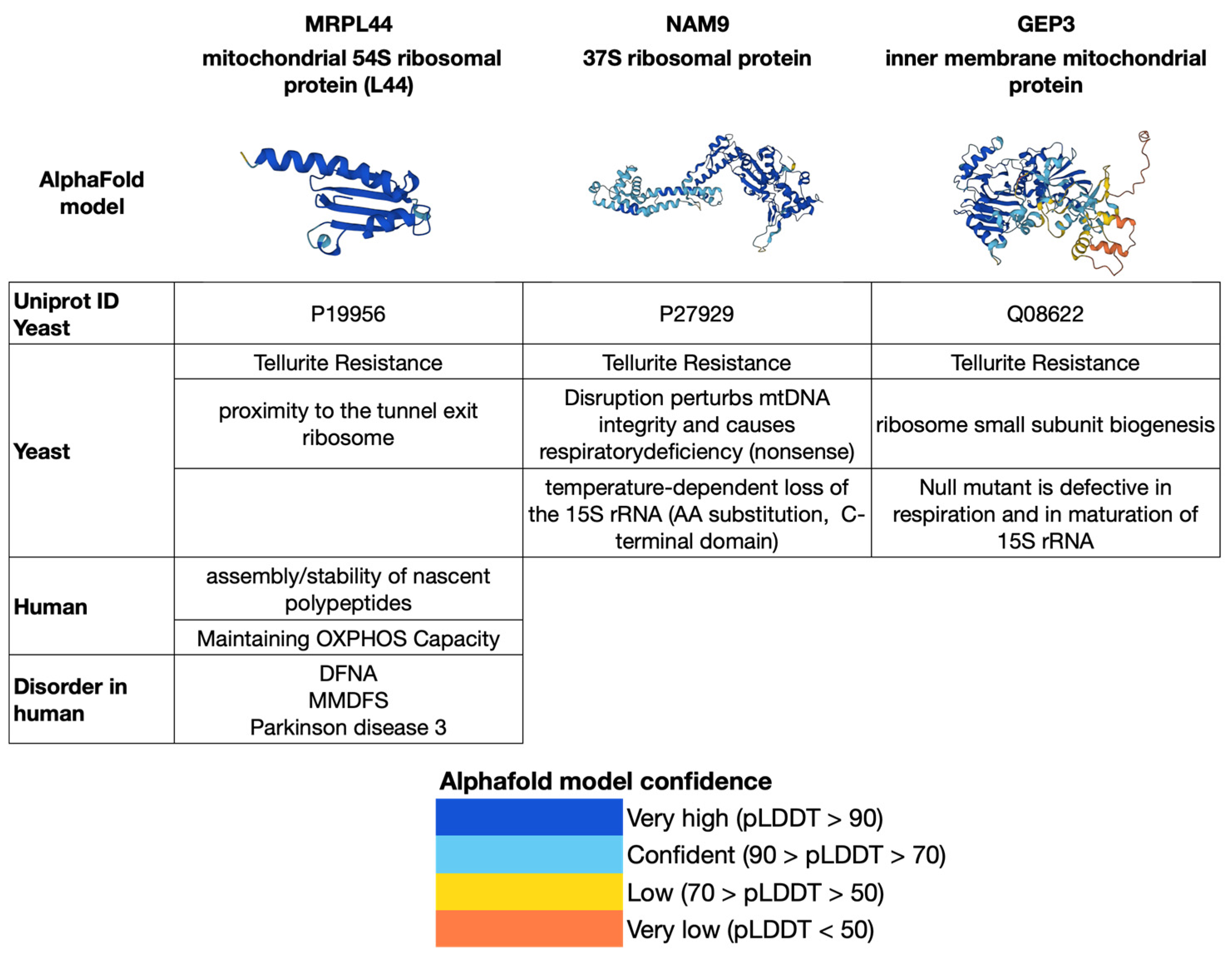
3. Mitochondrial Ribosomal Proteins Associated with Mitochondrial Neurodegenerative Diseases
4. Three Mitochondrial Ribosomal Proteins as Intermediate Stage in a Path Connected with Alzheimer’s and Parkinson’s
4.1. The Pathophysiology of Mitochondrial Diseases
4.2. The Connection between Mitochondrial Dysfunctions and the Effects of Heavy Metals and Metalloid Oxyanions
5. Experimental Hypothesis for the Isolation and Characterization of the Three Proteins MRPL44, NAM9 and GEP3
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luft, R.; Ikkos, D.; Palmieri, G.; Ernster, L.; Afzelius, B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: A correlated clinical, biochemical, and morphological study. J. Clin. Investig. 1962, 41, 1776–1804. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. The mitochondrial genome in human adaptive radiation and disease: On the road to therapeutics and performance enhancement. Gene 2005, 354, 169–180. [Google Scholar] [CrossRef]
- Longley, M.J.; Graziewicz, M.A.; Bienstock, R.J.; Copeland, W.C. Consequences of mutations in human DNA polymerase gamma. Gene 2005, 354, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Dujon, B. Mitochondrial genetics and functions. In Molecular Biology of the Yeast Saccharomyces cerevisiae: Life Cycle and Inheritance; Strathern, J.N., Jones, E.W., Broach, J.R., Eds.; Cold Spring Harbor Laboratory, Cold Spring Harbor: New York, NY, USA, 1981; pp. 505–635. [Google Scholar]
- Wolf, K.; Del Giudice, L. The variable mitochondrial genome of ascomycetes: Organization, mutational alterations, and expression. Adv. Genet. 1988, 25, 185–309. [Google Scholar]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Spinazzola, A.; Zeviani, M. Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J. Intern. Med. 2009, 265, 174–192. [Google Scholar] [CrossRef]
- Chen, C.; Chen, Y.; Guan, M.X. A peep into mitochondrial disorder: Multifaceted from mitochondrial DNA mutations to nuclear gene modulation. Protein Cell 2015, 6, 862–870. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, S.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Paul, G.K.; Chung, S.; Saleh, M.A.; Alshehri, S.; et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Curr. Issues Mol. Biol. 2022, 44, 1127–1148. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Scheibye-Alsing, K.; Canugovi, C.; Croteau, D.L.; Bohr, V.A. A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging 2013, 5, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP1 hyperactivation and NAD+/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, G.; Chinnery, P.F. Mitochondrial DNA polymerase gamma and human disease. Hum. Mol. Genet. 2006, 15, R244–R252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; Di Mauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Spinazzola, A.; Zeviani, M. Disorders of nuclear mitochondrial intergenomic signaling. Gene 2005, 354, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, P.F. Primary Mitochondrial Disorders Overview 1. In Clinical Characteristics of Mitochondrial Disorders; GeneReviews: Seattle, DC, USA, 2021; pp. 1–16. [Google Scholar]
- Singh, K.K. Mitochondrial dysfunction is a common phenotype in aging and cancer. Ann. N. Y. Acad. Sci. 2004, 1019, 260–264. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Will, Y.; Dykens, J.A. Mitochondrial Dysfunction by Drug and Environmental Toxicants; John Wiley & Sons Limited: New York, NY, USA, 2018. [Google Scholar]
- Meyer, J.N.; Hartman, J.H.; Mello, D.F. Mitochondrial Toxicity. Toxicol. Sci. 2018, 162, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodičková, A.; Koren, S.A.; Wojtovich, A.P. Site-specific mitochondrial dysfunction in neurodegeneration. Mitochondrion 2022, 64, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 122–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial diseases of the Brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Mochly-Rosen, D. Mortal engines: Mitochondrial bioenergetics and dysfunction in neurodegenerative diseases. Pharmacol. Res. 2018, 138, 2–15. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Lu, M.H.; Yuan, D.J.; Xu, D.E.; Yao, P.P.; Ji, W.L.; Chen, H.; Liu, W.L.; Yan, C.X.; Xia, Y.Y.; et al. Mitochondrial Dysfunction in Neural Injury. Front. Neurosci. 2019, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Ferramosca, A. Mitochondrial Protein Network: From Biogenesis to Bioenergetics in Health and Disease. Int. J. Mol. Sci. 2021, 22, 1. [Google Scholar] [CrossRef]
- Kreimendahl, S.; Schwichtenberg, J.; Günnewig, K.; Brandherm, L.; Rassow, J. The selectivity filter of the mitochondrial protein import machinery. J. BMC Biol. 2020, 18, 156. [Google Scholar] [CrossRef]
- Gupta, A.; Becker, T. Mechanisms and pathways of mitochondrial outer membrane protein biogenesis. Biochim. Biophys. Acta Bioenerg. 2020, 1862, 148323. [Google Scholar] [CrossRef]
- Horten, P.; Colina-Tenorio, L.; Rampelt, H. Biogenesis of Mitochondrial Metabolite Carriers. Biomolecules 2020, 10, 1008. [Google Scholar] [CrossRef]
- Mokranjac, D. How to get to the other side of the mitochondrial inner membrane—The protein import motor. Biol. Chem. 2020, 401, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, J.E.; Fischel-Ghodsian, N.; Mougey, E.B.; O’Brien, T.W. Mitochondrial ribosomal proteins: Candidate genes for mitochondrial disease. Genet. Med. 2004, 6, 73–80. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, T.W.; O’Brien, B.J.; Norman, R.A. Nuclear MRP genes and mitochondrial disease. Gene 2005, 354, 147–151. [Google Scholar] [CrossRef] [PubMed]
- De Silva, D.; Tu, Y.-T.; Amunts, A.; Fontanesi, F.; Barrientos, A. Mitochondrial ribosome assembly in health and disease. Cell Cycle 2015, 14, 2226–2250. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514, Erratum in Nature 2017, 551, 398. [Google Scholar] [CrossRef] [Green Version]
- Pontieri, P.; Hartings, H.; Salvo, M.D.; Massardo, D.R.; Stefano, M.D.; Pizzolante, G.; Romano, R.; Troisi, J.; Giudice, A.D.; Alifano, P.; et al. Mitochondrial ribosomal proteins involved in tellurite resistance in yeast Saccharomyces cerevisiae. Sci. Rep. 2018, 8, 12022. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, L.; Alifano, P.; Calcagnile, M.; Di Schiavi, E.; Bertapelle, C.; Aletta, M.; Pontieri, P. Mitochondrial ribosomal protein genes connected with Alzheimer’s and tellurite toxicity. Mitochondrion 2022, 64, 45–58. [Google Scholar] [CrossRef]
- Chinnery, P.; Howell, N.; Andrews, R.; Turnbull, D. Clinical mitochondrial genetics. J. Med. Genet. 1999, 36, 425–436. [Google Scholar]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.J.; Crameri, J.J.; Thorburn, D.R.; Frazier, A.E.; Stojanovski, D. Mitochondrial biology and dysfunction in secondary mitochondrial disease. Open Biol. 2022, 12, 220274. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Baker, B.M.; Haynes, C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011, 36, 254–261. [Google Scholar] [CrossRef]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [Green Version]
- Holt, I.; Harding, A.E.; Morgan-Hughes, J.A. Deletion of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Birtel, J.; von Landenberg, C.; Gliem, M.; Gliem, C.; Reimann, J.; Kunz, W.S.; Herrmann, P.; Betz, C.; Caswell, R.; Nesbitt, V.; et al. Mitochondrial Retinopathy. Ophthalmol. Retin. 2022, 6, 65–79. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.; Sharma, T. Mitochondrial Disorder: Kearns-Sayre Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 161–162. [Google Scholar]
- Zeviani, M.; Carelli, V. Mitochondrial Retinopathies. Int. J. Mol. Sci. 2022, 23, 210. [Google Scholar] [CrossRef] [PubMed]
- Cormier, V.; Rötig, A.; Quartino, A.R.; Forni, G.L.; Cerone, R.; Maier, M.; Saudubray, J.-M.; Munnich, A. Widespread multitissue deletions of the mitochondrial genome in Pearsons marrow-pancreas syndrome. J. Pediatr. 1990, 117, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Ishikawa, K.; Niemeyer, C.; Grünert, S.C. Pearson syndrome: A multisystem mitochondrial disease with bone marrow failure. Orphanet J. Rare Dis. 2022, 17, 379. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Shoffner, J.M.; Hedaya, E.V.; Trounce, I.; Polak, M.A.; Koontz, D.A.; Wallace, D.C. Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat. Genet. 1992, 1, 11–15. [Google Scholar] [CrossRef]
- Yang, M.; Xu, L.; Xu, C.; Cui, Y.; Jiang, S.; Dong, J.; Liao, L. The Mutations and Clinical Variability in Maternally Inherited Diabetes and Deafness: An Analysis of 161 Patients. Front. Endocrinol. 2021, 12, 728043. [Google Scholar] [CrossRef]
- Chalmers, R.M.; Brockington, M.; Howard, R.S.; Lecky, B.R.F.; Morgan-Hughes, J.A.; Harding, A.E. Mitochondrial encephalopathy with multiple mitochondrial DNA deletions. J. Neurol. Sci. 1996, 143, 41–45. [Google Scholar] [CrossRef]
- Borgione, E.; Giudice, M.L.; Paola, S.S.; Giuliano, M.; Di Blasi, F.D.; Di Stefano, V.; Lupica, A.; Brighina, F.; Pettinato, R.; Romano, C.; et al. The Mitochondrial tRNASer(UCN) Gene: A Novel m.7484A>G Mutation Associated with Mitochondrial Encephalomyopathy and Literature Review. Life 2023, 13, 554. [Google Scholar] [CrossRef]
- Ohno, K.; Tanaka, M.; Sahashi, K. Mitochondrial DNA deletions in inherited recurrent myoglobinuria. Ann. Neurol. 1991, 29, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Siciliani Scalco, R.; Gardiner, A.R.; Pitceathly, R.D.; Zanoteli, E.; Becker, J.; Holton, J.L.; Houlden, H.; Jungbluth, H.; Quinlivan, R. Rhabdomyolysis: A genetic perspective. Orphanet J. Rare Dis. 2015, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Fadic, R.; Russell, J.A.; Vedanarayanan, V.V.; Lehar, M.; Kuncl, R.W.; Johns, D.R. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology 1997, 49, 239–245. [Google Scholar] [CrossRef]
- Okun, M.S.; Bhatti, M.T. SANDO: Another presentation of mitochondrial disease. Am. J. Ophthalmol. 2004, 137, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy. 26 October 2000; Updated 11 March 2021. In GeneReviews® Internet; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1174/ (accessed on 23 February 2023).
- Manfredi, G.; Schon, E.A.; Moraes, C.T.; Bonilla, E.; Berry, G.T.; Sladky, J.T.; DiMauro, S. A new mutation associated with MELAS is located in a mitochondrial DNA polypeptide-coding gene. Neuromuscul. Disord. 1995, 5, 391–398. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Ng, Y.S.; Errington, L.; Blain, A.P.; McFarland, R.; Gorman, G.S. L-Arginine in Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes. A Systematic Review. Neurology 2022, 98, e2318–e2328. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, D.; Shanske, S.; Vazquez-Memije, M.; De Vivo, D.; DiMauro, S. A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann. Neurol. 1995, 38, 468–472. [Google Scholar] [CrossRef]
- Finsterer, J. Neuropathy, Ataxia, and Retinitis Pigmentosa Syndrome. J. Clin. Neuromuscul. Dis. 2023, 24, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Velez-Bartolomei, F.; Lee, C.; Enns, G. GeneReviews® Internet; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Taylor, R.W.; Chinnery, P.F.; Bates, M.J.; Jackson, M.J.; Johnson, M.A.; Andrews, R.M.; Turnbull, D.M. A novel mitochondrial DNA point mutation in the tRNA(Ile) gene: Studies in a patient presenting with chronic progressive external ophthalmoplegia and multiple sclerosis. Biochem. Biophys. Res. Commun. 1998, 243, 47–51. [Google Scholar] [CrossRef]
- Chalmers, R.M.; Lamont, P.J.; Nelson, I.; Ellison, D.W.; Thomas, N.H.; Harding, A.E.; Hammans, S.R. A mitochondrial DNA tRNA(Val) point mutation associated with adult-onset Leigh syndrome. Neurology 1997, 49, 589–592. [Google Scholar] [CrossRef]
- Rahman, S. Leigh syndrome. Handb. Clin. Neurol. 2023, 194, 43–63. [Google Scholar]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef]
- Jia, F.; Fellner, A.; Kumar, K.R. Monogenic Parkinson′s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes 2022, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Keita, M.; McIntyre, K.; Rodden, L.N.; Schadt, K.; Lynch, D.R. Friedreich ataxia: Clinical features and new developments. Neurodegener. Dis. Manag. 2022, 12, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington′s disease: Diagnosis and management. Pract. Neurol. 2022, 22, 32–41. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Hetland, L.E.; Susrud, K.S.; Lindahl, K.H.; Bygum, A. Henoch-Schönlein Purpura: A Literature Review. Acta Derm. Venereol. 2017, 97, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement 2013, 9, 63–75. [Google Scholar] [CrossRef]
- Prince, M.; Guerchet, M.; Prina, M. The Epidemiology and Impact of Dementia: Current State and Future Trends. 2015. Available online: http://www.who.int/mental_health/neurology/en/ (accessed on 26 March 2015).
- Norfray, J.F.; Provenzale, J.M. Alzheimer’s Disease: Neuropathologic Findings and Recent Advances in Imaging. Am. J. Roentgenol. 2004, 182, 3–13. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [Green Version]
- Seynnaeve, D.; Del Vecchio, M.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; IWinderickx, J.; Franssens, V. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. Int. J. Mol. Sci. 2018, 19, 1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer′s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Sarma, P.; Medhi, B.; Prakash, A.; Reddy, D.H. Recent Advances in Molecular Pathways and Therapeutic Implications Targeting Mitochondrial Dysfunction for Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 535–555. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimers 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, V.A.; Covache-Busuioc, R.A.; Mohan, A.G.; Costin, H.P.; Voicu, V. Alzheimer’s disease: 120 years of research and progress. J. Med. Life 2023, 16, 173–177. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson dis-ease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Surmeier, D.J. Determinants of Dopaminergic Neuron Loss in Parkinson′s Disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Checler, F.; Alves da Costa, C. Parkin as a Molecular Bridge Linking Alzheimer’s and Parkinson’s Diseases? Biomolecules 2022, 12, 559. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, N.; Dong, J.; Tian, W.; Chang, L.; Ma, J.; Guo, J.; Tan, J.; Dong, A.; He, K.; et al. Deficiency in endocannabinoid syn-thase DAGLB contributes to early onset Parkinsonism and murine nigral dopaminergic neuron dysfunction. Nat. Commun. 2022, 13, 3490. [Google Scholar] [CrossRef]
- Kumar, K.R.; Lohmann, K.; Klein, C. Genetics of Parkinson disease and other movement disorders. Curr. Opin. Neurol. 2012, 25, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Xiang, Y.; Song, T.; Zhao, Y.; Pan, H.; Xu, Q.; Chen, Y.; Sun, Q.; Wu, X.; Yan, X.; et al. Character-istics of fatigue in Parkinson’s disease: A longitudinal cohort study. Front. Aging Neurosci. 2023, 15, 1133705. [Google Scholar] [CrossRef] [PubMed]
- Scaltsoyiannes, V.; Corre, N.; Waltz, F.; Giegé, P. Types and Functions of Mitoribosome-Specific Ribosomal Proteins across Eukaryotes. Int. J. Mol. Sci. 2022, 23, 3474. [Google Scholar] [CrossRef]
- Sharma, M.R.; Booth, T.M.; Simpson, L.; Maslov, D.A.; Agrawal, R.K. Structure of a mitochondrial ribosome with minimal RNA. Proc. Natl. Acad. Sci. USA 2009, 106, 9637–9642. [Google Scholar] [CrossRef]
- Greber, B.J.; Bieri, P.; Leibundgut, M.; Leitner, A.; Aebersold, R.; Boehringer, D.; Ban, N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.J.; Ban, N. Structure and Function of the Mitochondrial Ribosome. Annu. Rev. Biochem. 2016, 85, 103–132. [Google Scholar] [CrossRef]
- Mai, N.; Chrzanowska-Lightowlers, Z.M.A.; Lightowlers, R.N. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 2017, 367, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Del’olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Horvath, R. Mitochondria: Impaired mitochondrial translation in human disease. Int. J. Biochem. Cell Biol. 2014, 48, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotig, A. Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta 2011, 1807, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar]
- Lopez Sanchez, M.I.G.; Krüger, A.; Shiriaev, D.I.; Liu, Y.; Rorbach, J. Human Mitoribosome Biogenesis and Its Emerging Links to Disease. Int. J. Mol. Sci. 2021, 22, 3827. [Google Scholar] [CrossRef]
- Greber, B.J.; Boehringer, D.; Leibundgut, M.; Bieri, P.; Leitner, A.; Schmitz, N.; Aebersold, R.; Ban, N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 2014, 515, 283–286. [Google Scholar] [CrossRef] [Green Version]
- Lake, N.J.; Bird, M.J.; Isohanni, P.; Paetau, A. Leigh Syndrome: Neuropathology and Pathogenesis. J. Neuropathol. Exp. Neurol. 2015, 74, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell Death: Critical Control Points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Scarpelli, M.; Todeschini, A.; Volonghi, I.; Padovani, A.; Filosto, M. Mitochondrial diseases: Advances and issues. Appl. Clin. Genet. 2017, 10, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Filosto, M.; Mancuso, M. Mitochondrial diseases: A nosological update. Acta Neurol. Scand. 2007, 115, 211–221. [Google Scholar] [CrossRef]
- Nafisinia, M.; Guo, Y.; Dang, X.; Li, J.; Chen, Y.; Zhang, J.; Lake, N.J.; Gold, W.A.; Riley, L.G.; Thorburn, D.R.; et al. Whole Exome Sequencing Identifies the Genetic Basis of Late-Onset Leigh Syndrome in a Patient with MRI but Little Biochemical Evidence of a Mitochondrial Disorder. JIMD Rep. 2017, 32, 117–124. [Google Scholar]
- Taylor, S.W.; Fahy, E.; Zhang, B.; Glenn, G.M.; Warnock, D.E.; Wiley, S.; Murphy, A.N.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003, 21, 281–286. [Google Scholar] [CrossRef]
- Lescuyer, P.; Strub, J.M.; Luche, S.; Diemer, H.; Martinez, P.; Van Dorsselaer, A.; Lunardi, J.; Rabilloud, T. Progress in the defi-nition of a reference human mitochondrial proteome. Proteomics 2003, 3, 157–167. [Google Scholar] [CrossRef]
- Uechi, T.; Tanaka, T.; Kenmochi, N. A complete map of the human ribosomal proteingenes: Assignment of 80 genes to the cytogenetic map and implications for human disorders. Genomics 2001, 72, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, S.; Ben-Shem, A.; De Loubresse, N.G.; Jenner, L.; Yusupova, G.; Yusupov, M. One core, two shells: Bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. [Google Scholar] [CrossRef]
- Kressler, D.; Hurt, E.; Baßler, J. A Puzzle of Life: Crafting Ribosomal Subunits. Trends Biochem. Sci. 2017, 42, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Sissler, M.; Hashem, Y. Mitoribosome assembly comes into view. Nat. Struct. Mol. Biol. 2021, 28, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Robles, P.; Quesada, V. Emerging Roles of Mitochondrial Ribosomal Proteins in Plant Development. Int. J. Mol. Sci. 2017, 18, 2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- O′Brien, T.W. Properties of Human Mitochondrial Ribosomes. IUBMB Life 2003, 55, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Kenmochi, N.; Suzuki, T.; Uechi, T.; Magoori, M.; Kuniba, M.; Higa, S.; Watanabe, K.; Tanaka, T. The Human Mitochondrial Ribosomal Protein Genes: Mapping of 54 Genes to the Chromosomes and Implications for Human Disorders. Genomics 2001, 77, 65–70. [Google Scholar] [CrossRef]
- Yeo, J.H.C.; Skinner, J.P.J.; Bird, M.; Formosa, L.E.; Zhang, J.-G.; Kluck, R.M.; Belz, G.T.; Chong, M.M.W. A Role for the Mitochondrial Protein Mrpl44 in Maintaining OXPHOS Capacity. PLoS ONE 2015, 10, e0134326. [Google Scholar] [CrossRef] [PubMed]
- Verity, M.A. Environmental neurotoxicity of chemicals and radiation. Curr. Opin. Neurol. Neurosurg. 1993, 6, 437–442. [Google Scholar] [PubMed]
- Larner, A.J. Alzheimer’s disease, Kuf ’s disease, tellurium and selenium. Med. Hypotheses 1996, 47, 73–75. [Google Scholar] [CrossRef]
- Larner, A.J. Biological effects of tellurium, A review. Trace Elem. Electrolytes 1995, 12, 26–31. [Google Scholar]
- Pontieri, P.; De Stefano, M.; Massardo, D.R.; Gunge, N.; Miyakawa, I.; Sando, N.; Pignone, D.; Pizzolante, G.; Romano, R.; Alifano, P.; et al. Tellurium as a valuable tool for studying the prokaryotic origins of mitochondria. Gene 2015, 559, 177–183. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Giudice, L.; Pontieri, P.; Aletta, M.; Calcagnile, M. Mitochondrial Neurodegenerative Diseases: Three Mitochondrial Ribosomal Proteins as Intermediate Stage in the Pathway That Associates Damaged Genes with Alzheimer’s and Parkinson’s. Biology 2023, 12, 972. https://doi.org/10.3390/biology12070972
Del Giudice L, Pontieri P, Aletta M, Calcagnile M. Mitochondrial Neurodegenerative Diseases: Three Mitochondrial Ribosomal Proteins as Intermediate Stage in the Pathway That Associates Damaged Genes with Alzheimer’s and Parkinson’s. Biology. 2023; 12(7):972. https://doi.org/10.3390/biology12070972
Chicago/Turabian StyleDel Giudice, Luigi, Paola Pontieri, Mariarosaria Aletta, and Matteo Calcagnile. 2023. "Mitochondrial Neurodegenerative Diseases: Three Mitochondrial Ribosomal Proteins as Intermediate Stage in the Pathway That Associates Damaged Genes with Alzheimer’s and Parkinson’s" Biology 12, no. 7: 972. https://doi.org/10.3390/biology12070972